Intermediate Filaments as Effectors of Cancer Development and Metastasis: A Focus on Keratins, Vimentin, and Nestin


Abstract
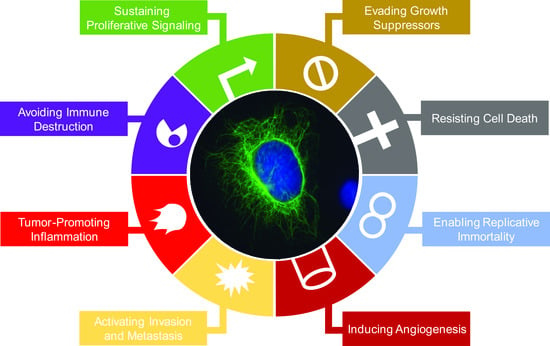
:
1. Introduction
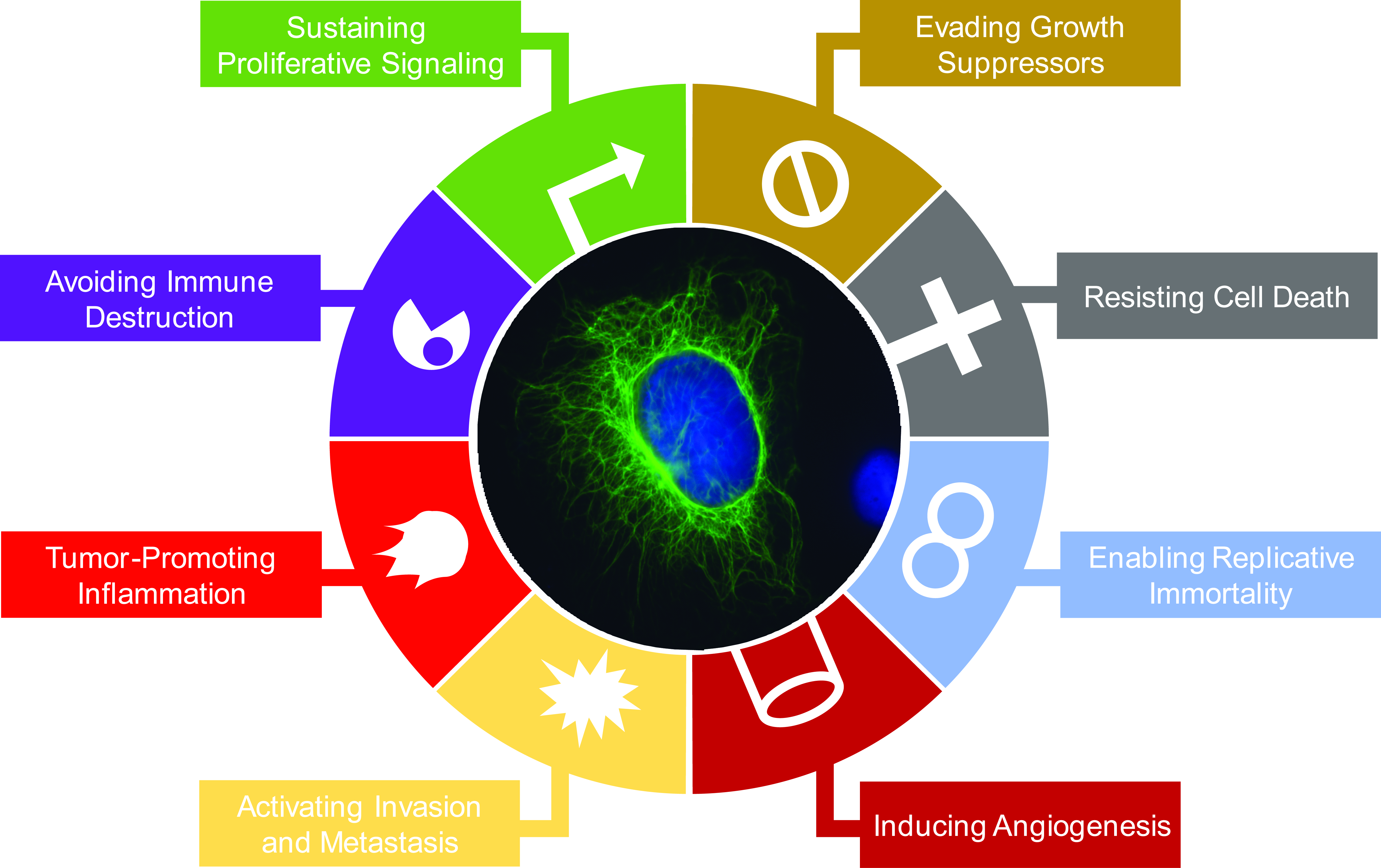
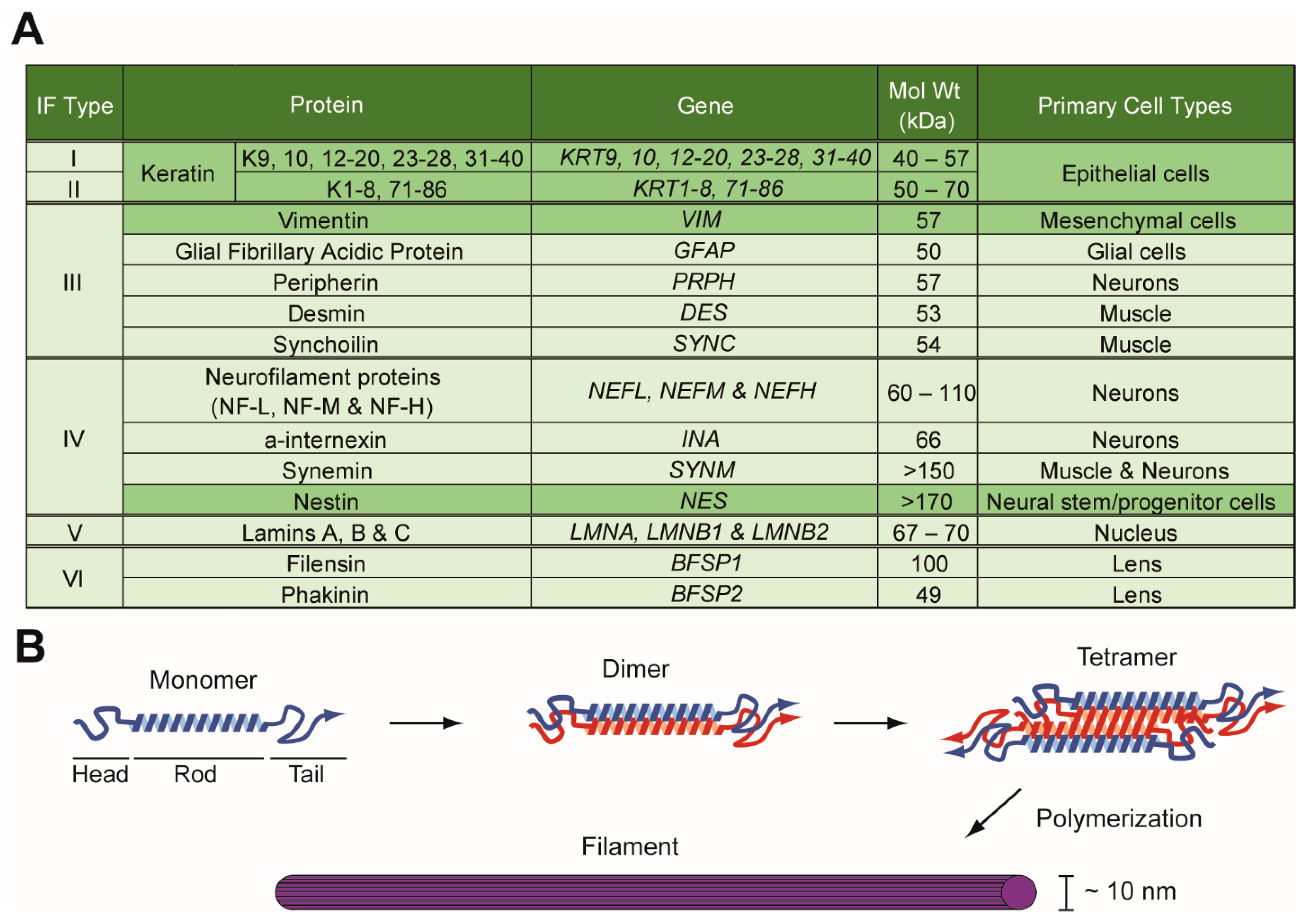
1.1. Intermediate Filament Family of Cytoskeletal Proteins
1.2. IF and Cancer—IFs as Diagnostic and Prognostic Markers of Cancer
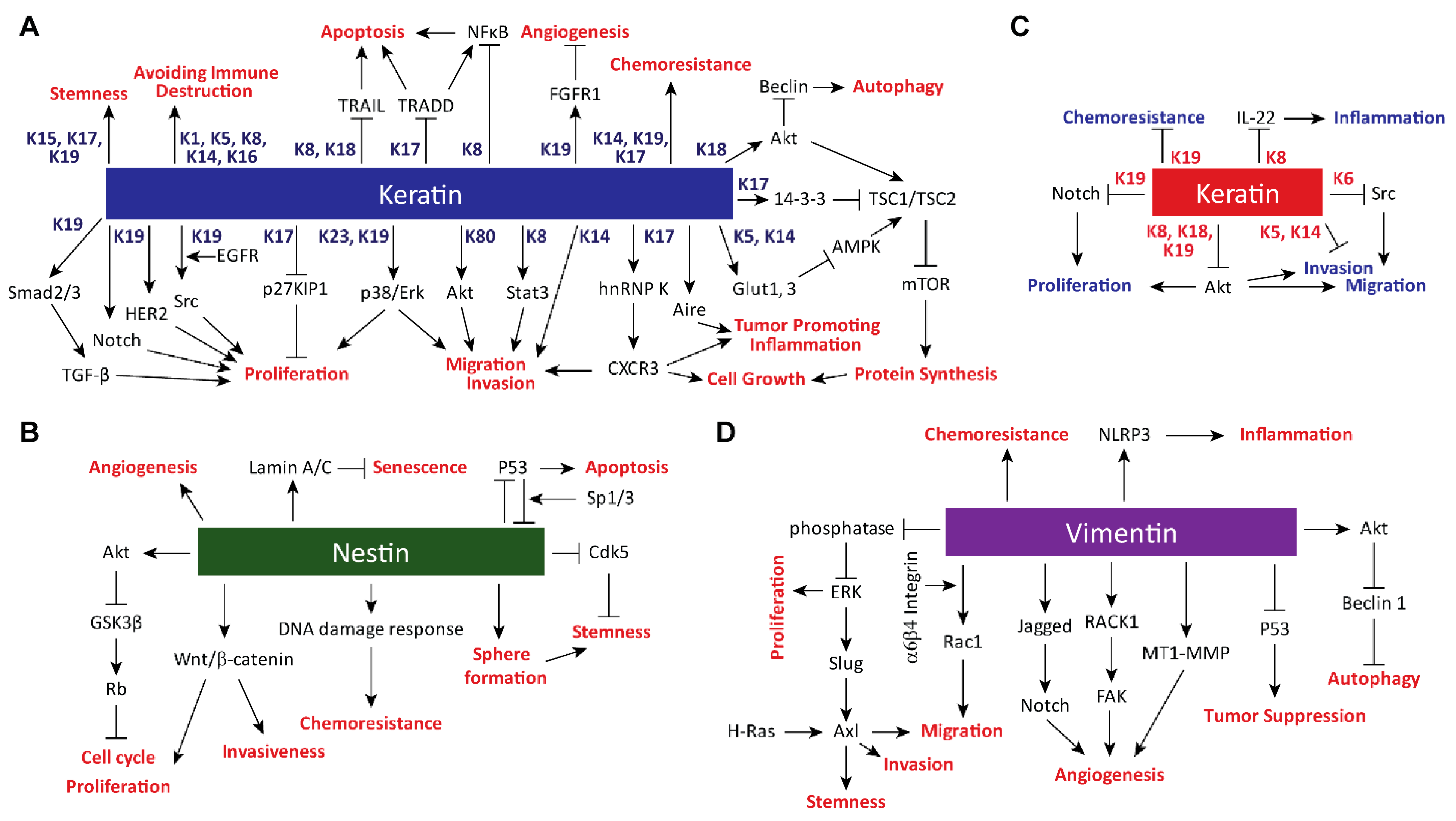
2. Impacts of Intermediate Filament Proteins on Cancer Hallmarks
2.1. Sustaining Proliferative Signaling
2.2. Evading Growth Suppressors
2.3. Resisting Cell Death
2.4. Enabling Replicative Immortality
2.5. Inducing Angiogenesis
2.6. Activating Invasion and Metastasis
2.7. Tumor-Promoting Inflammation
2.8. Avoiding Immune Destruction
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Omary, M.B. “IF-pathies”: A broad spectrum of intermediate filament-associated diseases. J. Clin. Invest. 2009, 119, 1756–1762. [Google Scholar] [CrossRef]
- Kim, S.; Coulombe, P.A. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007, 21, 1581–1597. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef]
- Gu, L.-H.; Coulombe, P.A. Keratin function in skin epithelia: A broadening palette with surprising shades. Curr. Opin. Cell Biol. 2007, 19, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Pallari, H.M.; Eriksson, J.E. Intermediate filaments as signaling platforms. Sci. STKE 2006, 2006, pe53. [Google Scholar] [CrossRef]
- Chung, B.M.; Rotty, J.D.; Coulombe, P.A. Networking galore: Intermediate filaments and cell migration. Curr. Opin. Cell Biol. 2013, 25, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Strnad, P.; Paschke, S.; Jang, K.H.; Ku, N.O. Keratins: Markers and modulators of liver disease. Curr. Opin. Gastroenterol. 2012, 28, 209–216. [Google Scholar] [CrossRef]
- Jin, L.; Wang, G. Keratin 17: A critical player in the pathogenesis of psoriasis. Med. Res. Rev. 2014, 34, 438–454. [Google Scholar] [CrossRef]
- Pan, X.; Hobbs, R.P.; Coulombe, P.A. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 2013, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Toivola, D.M.; Boor, P.; Alam, C.; Strnad, P. Keratins in health and disease. Curr. Opin. Cell Biol. 2015, 32, 73–81. [Google Scholar] [CrossRef]
- Moll, R.; Franke, W.W.; Schiller, D.L.; Geiger, B.; Krepler, R. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982, 31, 11–24. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivaska, J.; Pallari, H.M.; Nevo, J.; Eriksson, J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Lendahl, U.; Zimmerman, L.B.; McKay, R.D. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60, 585–595. [Google Scholar] [CrossRef]
- Tampaki, E.C.; Nakopoulou, L.; Tampakis, A.; Kontzoglou, K.; Weber, W.P.; Kouraklis, G. Nestin involvement in tissue injury and cancer—A potential tumor marker? Cell. Oncol. Dordr. 2014, 37, 305–315. [Google Scholar] [CrossRef]
- Matsuda, Y.; Kure, S.; Ishiwata, T. Nestin and other putative cancer stem cell markers in pancreatic cancer. Med. Mol. Morphol. 2012, 45, 59–65. [Google Scholar] [CrossRef]
- Narita, K.; Matsuda, Y.; Seike, M.; Naito, Z.; Gemma, A.; Ishiwata, T. Nestin regulates proliferation, migration, invasion and stemness of lung adenocarcinoma. Int. J. Oncol. 2014, 44, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Oshima, R.G. Intermediate filaments: A historical perspective. Exp. Cell Res. 2007, 313, 1981–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, V.; Goike, H.; Panaretakis, K.W.; Einarsson, R. Clinical utility of cytokeratins as tumor markers. Clin. Biochem. 2004, 37, 529–540. [Google Scholar] [CrossRef]
