Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer

 and
and 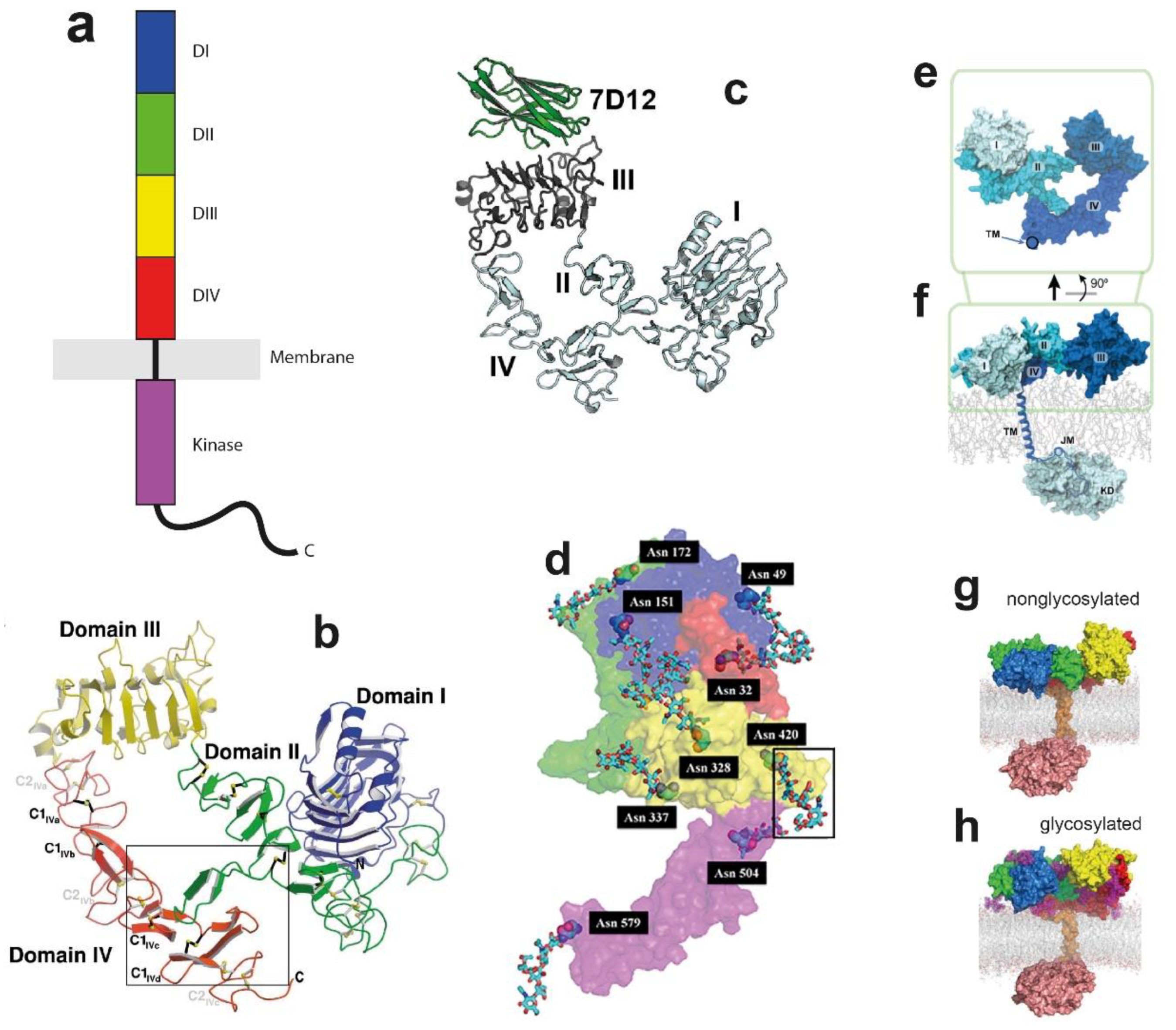{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. EGFR and Its Connection with Oncogenic Cell Growth
1.2. A Brief Description of Methods to Derive EGFR Structural Properties
1.3. MD Simulation Methods Applied to EGFR Research
2. The Glycosylated EGFR Monomer and Interactions with the Supporting Bilayer
2.1. The Ligand-Binding ECM Monomer
2.2. Glycosylation Regulates the Structure and Function of the ECM
2.3. Linking Across the Lipid Bilayer to the ICM
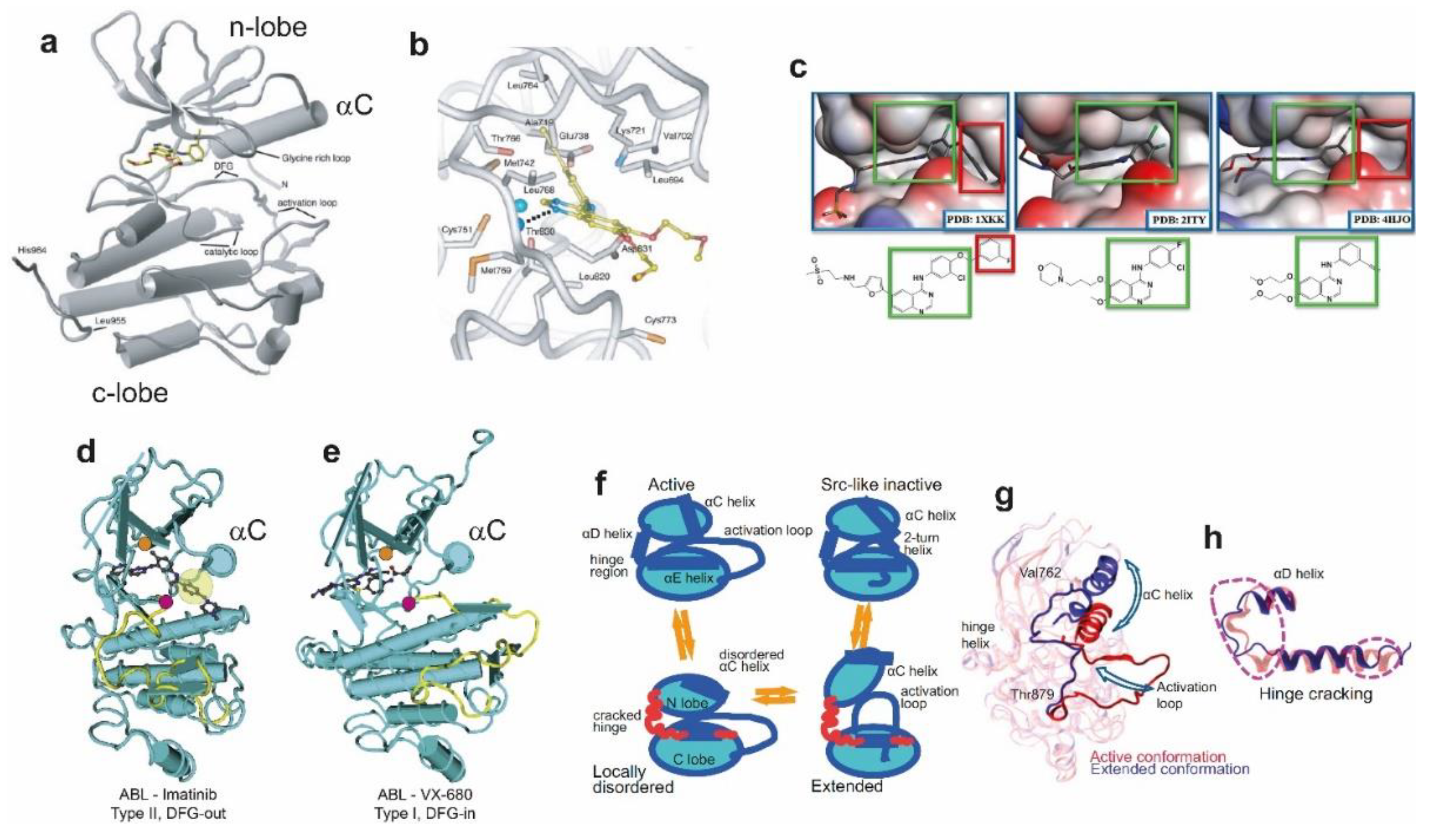
3. Kinase Domain Conformations and Their Coupling to Tyrosine Kinase Inhibitors (TKIs)
3.1. Topology of the Catalytic Kinase Domain
3.2. TKIs Bind the ATP-Binding Kinase Pocket and Stabilise Active and Inactive Conformations
3.3. NSCLC Oncogenic Mutations and Their Impact on Structure and TKI Interactions
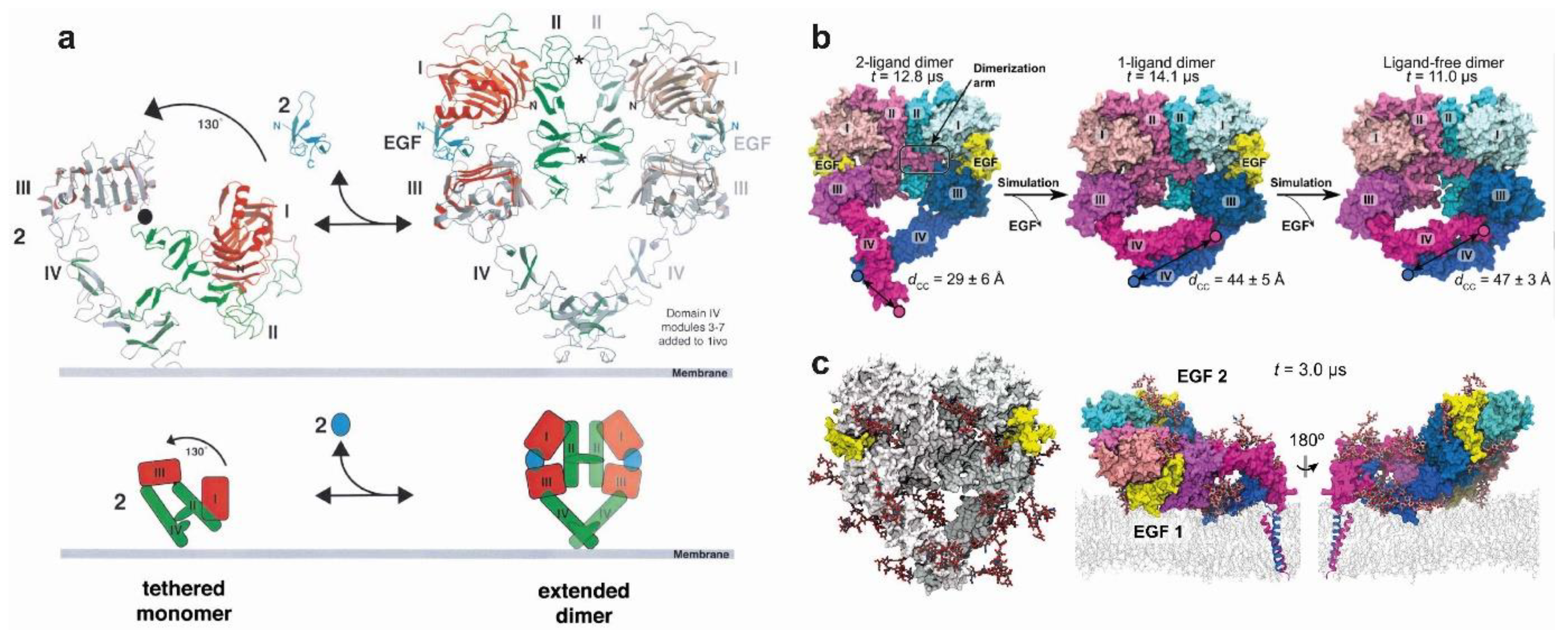
4. Regulation of EGFR Enzymatic Activation by Ligand-Induced Dimerisation
4.1. The Stable Heart-Shape of the Ligand-Bound Extracellular Dimer Module
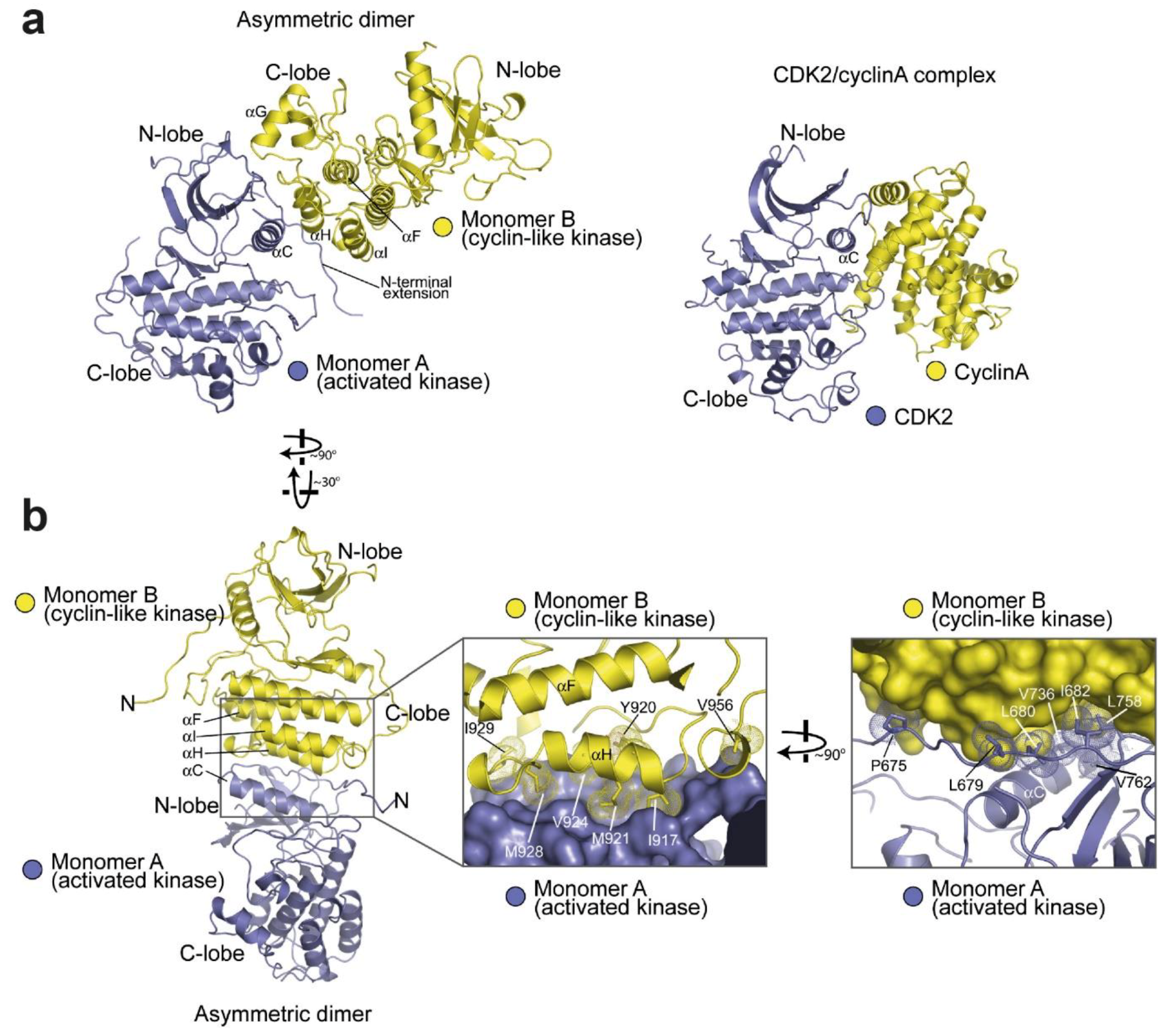
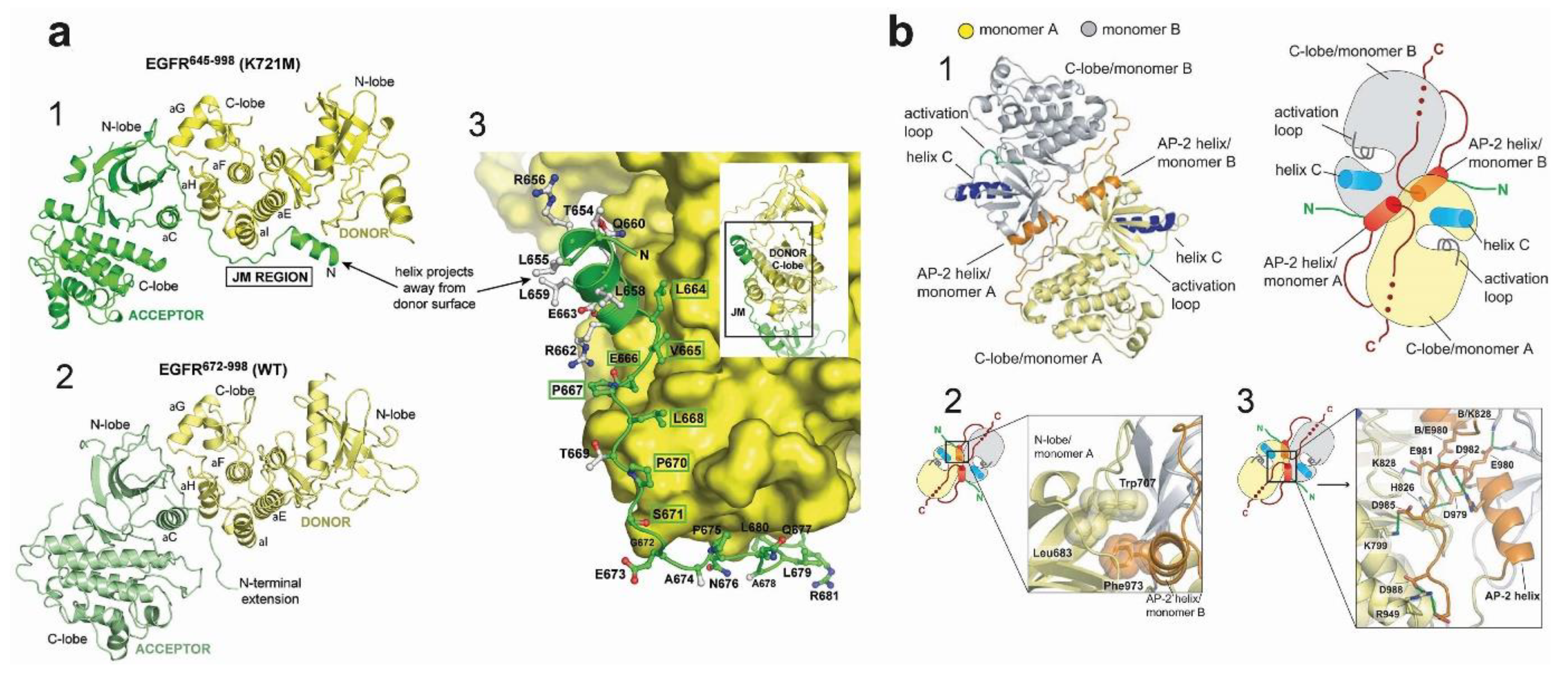
4.2. Active Kinase Dimers and Structural Cross-Talk Across the Asymmetric Interface
4.3. Active and Inactive Kinase Dimers and Structural Cross-Talk with the TM, JM and C-Terminal Tail Domains
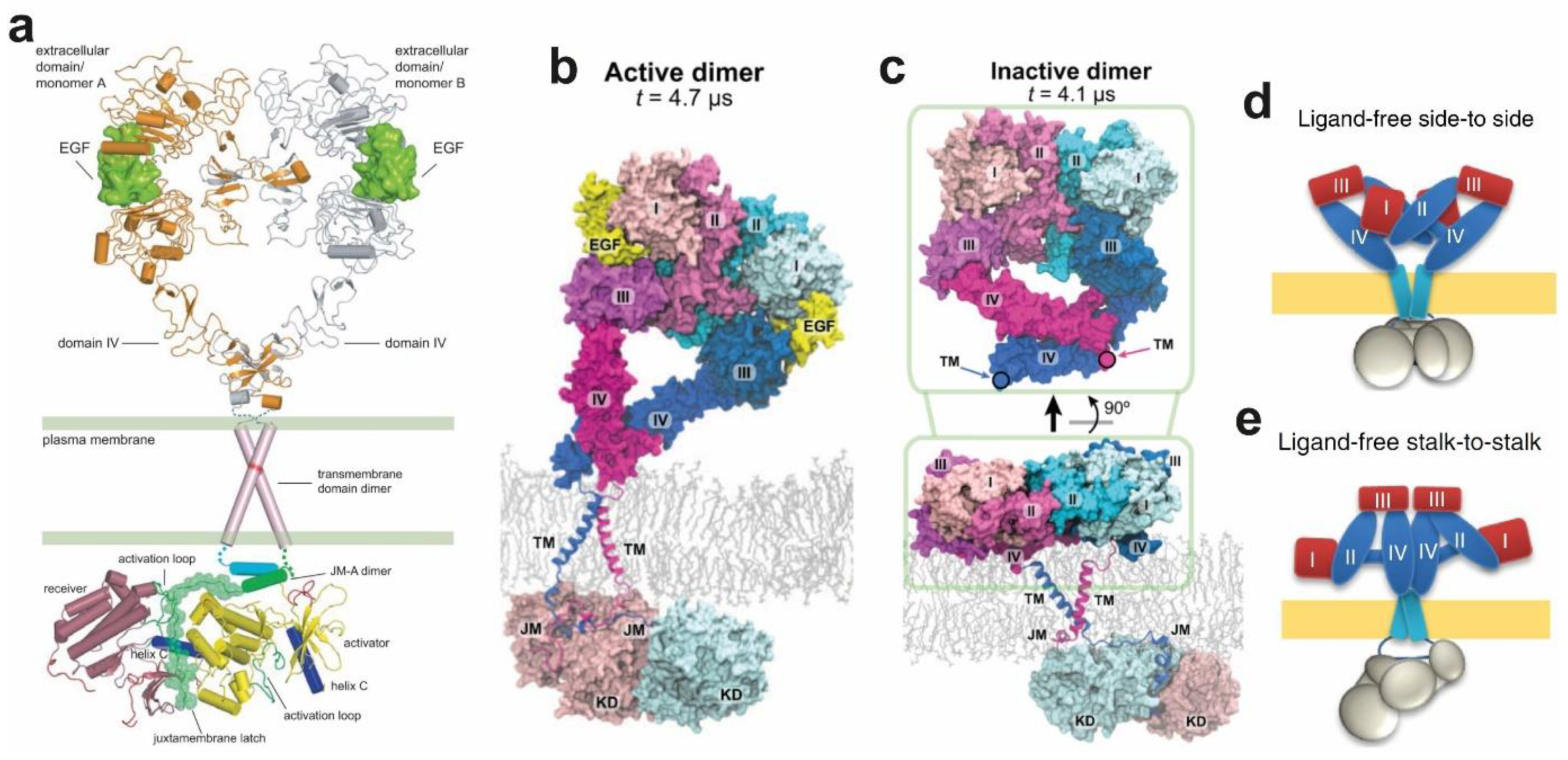
4.4. Structural Coupling Across the Plasma Membrane in Ligand-Bound Active Dimers
4.5. Structural Coupling Across the Plasma Membrane in Ligand-Free Inactive Dimers
4.6. Autoinhibition Mechanisms in Ligand-Free Dimers
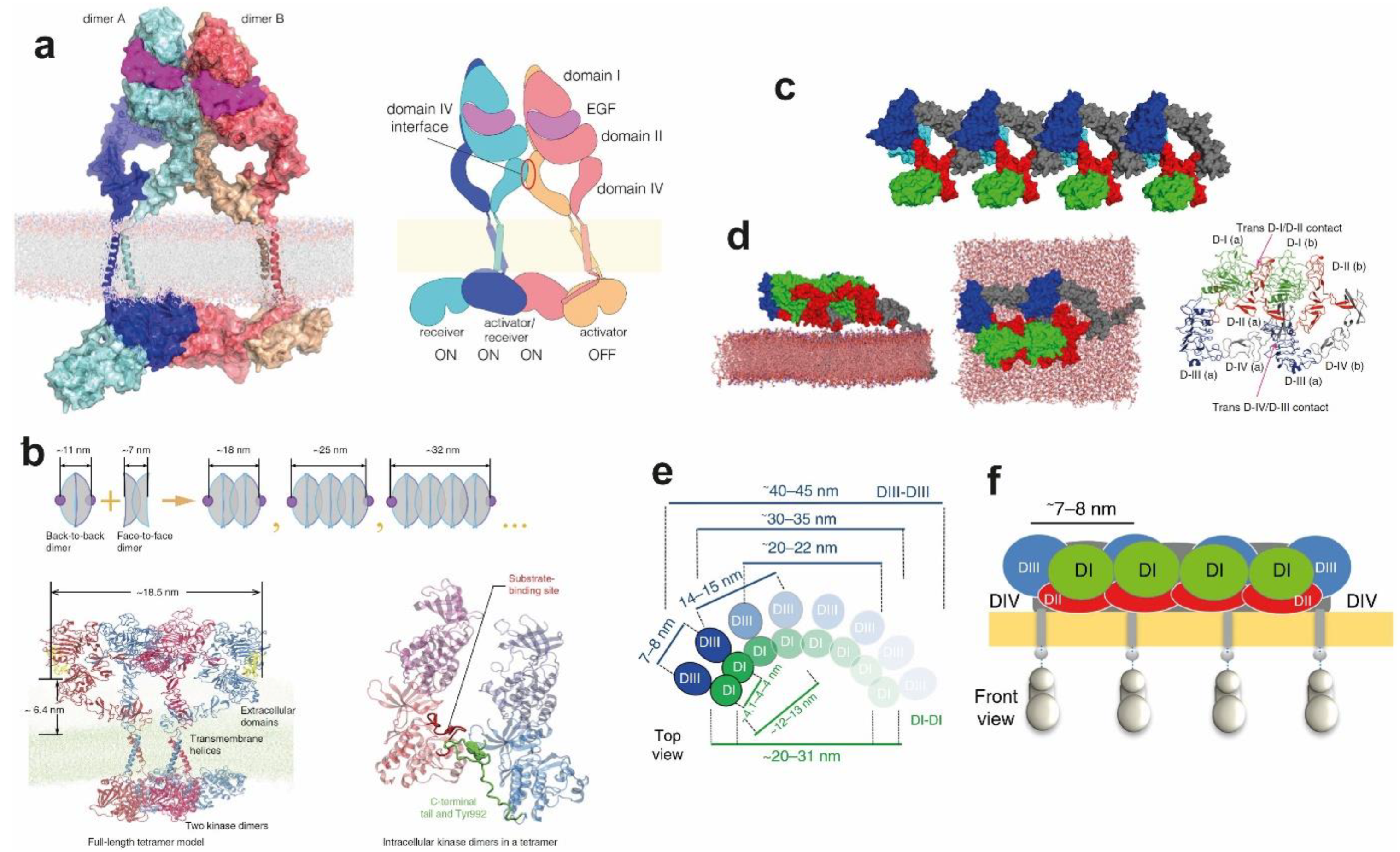
5. Ligand-Free and Ligand-Bound Multimeric EGFR Assemblies
6. Conclusions
Funding
Conflicts of Interest
References
- Cohen, S. Origins of growth factors: NGF and EGF. J. Biol. Chem. 2008, 283, 33793–33797. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; King, L.; Cohen, S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 1978, 276, 409–410. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, S.L.J.; Mac, A.S.W.; Henriksen, L.; van Deurs, B.; Grøvdal, L.M. EGFR signaling patterns are regulated by its different ligands. Growth Factors 2014, 32, 155–163. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef]
- Adrain, C.; Freeman, M. Regulation of receptor tyrosine kinase ligand processing. Cold Spring Harb. Perspect. Biol. 2014, 6, a008995. [Google Scholar] [CrossRef]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 1–13. [Google Scholar] [CrossRef]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5, 2270. [Google Scholar]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Downward, J.; Parker, P.; Waterfield, M.D. Autophosphorylation sites on the epidermal growth factor receptor. Nature 1984, 311, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; De Luca, A.; Caponigro, F.; Salomon, D.S. The ErbB receptors and their ligands in cancer: An overview. Curr. Drug Targets 2005, 6, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.S.; Lao, X.Q.; Ho, K.-F.; Goggins, W.B.; Tse, S.L.A. Incidence and mortality of lung cancer: Global trends and association with socioeconomic status. Sci. Rep. 2017, 7, 14300. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Marks, J.L.; Broderick, S.; Zhou, Q.; Chitale, D.; Li, A.R.; Zakowski, M.F.; Kris, M.G.; Rusch, V.W.; Azzoli, C.G.; Seshan, V.E.; et al. Prognostic and Therapeutic Implications of EGFR and KRAS Mutations in Resected Lung Adenocarcinoma. J. Thorac. Oncol. 2008, 3, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.-Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients With Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients With Non–Small Cell Lung Cancer. JAMA 2003, 290, 2149–2158. [Google Scholar]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in Previously Treated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Akram, A.; Khalil, S.; Halim, S.A.; Younas, H.; Iqbal, S.; Mehar, S. Therapeutic Uses of HSP90 Inhibitors in Non-Small Cell Lung Carcinoma (NSCLC). Curr. Drug Metab. 2018, 19, 335–341. [Google Scholar] [CrossRef]
- Ferraro, M.; D’Annessa, I.; Moroni, E.; Morra, G.; Paladino, A.; Rinaldi, S.; Compostella, F.; Colombo, G. Allosteric Modulators of HSP90 and HSP70: Dynamics Meets Function through Structure-Based Drug Design. J. Med. Chem. 2019, 62, 60–87. [Google Scholar] [CrossRef]
- Paladino, A.; Marchetti, F.; Ponzoni, L.; Colombo, G. The Interplay between Structural Stability and Plasticity Determines Mutation Profiles and Chaperone Dependence in Protein Kinases. J. Chem. Theory Comput. 2018, 14, 1059–1070. [Google Scholar] [CrossRef]
- Verba, K.A.; Agard, D.A. How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem. Sci. 2017, 42, 799–811. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Ahsan, A.; Ramanand, S.G.; Whitehead, C.; Hiniker, S.M.; Rehemtulla, A.; Pratt, W.B.; Jolly, S.; Gouveia, C.; Truong, K.; Van Waes, C.; et al. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 2012, 14, 670–677. [Google Scholar] [CrossRef]
- Protein Crystallography—Methods and Protocols; Wlodawer, A.; Dauter, Z.; Jakolski, M. (Eds.) Humana Press: New York, NY, USA, 2017; ISBN 9781493969982. [Google Scholar]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Littlefield, P.; Liu, L.; Mysore, V.; Shan, Y.; Shaw, D.E.; Jura, N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci. Signal. 2014, 7, ra114. [Google Scholar] [CrossRef]
- Shi, F.; Telesco, S.E.; Liu, Y.; Radhakrishnan, R.; Lemmon, M. A ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 7692–7697. [Google Scholar] [CrossRef]
- Jura, N.; Shan, Y.; Cao, X.; Shaw, D.E.; Kuriyan, J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. USA 2009, 106, 21608–21613. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Zhang, X.; Pickin, K.A.; Bose, R.; Jura, N.; Cole, P.A.; Kuriyan, J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 2007, 450, 741–744. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Sutto, L.; Saladino, G.; Nebreda, A.R.; Gervasio, F.L.; Orozco, M. Changes in the free-energy landscape of p38α MAP kinase through its canonical activation and binding events as studied by enhanced molecular dynamics simulations. Elife 2017, 6, e22175. [Google Scholar] [CrossRef]
- Opella, S.J. Solid-state NMR and membrane proteins. J. Magn. Reson. 2015, 253, 129–137. [Google Scholar] [CrossRef]
- Ghose, R. (Ed.) Protein NMR; Humana Press: New York, NY, USA, 2018; ISBN 978-1-4939-7386-6. [Google Scholar]
- Chait, B.T.; Cadene, M.; Olinares, P.D.; Rout, M.P.; Shi, Y. Revealing Higher Order Protein Structure Using Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 952–965. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef]
- Cheng, Y.; Grigorieff, N.; Penczek, P.A.; Walz, T. A primer to single-particle cryo-electron microscopy. Cell 2015, 161, 438–449. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Kim, E.T.; Shaw, D.E. Membrane interaction of bound ligands contributes to the negative binding cooperativity of the EGF receptor. PLoS Comput. Biol. 2014, 10, e1003742. [Google Scholar] [CrossRef]
- Stryer, L.; Haugland, R. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar] [CrossRef]
- Chen, L.-C.; Lloyd, W.R.; Chang, C.-W.; Sud, D.; Mycek, M.-A. Fluorescence Lifetime Imaging Microscopy for Quantitative Biological Imaging. In Methods in cell biolog; 2013; Volume 114, pp. 457–488. [Google Scholar]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Korovesis, D.; Needham, S.R.; Tynan, C.J.; Sagawa, S.; Roberts, S.K.; Kuzmanic, A.; Ortiz-Zapater, E.; Jain, P.; Roovers, R.C.; et al. The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nat. Commun. 2018, 9, 4325. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- Gelpi, J.; Hospital, A.; Goñi, R.; Orozco, M. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinforma. Chem. 2015, 8, 37. [Google Scholar] [CrossRef]
- Hermans, J.; Berendsen, H.J.C.; Van Gunsteren, W.F.; Postma, J.P.M. A consistent empirical potential for water-protein interactions. Biopolymers 1984, 23, 1513–1518. [Google Scholar] [CrossRef]
- Ott, K.-H.; Meyer, B. Parametrization of GROMOS force field for oligosaccharides and assessment of efficiency of molecular dynamics simulations. J. Comput. Chem. 1996, 17, 1068–1084. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Wiorkiewicz-Kuczera, J.; Karplus, M. An all-atom empirical energy function for the simulation of nucleic acids. J. Am. Chem. Soc. 1995, 117, 11946–11975. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC14: International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, Louisana, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Cavalli, A.; Spitaleri, A.; Saladino, G.; Gervasio, F.L. Investigating Drug–Target Association and Dissociation Mechanisms Using Metadynamics-Based Algorithms. Acc. Chem. Res. 2015, 48, 277–285. [Google Scholar] [CrossRef]