- Riethdorf, S.; Fritsche, H.; Muller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Janicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: A validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells—Mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Mostert, B.; Bolt-de Vries, J.; Peeters, D.; de Jongh, F.E.; Stouthard, J.M.; Dirix, L.Y.; van Dam, P.A.; Van Galen, A.; de Weerd, V.; et al. mRNA and microRNA expression profiles in circulating tumor cells and primary tumors of metastatic breast cancer patients. Clin. Cancer Res. 2011, 17, 3600–3618. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef]
- Woelfle, U.; Sauter, G.; Santjer, S.; Brakenhoff, R.; Pantel, K. Down-regulated expression of cytokeratin 18 promotes progression of human breast cancer. Clin. Cancer Res. 2004, 10, 2670–2674. [Google Scholar] [CrossRef]
- Laakso, M.; Loman, N.; Borg, A.; Isola, J. Cytokeratin 5/14-positive breast cancer: True basal phenotype confined to BRCA1 tumors. Mod. Pathol. 2005, 18, 1321–1328. [Google Scholar] [CrossRef]
- van de Rijn, M.; Perou, C.M.; Tibshirani, R.; Haas, P.; Kallioniemi, O.; Kononen, J.; Torhorst, J.; Sauter, G.; Zuber, M.; Kochli, O.R.; et al. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am. J. Pathol. 2002, 161, 1991–1996. [Google Scholar] [CrossRef]
- de Silva Rudland, S.; Platt-Higgins, A.; Winstanley, J.H.; Jones, N.J.; Barraclough, R.; West, C.; Carroll, J.; Rudland, P.S. Statistical association of basal cell keratins with metastasis-inducing proteins in a prognostically unfavorable group of sporadic breast cancers. Am. J. Pathol. 2011, 179, 1061–1072. [Google Scholar] [CrossRef]
- Cen, D.; Chen, J.; Li, Z.; Zhao, J.; Cai, X. Prognostic significance of cytokeratin 19 expression in pancreatic neuroendocrine tumor: A meta-analysis. PLoS ONE 2017, 12, e0187588. [Google Scholar] [CrossRef]
- Choi, I.; Gudas, L.J.; Katzenellenbogen, B.S. Regulation of keratin 19 gene expression by estrogen in human breast cancer cells and identification of the estrogen responsive gene region. Mol. Cell. Endocrinol. 2000, 164, 225–237. [Google Scholar] [CrossRef]
- Callahan, C.A.; Ofstad, T.; Horng, L.; Wang, J.K.; Zhen, H.H.; Coulombe, P.A.; Oro, A.E. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004, 18, 2724–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Chen, S.; Han, J.-X.; Qian, B.; Wang, X.-R.; Zhong, W.-L.; Qin, Y.; Zhang, H.; Gao, W.-F.; Lei, Y.-Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, Y.; Sui, Z.; Zhang, Y.; Liu, M.; Tang, H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget 2017, 8, 48725–48736. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, P.; Su, X.-J.; Zhang, B. The ubiquitin ligase TRIM56 inhibits ovarian cancer progression by targeting vimentin. J. Cell. Physiol. 2018, 233, 2420–2425. [Google Scholar] [CrossRef]
- Stacey, S.N.; Sulem, P.; Masson, G.; Gudjonsson, S.A.; Thorleifsson, G.; Jakobsdottir, M.; Sigurdsson, A.; Gudbjartsson, D.F.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. New common variants affecting susceptibility to basal cell carcinoma. Nat. Genet. 2009, 41, 909–914. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Danielsson, F.; Peterson, M.K.; Caldeira Araújo, H.; Lautenschläger, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Krupkova, O., Jr.; Loja, T.; Redova, M.; Neradil, J.; Zitterbart, K.; Sterba, J.; Veselska, R. Analysis of nuclear nestin localization in cell lines derived from neurogenic tumors. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2011, 32, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Kim, S.; Wong, P.; Coulombe, P.A. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006, 441, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraj, P.; Kroger, C.; Reuter, U.; Windoffer, R.; Leube, R.E.; Magin, T.M. Keratins regulate protein biosynthesis through localization of GLUT1 and -3 upstream of AMP kinase and Raptor. J. Cell Biol. 2009, 187, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, J.; Cai, L.; Zhong, B.; Luo, H.; Hao, Y.; Yu, W.; Wang, B.; Su, C.; Lei, Y.; et al. Role of the stem cell-associated intermediate filament nestin in malignant proliferation of non-small cell lung cancer. PLoS ONE 2014, 9, e85584. [Google Scholar] [CrossRef]