- Saladino, G.; Estarellas, C.; Gervasio, F.L. Recent Progress in Free Energy Methods. Compr. Med. Chem. III 2017, 34–50. [Google Scholar]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Reports Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Dellago, C.; Bolhuis, P.G. Transition Path Sampling and Other Advanced Simulation Techniques for Rare Events. In Advanced Computer Simulation Approaches for Soft Matter Sciences III; Springer: Berlin/Heidelberg, Germany, 2009; pp. 167–233. [Google Scholar]
- Sutto, L.; Marsili, S.; Gervasio, F.L. New advances in metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 771–779. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Das, R.; Endres, N.F.; Eastwood, M.P.; Wemmer, D.E.; Kuriyan, J.; Shaw, D.E. Architecture and Membrane Interactions of the EGF Receptor. Cell 2013, 152, 557–569. [Google Scholar] [CrossRef]
- Pan, A.C.; Weinreich, T.M.; Piana, S.; Shaw, D.E. Demonstrating an Order-of-Magnitude Sampling Enhancement in Molecular Dynamics Simulations of Complex Protein Systems. J. Chem. Theory Comput. 2016, 12, 1360–1367. [Google Scholar] [CrossRef]
- Free Energy Calculations; Chipot, C.; Pohorille, A. (Eds.) Springer: Berlin/Heidelberg, Germany, 2007; ISBN 978-3-540-38447-2. [Google Scholar]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef]
- Perez, A.; Morrone, J.A.; Simmerling, C.; Dill, K.A. Advances in free-energy-based simulations of protein folding and ligand binding. Curr. Opin. Struct. Biol. 2016, 36, 25–31. [Google Scholar] [CrossRef]
- Banavali, N.K.; Roux, B. Anatomy of a structural pathway for activation of the catalytic domain of Src kinase Hck. Proteins Struct. Funct. Bioinforma. 2007, 67, 1096–1112. [Google Scholar] [CrossRef]
- Meng, Y.; Roux, B. Locking the Active Conformation of c-Src Kinase through the Phosphorylation of the Activation Loop. J. Mol. Biol. 2014, 426, 423–435. [Google Scholar] [CrossRef]
- Berteotti, A.; Cavalli, A.; Branduardi, D.; Gervasio, F.L.; Recanatini, M.; Parrinello, M. Protein Conformational Transitions: The Closure Mechanism of a Kinase Explored by Atomistic Simulations. J. Am. Chem. Soc. 2009, 131, 244–250. [Google Scholar] [CrossRef]
- Sultan, M.M.; Kiss, G.; Pande, V.S. Towards simple kinetic models of functional dynamics for a kinase subfamily. Nat. Chem. 2018, 10, 903–909. [Google Scholar] [CrossRef]
- Bussi, G.; Gervasio, F.L.; Laio, A.; Parrinello, M. Free-Energy Landscape for β Hairpin Folding from Combined Parallel Tempering and Metadynamics. J Am Chem Soc 2006, 128, 13435–13441. [Google Scholar] [CrossRef]
- Bunney, T.D.; Wan, S.; Thiyagarajan, N.; Sutto, L.; Williams, S.V.; Ashford, P.; Koss, H.; Knowles, M.A.; Gervasio, F.L.; Coveney, P.V.; et al. The Effect of Mutations on Drug Sensitivity and Kinase Activity of Fibroblast Growth Factor Receptors: A Combined Experimental and Theoretical Study. EBioMedicine 2015, 2, 194–204. [Google Scholar] [CrossRef]
- Lovera, S.; Sutto, L.; Boubeva, R.; Scapozza, L.; Dölker, N.; Gervasio, F.L. The Different Flexibility of c-Src and c-Abl Kinases Regulates the Accessibility of a Druggable Inactive Conformation. J. Am. Chem. Soc. 2012, 134, 2496–2499. [Google Scholar] [CrossRef]
- Pucheta-Martínez, E.; Saladino, G.; Morando, M.A.; Martinez-Torrecuadrada, J.; Lelli, M.; Sutto, L.; D’Amelio, N.; Gervasio, F.L. An Allosteric Cross-Talk Between the Activation Loop and the ATP Binding Site Regulates the Activation of Src Kinase. Sci. Rep. 2016, 6, 24235. [Google Scholar] [CrossRef]
- Sutto, L.; Gervasio, F. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef]
- Marino, K.A.; Sutto, L.; Gervasio, F.L. The Effect of a Widespread Cancer-Causing Mutation on the Inactive to Active Dynamics of the B-Raf Kinase. J. Am. Chem. Soc. 2015, 137, 5280–5283. [Google Scholar] [CrossRef]