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.C.; Dlugosz, A.A. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217. [Google Scholar] [CrossRef]
- DePianto, D.; Kerns, M.L.; Dlugosz, A.A.; Coulombe, P.A. Keratin 17 promotes epithelial proliferation and tumor growth by polarizing the immune response in skin. Nat. Genet. 2010, 42, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, R.P.; DePianto, D.J.; Jacob, J.T.; Han, M.C.; Chung, B.M.; Batazzi, A.S.; Poll, B.G.; Guo, Y.; Han, J.; Ong, S.; et al. Keratin-dependent regulation of Aire and gene expression in skin tumor keratinocytes. Nat. Genet. 2015, 47, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, S.; Tanner, J.M.; Bell, R.; Chaturvedi, A.; Randall, R.L.; Beckerle, M.C.; Lessnick, S.L. A novel role for keratin 17 in coordinating oncogenic transformation and cellular adhesion in Ewing sarcoma. Mol. Cell. Biol. 2013, 33, 4448–4460. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Hoyos, L.F.; Shah, R.; Roa-Pena, L.; Vanner, E.A.; Najafian, N.; Banach, A.; Nielsen, E.; Al-Khalil, R.; Akalin, A.; Talmage, D.; et al. Keratin-17 promotes p27KIP1 nuclear export and degradation and offers potential prognostic utility. Cancer Res. 2015, 75, 3650–3662. [Google Scholar] [CrossRef]
- Santos, M.; Paramio, J.M.; Bravo, A.; Ramirez, A.; Jorcano, J.L. The expression of keratin k10 in the basal layer of the epidermis inhibits cell proliferation and prevents skin tumorigenesis. J. Biol. Chem. 2002, 277, 19122–19130. [Google Scholar] [CrossRef]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-H.; Oh, S.; Lee, K.-M.; Yang, W.; Nam, K.S.; Moon, H.-G.; Noh, D.-Y.; Kim, C.G.; Park, G.; Park, J.B.; et al. Cytokeratin19 induced by HER2/ERK binds and stabilizes HER2 on cell membranes. Cell Death Differ. 2015, 22, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-H.; Yang, W.; Lee, K.-M.; Oh, S.; Nam, K.; Shim, S.; Shin, S.Y.; Gye, M.C.; Chu, I.-S.; Shin, I. Regulation of cell proliferation and migration by keratin19-induced nuclear import of early growth response-1 in breast cancer cells. Clin. Cancer Res. 2013, 19, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Alam, H.; Gangadaran, P.; Bhate, A.V.; Chaukar, D.A.; Sawant, S.S.; Tiwari, R.; Bobade, J.; Kannan, S.; D’cruz, A.K.; Kane, S.; et al. Loss of keratin 8 phosphorylation leads to increased tumor progression and correlates with clinico-pathological parameters of OSCC patients. PLoS ONE 2011, 6, e27767. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Langa, F.; Kress, C.; Colucci-Guyon, E.; Khun, H.; Vandormael-Pournin, S.; Huerre, M.; Babinet, C. Teratocarcinomas induced by embryonic stem (ES) cells lacking vimentin: An approach to study the role of vimentin in tumorigenesis. J. Cell Sci. 2000, 113 Pt 19, 3463–3472. [Google Scholar]
- Zelenko, Z.; Gallagher, E.J.; Tobin-Hess, A.; Belardi, V.; Rostoker, R.; Blank, J.; Dina, Y.; LeRoith, D. Silencing vimentin expression decreases pulmonary metastases in a pre-diabetic mouse model of mammary tumor progression. Oncogene 2017, 36, 1394–1403. [Google Scholar] [CrossRef]
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef]
- Ma, J.; Sun, F.; Li, C.; Zhang, Y.; Xiao, W.; Li, Z.; Pan, Q.; Zeng, H.; Xiao, G.; Yao, K.; et al. Depletion of intermediate filament protein Nestin, a target of microRNA-940, suppresses tumorigenesis by inducing spontaneous DNA damage accumulation in human nasopharyngeal carcinoma. Cell Death Dis. 2014, 5, e1377. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Hagio, M.; Arai, T. Inhibition of nestin suppresses stem cell phenotype of glioblastomas through the alteration of post-translational modification of heat shock protein HSPA8/HSC71. Cancer Lett. 2015, 357, 602–611. [Google Scholar] [CrossRef]
- Matsuda, Y.; Yoshimura, H.; Ueda, J.; Naito, Z.; Korc, M.; Ishiwata, T. Nestin delineates pancreatic cancer stem cells in metastatic foci of NOD/Shi-scid IL2Rgamma(null) (NOG) mice. Am. J. Pathol. 2014, 184, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Yamahatsu, K.; Matsuda, Y.; Ishiwata, T.; Uchida, E.; Naito, Z. Nestin as a novel therapeutic target for pancreatic cancer via tumor angiogenesis. Int. J. Oncol. 2012, 40, 1345–1357. [Google Scholar] [Green Version]
- Li, B.; Antonyak, M.A.; Druso, J.E.; Cheng, L.; Nikitin, A.Y.; Cerione, R.A. EGF potentiated oncogenesis requires a tissue transglutaminase-dependent signaling pathway leading to Src activation. Proc. Natl. Acad. Sci. USA 2010, 107, 1408–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Zhuo, H.; Zhang, X.; Jiang, R.; Ji, J.; Deng, L.; Qian, X.; Zhang, F.; Sun, B. A novel biomarker Linc00974 interacting with KRT19 promotes proliferation and metastasis in hepatocellular carcinoma. Cell Death Dis. 2014, 5, e1549. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Choi, H.Y.; Kim, B.W.; Dayem, A.A.; Yang, G.-M.; Kim, K.S.; Yin, Y.F.; Cho, S.-G. KRT19 directly interacts with β-catenin/RAC1 complex to regulate NUMB-dependent NOTCH signaling pathway and breast cancer properties. Oncogene 2016. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, R.; Zou, K.; Yu, W.; Guo, W.; Gao, Y.; Li, J.; Li, M.; Tai, Y.; Huang, W.; et al. Keratin 23 promotes telomerase reverse transcriptase expression and human colorectal cancer growth. Cell Death Dis. 2017, 8, e2961. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindström, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-β-Slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef]
- Chung, B.M.; Arutyunov, A.; Ilagan, E.; Yao, N.; Wills-Karp, M.; Coulombe, P.A. Regulation of C-X-C chemokine gene expression by keratin 17 and hnRNP K in skin tumor keratinocytes. J. Cell Biol. 2015, 208, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Rotty, J.D.; Coulombe, P.A. A wound-induced keratin inhibits Src activity during keratinocyte migration and tissue repair. J. Cell Biol. 2012, 197, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin-ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Sembritzki, O.; Hagel, C.; Lamszus, K.; Deppert, W.; Bohn, W. Cytoplasmic localization of wild-type p53 in glioblastomas correlates with expression of vimentin and glial fibrillary acidic protein. Neuro-Oncol. 2002, 4, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Malminen, M.; Peltonen, S.; Koivunen, J.; Peltonen, J. Functional expression of NF1 tumor suppressor protein: Association with keratin intermediate filaments during the early development of human epidermis. BMC Dermatol. 2002, 2, 10. [Google Scholar] [CrossRef]
- Ming, M.; Qiang, L.; Zhao, B.; He, Y.Y. Mammalian SIRT2 inhibits keratin 19 expression and is a tumor suppressor in skin. Exp. Dermatol. 2014, 23, 207–209. [Google Scholar] [CrossRef]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar] [CrossRef] [Green Version]