- Lovera, S.; Morando, M.; Pucheta-Martinez, E.; Martinez-Torrecuadrada, J.L.; Saladino, G.; Gervasio, F.L. Towards a Molecular Understanding of the Link between Imatinib Resistance and Kinase Conformational Dynamics. PLOS Comput. Biol. 2015, 11, e1004578. [Google Scholar] [CrossRef]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.-J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef]
- Lax, I.; Bellot, F.; Howk, R.; Ullrich, A.; Givol, D.; Schlessinger, J. Functional analysis of the ligand binding site of EGF-receptor utilizing chimeric chicken/human receptor molecules. EMBO J. 1989, 8, 421–427. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Bouyain, S.; Longo, P.A.; Li, S.; Ferguson, K.M.; Leahy, D.J. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 15024–15029. [Google Scholar] [CrossRef]
- Dawson, J.P.; Bu, Z.; Lemmon, M. A Ligand-induced structural transitions in ErbB receptor extracellular domains. Structure 2007, 15, 942–954. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; Van Bergen, P.M.P.; Ferguson, K.M. Structural Ealuation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure 2014, 21, 1214–1224. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-Based View of Epidermal Growth Factor Receptor Regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef]
- Mattoon, D.; Klein, P.; Lemmon, M.A.; Lax, I.; Schlessinger, J. The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function. Proc. Natl. Acad. Sci. USA 2004, 101, 923–928. [Google Scholar] [CrossRef]
- Özcan, F.; Klein, P.; Lemmon, M.A.; Lax, I.; Schlessinger, J. On the nature of low- and high-affinity EGF receptors on living cells. Proc. Nat. Acad. Sci. 2006, 103, 5735–5740. [Google Scholar]
- Azimzadeh Irani, M. Correlation between experimentally indicated and atomistically simulated roles of EGFR N-glycosylation. Mol. Simul. 2018, 44, 743–748. [Google Scholar] [CrossRef]
- Kaszuba, K.; Grzybek, M.; Orłowski, A.; Danne, R.; Róg, T.; Simons, K.; Coskun, Ü.; Vattulainen, I. N-Glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc. Natl. Acad. Sci. USA 2015, 112, 4334–4339. [Google Scholar] [CrossRef]
- Kaplan, M.; Narasimhan, S.; de Heus, C.; Mance, D.; van Doorn, S.; Houben, K.; Popov-Čeleketić, D.; Damman, R.; Katrukha, E.A.; Jain, P.; et al. EGFR Dynamics Change during Activation in Native Membranes as Revealed by NMR. Cell 2016, 167, 1241–1251. [Google Scholar] [CrossRef]
- Lu, C.; Mi, L.-Z.; Grey, M.J.; Zhu, J.; Graef, E.; Yokoyama, S.; Springer, T.A. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol. Cell. Biol. 2010, 30, 5432–5443. [Google Scholar] [CrossRef]
- Slieker, L.J.; Martensen, T.M.; Lane, M.D. Synthesis of epidermal growth factor receptor in human A431 cells. Glycosylation-dependent acquisition of ligand binding activity occurs post-translationally in the endoplasmic reticulum. J. Biol. Chem. 1986, 261, 15233–15241. [Google Scholar]
- Matsumoto, K.; Yokote, H.; Arao, T.; Maegawa, M.; Tanaka, K.; Fujita, Y.; Shimizu, C.; Hanafusa, T.; Fujiwara, Y.; Nishio, K. N -Glycan fucosylation of epidermal growth factor receptor modulates receptor activity and sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2008, 99, 1611–1617. [Google Scholar] [CrossRef]
- Takahashi, M.; Yokoe, S.; Asahi, M.; Lee, S.H.; Li, W.; Osumi, D.; Miyoshi, E.; Taniguchi, N. N-glycan of ErbB family plays a crucial role in dimer formation and tumor promotion. Biochim. Biophys. Acta 2008, 1780, 520–524. [Google Scholar] [CrossRef]
- Tsuda, T.; Ikeda, Y.; Taniguchi, N. The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J. Biol. Chem. 2000, 275, 21988–21994. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Liu, Y.-C.; Chen, N.-Y.; Tsai, C.-F.; Wang, Y.-T.; Chen, Y.-J.; Hsu, T.-L.; Yang, P.-C.; Wong, C.-H. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 6955–6960. [Google Scholar] [CrossRef]
- Ling, Y.-H.; Li, T.; Perez-Soler, R.; Haigentz, M. Activation of ER stress and inhibition of EGFR N-glycosylation by tunicamycin enhances susceptibility of human non-small cell lung cancer cells to erlotinib. Cancer Chemother. Pharmacol. 2009, 64, 539–548. [Google Scholar] [CrossRef]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F.; Gomes Ferreira, I.; Pucci, M.; Venturi, G.; Malagolini, N.; et al. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef]
- Mariño, K.; Bones, J.; Kattla, J.J.; Rudd, P.M. A systematic approach to protein glycosylation analysis: A path through the maze. Nat. Chem. Biol. 2010, 6, 713–723. [Google Scholar] [CrossRef]
- Taylor, E.S.; Pol-Fachin, L.; Lins, R.D.; Lower, S.K. Conformational stability of the epidermal growth factor (EGF) receptor as influenced by glycosylation, dimerization and EGF hormone binding. Proteins 2017, 85, 561–570. [Google Scholar] [CrossRef]
- Sato, C.; Kim, J.H.; Abe, Y.; Saito, K.; Yokoyama, S.; Kohda, D. Characterization of the N-oligosaccharides attached to the atypical Asn-X-Cys sequence of recombinant human epidermal growth factor receptor. J. Biochem. 2000, 127, 65–72. [Google Scholar] [CrossRef]
- Smith, K.D.; Davies, M.J.; Bailey, D.; Renouf, D.V.; Hounsell, E.F. Analysis of the glycosylation patterns of the extracellular domain of the epidermal growth factor receptor expressed in Chinese hamster ovary fibroblasts. Growth Factors 1996, 13, 121–132. [Google Scholar] [CrossRef]
- Xu, G.; Abad, M.C.; Connolly, P.J.; Neeper, M.P.; Struble, G.T.; Springer, B.A.; Emanuel, S.L.; Pandey, N.; Gruninger, R.H.; Adams, M.; et al. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4615–4619. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Yen, H.-Y.; Chen, C.-Y.; Chen, C.-H.; Cheng, P.-F.; Juan, Y.-H.; Chen, C.-H.; Khoo, K.-H.; Yu, C.-J.; Yang, P.-C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef]
- Zhen, Y.; Caprioli, R.M.; Staros, J.V. Characterization of Glycosylation Sites of the Epidermal Growth Factor Receptor †. Biochemistry 2003, 42, 5478–5492. [Google Scholar] [CrossRef]
- Whitson, K.B.; Whitson, S.R.; Red-brewer, M.L.; Mccoy, A.J.; Vitali, A.A.; Walker, F.; Johns, T.G.; Beth, A.H.; Staros, J.V. Functional Effects of Glycosylation at Asn-579 of the Epidermal Growth Factor. Biochemistry 2005, 44, 14920–14931. [Google Scholar] [CrossRef]
- Azimzadeh Irani, M.; Kannan, S.; Verma, C. Role of N-glycosylation in EGFR ectodomain ligand binding. Proteins Struct. Funct. Bioinforma. 2017, 85, 1529–1549. [Google Scholar] [CrossRef]
- Ziomkiewicz, I.; Loman, A.; Klement, R.; Fritsch, C.; Klymchenko, A.S.; Bunt, G.; Jovin, T.M.; Arndt-Jovin, D.J. Dynamic conformational transitions of the EGF receptor in living mammalian cells determined by FRET and fluorescence lifetime imaging microscopy. Cytometry. A 2013, 83, 794–805. [Google Scholar] [CrossRef]
- Kozer, N.; Henderson, C.; Jackson, J.T.; Nice, E.C.; Burgess, A.W.; Clayton, A.H. Evidence for extended YFP-EGFR dimers in the absence of ligand on the surface of living cells. Phys. Biol. 2011, 8, 066002. [Google Scholar] [CrossRef]
- Valley, C.C.; Arndt-Jovin, D.J.; Karedla, N.; Steinkamp, M.P.; Chizhik, A.I.; Hlavacek, W.S.; Wilson, B.S.; Lidke, K.A.; Lidke, D.S. Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol Biol. Cell. 2015, 26, 4087–4099. [Google Scholar] [CrossRef]
- Coskun, Ü.; Grzybek, M.; Drechsel, D.; Simons, K. Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. USA 2011, 108, 9044–9048. [Google Scholar] [CrossRef]
- Hunter, T. The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 2014, 6, a020644. [Google Scholar] [CrossRef]
- Scaltriti, M.; Balsega, J. The Epidermal Growth Factor Receptor Pathway: A Model for Targeted Therapy. Clin. Cancer Res. 2006, 12, 5368–5372. [Google Scholar] [CrossRef]
- Mirza, A.; Mustafa, M.; Talevich, E.; Kannan, N. Co-conserved features associated with cis regulation of ErbB tyrosine kinases. PLoS ONE 2010, 5, e14310. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Jura, N.; Zhang, X.; Endres, N.; Seeliger, M. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef]
- Gajiwala, K.S. EGFR: Tale of the C-terminal tail. Protein Sci. 2013, 22, 995–999. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; Mcdonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib) relationships among protein conformation, inhibitor off-rate, and receptor activity. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar]
- Treiber, D.K.; Shah, N.P. Ins and Outs of Kinase DFG Motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef]
- Young, M.A.; Shah, N.P.; Chao, L.H.; Seeliger, M.; Milanov, Z.V.; Biggs, W.H.; Treiber, D.K.; Patel, H.K.; Zarrinkar, P.P.; Lockhart, D.J.; et al. Structure of the Kinase Domain of an Imatinib-Resistant Abl Mutant in Complex with the Aurora Kinase Inhibitor VX-680. Cancer Res. 2006, 66, 1007–1014. [Google Scholar] [CrossRef]