- Michel, M.; Torok, N.; Godbout, M.J.; Lussier, M.; Gaudreau, P.; Royal, A.; Germain, L. Keratin 19 as a biochemical marker of skin stem cells in vivo and in vitro: Keratin 19 expressing cells are differentially localized in function of anatomic sites, and their number varies with donor age and culture stage. J. Cell Sci. 1996, 109 Pt 5, 1017–1028. [Google Scholar] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Dittmer, J.; Rody, A. Cancer stem cells in breast cancer. Histol. Histopathol. 2013, 28, 827–838. [Google Scholar] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, J.; Miyata, H.; Yamasaki, M.; Sugimura, K.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; Mori, M.; Doki, Y. Mesenchymal phenotype after chemotherapy is associated with chemoresistance and poor clinical outcome in esophageal cancer. Oncol. Rep. 2014, 31, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [Green Version]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; von Burstin, J.; Mastracci, T.L.; Worthley, D.L.; et al. Krt19(+)/Lgr5(-) Cells Are Radioresistant Cancer-Initiating Stem Cells in the Colon and Intestine. Cell Stem Cell 2015, 16, 627–638. [Google Scholar] [CrossRef]
- Bambang, I.F.; Lu, D.; Li, H.; Chiu, L.-L.; Lau, Q.C.; Koay, E.; Zhang, D. Cytokeratin 19 regulates endoplasmic reticulum stress and inhibits ERp29 expression via p38 MAPK/XBP-1 signaling in breast cancer cells. Exp. Cell Res. 2009, 315, 1964–1974. [Google Scholar] [CrossRef]
- Tong, X.; Coulombe, P.A. Keratin 17 modulates hair follicle cycling in a TNFalpha-dependent fashion. Genes Dev. 2006, 20, 1353–1364. [Google Scholar] [CrossRef]
- Tiwari, R.; Sahu, I.; Soni, B.L.; Sathe, G.J.; Thapa, P.; Patel, P.; Sinha, S.; Vadivel, C.K.; Patel, S.; Jamghare, S.N.; et al. Depletion of keratin 8/18 modulates oncogenic potential by governing multiple signaling pathways. FEBS J. 2018, 285, 1251–1276. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.B.; Ku, N.O.; Toivola, D.M. Keratins: Guardians of the liver. Hepatology 2002, 35, 251–257. [Google Scholar] [CrossRef]
- Ku, N.-O.; Strnad, P.; Bantel, H.; Omary, M.B. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 2016, 64, 966–976. [Google Scholar] [CrossRef]
- Bozza, W.P.; Zhang, Y.; Zhang, B. Cytokeratin 8/18 protects breast cancer cell lines from TRAIL-induced apoptosis. Oncotarget 2018, 9, 23264–23273. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Krüger, K.; Wik, E.; Knutsvik, G.; Nalwoga, H.; Klingen, T.A.; Arnes, J.B.; Chen, Y.; Mannelqvist, M.; Dimitrakopoulou, K.; Stefansson, I.M.; et al. Expression of Nestin associates with BRCA1 mutations, a basal-like phenotype and aggressive breast cancer. Sci. Rep. 2017, 7, 1089. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Naito, Z. Nestin in gastrointestinal and other cancers: Effects on cells and tumor angiogenesis. World J. Gastroenterol. 2011, 17, 409–418. [Google Scholar] [CrossRef]
- Pallari, H.M.; Lindqvist, J.; Torvaldson, E.; Ferraris, S.E.; He, T.; Sahlgren, C.; Eriksson, J.E. Nestin as a regulator of Cdk5 in differentiating myoblasts. Mol. Biol. Cell 2011, 22, 1539–1549. [Google Scholar] [CrossRef]
- Sahlgren, C.M.; Pallari, H.M.; He, T.; Chou, Y.H.; Goldman, R.D.; Eriksson, J.E. A nestin scaffold links Cdk5/p35 signaling to oxidant-induced cell death. EMBO J. 2006, 25, 4808–4819. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Huang, W.; Cai, J.; Ba, J.; Wang, Y.; Ke, Q.; Huang, Y.; Liu, X.; Qiu, Y.; et al. Nuclear Nestin deficiency drives tumor senescence via lamin A/C-dependent nuclear deformation. Nat. Commun. 2018, 9, 3613. [Google Scholar] [CrossRef]
- Wang, G.Y.; Wang, J.; Mancianti, M.L.; Epstein, E.H., Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/-) mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef]