- Shan, Y.; Arkhipov, A.; Kim, E.T.; Pan, A.C.; Shaw, D.E. Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 7270–7275. [Google Scholar] [CrossRef]
- Ranson, M. Epidermal growth factor receptor tyrosine kinase inhibitors. Br. J. Cancer 2004, 90, 2250–2255. [Google Scholar] [CrossRef]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Miranda, O.; Farooqui, M.; Siegfried, J.M. Status of agents targeting the HGF/c-Met axis in lung cancer. Cancers 2018, 10, 280. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, Y.; James, M.; Jeong, J.H.; You, M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 2016, 55, 991–1001. [Google Scholar] [CrossRef]
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta Pharm. Sin. B 2018, 8, 503–510. [Google Scholar] [CrossRef]
- Yoneda, K.; Imanishi, N.; Ichiki, Y.; Tanaka, F. Immune Checkpoint Inhibitors (ICIs) in Non-Small Cell Lung Cancer (NSCLC). J uoeh 2018, 40, 173–189. [Google Scholar] [CrossRef]
- Qin, H.; Wang, F.; Liu, H.; Zeng, Z.; Wang, S.; Pan, X.; Gao, H. New advances in immunotherapy for non-small cell lung cancer. Am. J. Transl. Res. 2018, 10, 2234–2245. [Google Scholar]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Mohammadi, M.; McMahon, G.; Sun, L.; Tang, C.; Hirth, P.; Yeh, B.K.; Hubbard, S.R.; Schlessinger, J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276, 955–960. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Baselga, J. Targeting Tyrosine Kinases in Cancer: The Second Wave. Science 2006, 312, 1175–1178. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Kleiman, L.B.; Maiwald, T.; Conzelmann, H.; Lauffenburger, D.A.; Sorger, P.K. Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 2011, 43, 723–737. [Google Scholar] [CrossRef]
- Carmi, C.; Galvani, E.; Vacondio, F.; Rivara, S.; Lodola, A.; Russo, S.; Aiello, S.; Bordi, F.; Costantino, G.; Cavazzoni, A.; et al. Irreversible Inhibition of Epidermal Growth Factor Receptor Activity by 3-Aminopropanamides. J. Med. Chem. 2012, 55, 2251–2264. [Google Scholar] [CrossRef]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef]
- Ferby, I.; Reschke, M.; Kudlacek, O.; Knyazev, P.; Pantè, G.; Amann, K.; Sommergruber, W.; Kraut, N.; Ullrich, A.; Fässler, R.; et al. Mig6 is a negative regulator of EGF receptor–mediated skin morphogenesis and tumor formation. Nat. Med. 2006, 12, 568–573. [Google Scholar] [CrossRef]
- Hasenahuer, M.A.; Barletta, G.P.; Fernandez-Alberti, S.; Parisi, G.; Fornasari, M.S. Pockets as structural descriptors of EGFR kinase conformations. PLoS ONE 2017, 12, e0189147. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. 1), S24–S31. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef]
- Liu, B.; Bernard, B.; Wu, J.H. Impact of EGFR point mutations on the sensitivity to gefitinib: Insights from comparative structural analyses and molecular dynamics simulations. Proteins Struct. Funct. Bioinforma. 2006, 65, 331–346. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Kukimoto-Niino, M.; Parker, L.; Handa, N.; Terada, T.; Fujimoto, T.; Terazawa, Y.; Wakiyama, M.; Sato, M.; Sano, S.; et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 2012, 32, 27–38. [Google Scholar] [CrossRef]
- Red Brewer, M.; Yun, C.-H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef]
- Ribaudo, G.; Zanforlin, E.; Zagotto, G. Overcoming resistance in non-small-cell lung cancer: A practical lesson for the medicinal chemist. Arch. Pharm. (Weinheim). 2018, 351, 1–8. [Google Scholar] [CrossRef]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C.; et al. Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients With Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef]
- Sullivan, I.; Planchard, D. Next-Generation EGFR Tyrosine Kinase Inhibitors for Treating EGFR-Mutant Lung Cancer beyond First Line. Front. Med. 2017, 3, 1–13. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef]
- Shan, Y.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.; Jensen, M.O.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 139–144. [Google Scholar] [CrossRef]
- Park, J.H.; Lemmon, M.K. Occupy EGFR. Cancer Discov. 2012, 2, 398–400. [Google Scholar]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Ma, W.; Dong, Z. Conformational Transition Pathways of Epidermal Growth Factor Receptor Kinase Domain from Multiple Molecular Dynamics Simulations and Bayesian Clustering. J. Chem. Theory Comput. 2014, 10, 3503–3511. [Google Scholar] [CrossRef]