- White, A.C.; Tran, K.; Khuu, J.; Dang, C.; Cui, Y.; Binder, S.W.; Lowry, W.E. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 7425–7430. [Google Scholar] [CrossRef] [Green Version]
- Smedts, F.; Ramaekers, F.; Troyanovsky, S.; Pruszczynski, M.; Link, M.; Lane, B.; Leigh, I.; Schijf, C.; Vooijs, P. Keratin expression in cervical cancer. Am. J. Pathol. 1992, 141, 497–511. [Google Scholar]
- Hobbs, R.P.; Batazzi, A.S.; Han, M.C.; Coulombe, P.A. Loss of Keratin 17 induces tissue-specific cytokine polarization and cellular differentiation in HPV16-driven cervical tumorigenesis in vivo. Oncogene 2016, 35, 5653–5662. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Han, L.; Zhou, C.; Wei, W.; Chen, X.; Yi, H.; Wu, X.; Bai, X.; Guo, S.; Yu, Y.; et al. TGF-β1-induced CK17 enhances cancer stem cell-like properties rather than EMT in promoting cervical cancer metastasis via the ERK1/2-MZF1 signaling pathway. FEBS J. 2017, 284, 3000–3017. [Google Scholar] [CrossRef]
- Bhagirath, D.; Zhao, X.; West, W.W.; Qiu, F.; Band, H.; Band, V. Cell type of origin as well as genetic alterations contribute to breast cancer phenotypes. Oncotarget 2015, 6, 9018–9030. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.W.; Polyak, K. Stem cells in the human breast. Cold Spring Harb. Perspect. Biol. 2010, 2, a003160. [Google Scholar] [CrossRef]
- Fu, C.-H.; Lin, R.-J.; Yu, J.; Chang, W.-W.; Liao, G.-S.; Chang, W.-Y.; Tseng, L.-M.; Tsai, Y.-F.; Yu, J.-C.; Yu, A.L. A novel oncogenic role of inositol phosphatase SHIP2 in ER-negative breast cancer stem cells: Involvement of JNK/vimentin activation. Stem Cells 2014, 32, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Hagio, M.; Ishiwata, T. Nestin: A novel angiogenesis marker and possible target for tumor angiogenesis. World J. Gastroenterol. 2013, 19, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.I.; Kang, H.; Dave, J.M.; Mendoza, E.A.; Su, S.C.; Maxwell, S.A.; Bayless, K.J. Calpain-mediated vimentin cleavage occurs upstream of MT1-MMP membrane translocation to facilitate endothelial sprout initiation. Angiogenesis 2012, 15, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Dave, J.M.; Kang, H.; Abbey, C.A.; Maxwell, S.A.; Bayless, K.J. Proteomic profiling of endothelial invasion revealed receptor for activated C kinase 1 (RACK1) complexed with vimentin to regulate focal adhesion kinase (FAK). J. Biol. Chem. 2013, 288, 30720–30733. [Google Scholar] [CrossRef]
- Antfolk, D.; Sjöqvist, M.; Cheng, F.; Isoniemi, K.; Duran, C.L.; Rivero-Muller, A.; Antila, C.; Niemi, R.; Landor, S.; Bouten, C.V.C.; et al. Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc. Natl. Acad. Sci. USA 2017, 114, E4574–E4581. [Google Scholar] [CrossRef] [Green Version]
- Takano, M.; Shimada, K.; Fujii, T.; Morita, K.; Takeda, M.; Nakajima, Y.; Nonomura, A.; Konishi, N.; Obayashi, C. Keratin 19 as a key molecule in progression of human hepatocellular carcinomas through invasion and angiogenesis. BMC Cancer 2016, 16, 903. [Google Scholar] [CrossRef]
- Esue, O.; Carson, A.A.; Tseng, Y.; Wirtz, D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem. 2006, 281, 30393–30399. [Google Scholar] [CrossRef]
- Hookway, C.; Ding, L.; Davidson, M.W.; Rappoport, J.Z.; Danuser, G.; Gelfand, V.I. Microtubule-dependent transport and dynamics of vimentin intermediate filaments. Mol. Biol. Cell 2015, 26, 1675–1686. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pascalis, C.; Pérez-González, C.; Seetharaman, S.; Boëda, B.; Vianay, B.; Burute, M.; Leduc, C.; Borghi, N.; Trepat, X.; Etienne-Manneville, S. Intermediate filaments control collective migration by restricting traction forces and sustaining cell-cell contacts. J. Cell Biol. 2018. [Google Scholar] [CrossRef]