- Shukla, D.; Meng, Y.; Roux, B.; Pande, V.S. Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun. 2014, 5, 3397. [Google Scholar] [CrossRef]
- Kannan, S.; Venkatachalam, G.; Lim, H.H.; Surana, U.; Verma, C. Conformational landscape of the epidermal growth factor receptor kinase reveals a mutant specific allosteric pocket. Chem. Sci. 2018, 9, 5212–5222. [Google Scholar] [CrossRef]
- Paladino, A.; Morra, G.; Colombo, G. Structural Stability and Flexibility Direct the Selection of Activating Mutations in Epidermal Growth Factor Receptor Kinase. J. Chem. Inf. Model. 2015, 55, 1377–1387. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Burgess, A.W.; Cho, H.-S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.J.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar] [CrossRef]
- Mi, L.-Z.; Lu, C.; Li, Z.; Nishida, N.; Walz, T.; Springer, T. Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat. Struct. Mol. Biol. 2011, 18, 984–989. [Google Scholar] [CrossRef]
- Macdonald, J.L.; Pike, L.J. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. USA 2008, 105, 112–117. [Google Scholar] [CrossRef]
- Alvarado, D.; Klein, D.E.; Lemmon, M.A. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 2009, 461, 287–291. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A Structural Perspective on the Regulation of the Epidermal Growth Factor Receptor. Annu. Rev. Biochem. 2015, 1–26. [Google Scholar] [CrossRef]
- Kidd, B.A.; Baker, D.; Thomas, W.E. Computation of Conformational Coupling in Allosteric Proteins. PLoS Comput. Biol. 2009, 5, e1000484. [Google Scholar] [CrossRef]
- Dixit, A.; Verkhivker, G.M. Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLoS Comput. Biol. 2011, 7, e1002179. [Google Scholar] [CrossRef]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009, 137, 1293–1307. [Google Scholar] [CrossRef]
- Wood, E.R.; Shewchuk, L.M.; Ellis, B.; Brignola, P.; Brashear, R.L.; Caferro, T.R.; Dickerson, S.H.; Dickson, H.D.; Donaldson, K.H.; Gaul, M.; et al. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 2773–2778. [Google Scholar] [CrossRef]
- Red Brewer, M.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef]
- Kovacs, E.; Das, R.; Wang, Q.; Collier, T.S.; Cantor, A.; Huang, Y.; Wong, K.; Mirza, A.; Barros, T.; Grob, P.; et al. Analysis of the role of the C-terminal tail in the regulation of the epidermal growth factor receptor. Mol. Cell. Biol. 2015, 1162, MCB.00248-15. [Google Scholar] [CrossRef]
- Sorkin, A.; Goh, L. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 2009, 314, 3093–3106. [Google Scholar] [CrossRef]
- Mustafa, M.; Mirza, A.; Kannan, N. Conformational regulation of the EGFR kinase core by the juxtamembrane and C-terminal tail: A molecular dynamics study. Proteins 2011, 79, 99–114. [Google Scholar] [CrossRef]
- Adak, S.; Yang, K.S.; Macdonald-Obermann, J.; Pike, L.J. The Membrane-proximal Intracellular Domain of the Epidermal Growth Factor Receptor Underlies Negative Cooperativity in Ligand Binding. J. Biol. Chem. 2011, 286, 45146–45155. [Google Scholar] [CrossRef]
- Doerner, A.; Scheck, R.; Schepartz, A. Growth Factor Identity Is Encoded by Discrete Coiled-Coil Rotamers in the EGFR Juxtamembrane Region. Chem. Biol. 2015, 22, 776–784. [Google Scholar] [CrossRef]
- Choowongkomon, K.; Carlin, C.R.; Sönnichsen, F.D. A structural model for the membrane-bound form of the juxtamembrane domain of the epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 24043–24052. [Google Scholar] [CrossRef]
- Endres, N.F.; Das, R.; Smith, A.W.; Arkhipov, A.; Kovacs, E.; Huang, Y.; Pelton, J.G.; Shan, Y.; Shaw, D.E.; Wemmer, D.E.; et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 2013, 152, 543–556. [Google Scholar] [CrossRef]
- Yu, X.; Sharma, K.D.; Takahashi, T.; Iwamoto, R.; Mekada, E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 2002, 13, 2547–2557. [Google Scholar] [CrossRef]
- Low-Nam, S.T.; Lidke, K.A.; Cutler, P.J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Wilson, B.S.; Lidke, D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol. 2011, 18, 1244–1249. [Google Scholar] [CrossRef]
- Clayton, A.H.A.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef]
- Freed, D.M.; Alvarado, D.; Lemmon, M.A. Ligand regulation of a constitutively dimeric EGF receptor. Nat. Commun. 2015, 6, 7380. [Google Scholar] [CrossRef]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef]
- Saffarian, S.; Li, Y.; Elson, E.L.; Pike, L.J. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 2007, 93, 1021–1031. [Google Scholar] [CrossRef]
- Clayton, A.H.A.; Tavarnesi, M.L.; Johns, T.G. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry 2007, 46, 4589–4597. [Google Scholar] [CrossRef]
- Moriki, T.; Maruyama, H.; Maruyama, I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001, 311, 1011–1026. [Google Scholar] [CrossRef]
- Liu, P.; Sudhaharan, T.; Koh, R.M.L.; Hwang, L.C.; Ahmed, S.; Maruyama, I.N.; Wohland, T. Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 2007, 93, 684–698. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Lesovoy, D.M.; Pavlov, K.V.; Pustovalova, Y.E.; Bocharova, O.V.; Arseniev, A.S. Alternative packing of EGFR transmembrane domain suggests that protein-lipid interactions underlie signal conduction across membrane. Biochim. Biophys. Acta 2016, 1858, 1254–1261. [Google Scholar] [CrossRef]
- Lelimousin, M.; Limongelli, V.; Sansom, M.S.P. Conformational Changes in the Epidermal Growth Factor Receptor: Role of the Transmembrane Domain Investigated by Coarse-Grained MetaDynamics Free Energy Calculations. J. Am. Chem. Soc. 2016, 138, 10611–10622. [Google Scholar] [CrossRef]
- Abd Halim, K.B.; Koldsø, H.; Sansom, M.S.P. Interactions of the EGFR juxtamembrane domain with PIP2-containing lipid bilayers: Insights from multiscale molecular dynamics simulations. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 1017–1025. [Google Scholar] [CrossRef]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef]
- Liu, P.; Cleveland, T.E.; Bouyain, S.; Byrne, P.O.; Longo, P.A.; Leahy, D.J. A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. USA 2012, 2012. [Google Scholar] [CrossRef]
- Westover, E.J.; Covey, D.F.; Brockman, H.L.; Brown, R.E.; Pike, L.J. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J. Biol. Chem. 2003, 278, 51125–51133. [Google Scholar] [CrossRef]
- Tynan, C.J.; Lo Schiavo, V.; Zanetti-Domingues, L.; Needham, S.R.; Roberts, S.K.; Hirsch, M.; Rolfe, D.J.; Korovesis, D.; Clarke, D.T.; Martin-Fernandez, M.L. A tale of the epidermal growth factor receptor: The quest for structural resolution on cells. Methods 2016, 95, 86–93. [Google Scholar] [CrossRef]
- Clarke, D.T.; Martin-Fernandez, M.L.; Clarke, D.T.; Martin-Fernandez, M.L. A Brief History of Single-Particle Tracking of the Epidermal Growth Factor Receptor. Methods Protoc. 2019, 2, 12. [Google Scholar] [CrossRef]
- Huang, Y.; Bharill, S.; Karandur, D.; Peterson, S.M.; Marita, M.; Shi, X.; Kaliszewski, M.J.; Smith, A.W.; Isacoff, E.Y.; Kuriyan, J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife 2016, 5, e14107. [Google Scholar] [CrossRef]
- Wikstrand, C.J.; Hale, L.P.; Batra, S.K.; Hill, M.L.; Humphrey, P.A.; Kurpad, S.N.; McLendon, R.E.; Moscatello, D.; Pegram, C.N.; Reist, C.J. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995, 55, 3140–3148. [Google Scholar]
- Stan, R.V. Structure of caveolae. Biochim. Biophys. Acta. 2005, 1746, 334–348. [Google Scholar] [CrossRef]
- Wu, H. Higher-order assemblies in a new paradigm of signal transduction. Cell 2013, 153, 287–292. [Google Scholar] [CrossRef]
- Harding, A.S.; Hancock, J.F. Using plasma membrane nanoclusters to build better signaling circuits. Trends Cell Biol. 2008, 18, 364–371. [Google Scholar] [CrossRef]
- Nussinov, R.; Jang, H.; Tsai, C.-J. Oligomerization and nanocluster organization render specificity. Biol. Rev. 2015, 90, 587–598. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8040316
Martin-Fernandez ML, Clarke DT, Roberts SK, Zanetti-Domingues LC, Gervasio FL. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells. 2019; 8(4):316. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8040316
Chicago/Turabian StyleMartin-Fernandez, Marisa L., David T. Clarke, Selene K. Roberts, Laura C. Zanetti-Domingues, and Francesco L. Gervasio. 2019. "Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer" Cells 8, no. 4: 316. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8040316