- Chu, Y.W.; Seftor, E.A.; Romer, L.H.; Hendrix, M.J. Experimental coexpression of vimentin and keratin intermediate filaments in human melanoma cells augments motility. Am. J. Pathol. 1996, 148, 63–69. [Google Scholar]
- Havel, L.S.; Kline, E.R.; Salgueiro, A.M.; Marcus, A.I. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene 2015, 34, 1979–1990. [Google Scholar] [CrossRef]
- Kleeberger, W.; Bova, G.S.; Nielsen, M.E.; Herawi, M.; Chuang, A.Y.; Epstein, J.I.; Berman, D.M. Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res. 2007, 67, 9199–9206. [Google Scholar] [CrossRef]
- Leduc, C.; Etienne-Manneville, S. Intermediate filaments in cell migration and invasion: The unusual suspects. Curr. Opin. Cell Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Colburn, Z.T.; Jones, J.C.R. Complexes of α6β4 integrin and vimentin act as signaling hubs to regulate epithelial cell migration. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Seltmann, K.; Fritsch, A.W.; Kas, J.A.; Magin, T.M. Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 18507–18512. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.-L.; Wu, J.-S.; Cao, M.-X.; Gao, S.-Y.; Cen, X.; Jiang, Y.-P.; Wang, S.-S.; Tang, Y.-J.; Chen, Q.-M.; Liang, X.-H.; et al. Cytokeratin-14 contributes to collective invasion of salivary adenoid cystic carcinoma. PLoS ONE 2017, 12, e0171341. [Google Scholar] [CrossRef]
- Crowe, D.L.; Milo, G.E.; Shuler, C.F. Keratin 19 downregulation by oral squamous cell carcinoma lines increases invasive potential. J. Dent. Res. 1999, 78, 1256–1263. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Vendrell, J.P.; Slijper, M.; Pelle, O.; Barbotte, E.; Mercier, G.; Jacot, W.; Fabbro, M.; Pantel, K. Full-length cytokeratin-19 is released by human tumor cells: A potential role in metastatic progression of breast cancer. Breast Cancer Res. 2009, 11, R39. [Google Scholar] [CrossRef]
- Ding, S.J.; Li, Y.; Tan, Y.X.; Jiang, M.R.; Tian, B.; Liu, Y.K.; Shao, X.X.; Ye, S.L.; Wu, J.R.; Zeng, R.; et al. From proteomic analysis to clinical significance: Overexpression of cytokeratin 19 correlates with hepatocellular carcinoma metastasis. Mol. Cell. Proteomics 2004, 3, 73–81. [Google Scholar] [CrossRef]
- Kabir, N.N.; Ronnstrand, L.; Kazi, J.U. Keratin 19 expression correlates with poor prognosis in breast cancer. Mol. Biol. Rep. 2014. [Google Scholar] [CrossRef]
- Kim, H.; Choi, G.H.; Na, D.C.; Ahn, E.Y.; Kim, G.I.; Lee, J.E.; Cho, J.Y.; Yoo, J.E.; Choi, J.S.; Park, Y.N. Human hepatocellular carcinomas with “Stemness”-related marker expression: Keratin 19 expression and a poor prognosis. Hepatology 2011, 54, 1707–1717. [Google Scholar] [CrossRef]
- Saloustros, E.; Perraki, M.; Apostolaki, S.; Kallergi, G.; Xyrafas, A.; Kalbakis, K.; Agelaki, S.; Kalykaki, A.; Georgoulias, V.; Mavroudis, D. Cytokeratin-19 mRNA-positive circulating tumor cells during follow-up of patients with operable breast cancer: Prognostic relevance for late relapse. Breast Cancer Res. 2011, 13, R60. [Google Scholar] [CrossRef]
- Govaere, O.; Petz, M.; Wouters, J.; Vandewynckel, Y.-P.; Scott, E.J.; Topal, B.; Nevens, F.; Verslype, C.; Anstee, Q.M.; Van Vlierberghe, H.; et al. The PDGFRα-laminin B1-keratin 19 cascade drives tumor progression at the invasive front of human hepatocellular carcinoma. Oncogene 2017. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Liu, Y.; Liu, X.; Wang, R.; Liao, J.; Wu, S.; Fan, J.; Peng, Z.; Li, B.; et al. Keratin 80 promotes migration and invasion of colorectal carcinoma by interacting with PRKDC via activating the AKT pathway. Cell Death Dis. 2018, 9, 1009. [Google Scholar] [CrossRef]
- Bordeleau, F.; Galarneau, L.; Gilbert, S.; Loranger, A.; Marceau, N. Keratin 8/18 modulation of protein kinase C-mediated integrin-dependent adhesion and migration of liver epithelial cells. Mol. Biol. Cell 2010, 21, 1698–1713. [Google Scholar] [CrossRef]
- Fortier, A.-M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef]
- Omary, M.B.; Ku, N.-O.; Strnad, P.; Hanada, S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Invest. 2009, 119, 1794–1805. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.-S.; Jiang, W.-H.; He, Y.; Wang, D.-S.; Wu, Z.-J.; Wu, D.-S.; Gao, L.; Bao, Y.; Shi, J.-Z.; Liu, B.; et al. KRT8 upregulation promotes tumor metastasis and is predictive of a poor prognosis in clear cell renal cell carcinoma. Oncotarget 2017. [Google Scholar] [CrossRef]
- Ku, N.-O.; Azhar, S.; Omary, M.B. Keratin 8 phosphorylation by p38 kinase regulates cellular keratin filament reorganization: Modulation by a keratin 1-like disease causing mutation. J. Biol. Chem. 2002, 277, 10775–10782. [Google Scholar] [CrossRef]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Baribault, H.; Penner, J.; Iozzo, R.V.; Wilson-Heiner, M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994, 8, 2964–2973. [Google Scholar] [CrossRef] [PubMed]
- Misiorek, J.O.; Lahdeniemi, I.A.; Nystrom, J.H.; Paramonov, V.M.; Gullmets, J.A.; Saarento, H.; Rivero-Muller, A.; Husoy, T.; Taimen, P.; Toivola, D.M. Keratin 8-deletion induced colitis predisposes to murine colorectal cancer enforced by the inflammasome and IL-22 pathway. Carcinogenesis 2016, 37, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, D.W.; Wilson, N.J.; Hill, A.J.; Rugg, E.L.; Porter, R.M.; Hutcheson, A.M.; Quinlan, R.A.; van Heel, D.; Parkes, M.; Jewell, D.P.; et al. Human keratin 8 mutations that disturb filament assembly observed in inflammatory bowel disease patients. J. Cell Sci. 2004, 117, 1989–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6574. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lan, F.; He, Q.; Lee, A.; Tang, C.Z.; Dong, L.; Lan, B.; Ma, X.; Wu, J.C.; Shen, L. Inducible expression of stem cell associated intermediate filament nestin reveals an important role in glioblastoma carcinogenesis. Int. J. Cancer 2011, 128, 343–351. [Google Scholar] [CrossRef]
- Rutka, J.T.; Ivanchuk, S.; Mondal, S.; Taylor, M.; Sakai, K.; Dirks, P.; Jun, P.; Jung, S.; Becker, L.E.; Ackerley, C. Co-expression of nestin and vimentin intermediate filaments in invasive human astrocytoma cells. Int. J. Dev. Neurosci. 1999, 17, 503–515. [Google Scholar] [CrossRef]
- Roth, W.; Kumar, V.; Beer, H.D.; Richter, M.; Wohlenberg, C.; Reuter, U.; Thiering, S.; Staratschek-Jox, A.; Hofmann, A.; Kreusch, F.; et al. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J. Cell Sci. 2012, 125, 5269–5279. [Google Scholar] [CrossRef]
- Lu, H.; Chen, J.; Planko, L.; Zigrino, P.; Klein-Hitpass, L.; Magin, T.M. Induction of inflammatory cytokines by a keratin mutation and their repression by a small molecule in a mouse model for EBS. J. Invest. Dermatol. 2007, 127, 2781–2789. [Google Scholar] [CrossRef]
- Lessard, J.C.; Pina-Paz, S.; Rotty, J.D.; Hickerson, R.P.; Kaspar, R.L.; Balmain, A.; Coulombe, P.A. Keratin 16 regulates innate immunity in response to epidermal barrier breach. Proc. Natl. Acad. Sci. USA 2013, 110, 19537–19542. [Google Scholar] [CrossRef] [Green Version]
- Cheah, M.T.; Chen, J.Y.; Sahoo, D.; Contreras-Trujillo, H.; Volkmer, A.K.; Scheeren, F.A.; Volkmer, J.-P.; Weissman, I.L. CD14-expressing cancer cells establish the inflammatory and proliferative tumor microenvironment in bladder cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 4725–4730. [Google Scholar] [CrossRef] [Green Version]
- McGowan, K.M.; Coulombe, P.A. Onset of keratin 17 expression coincides with the definition of major epithelial lineages during skin development. J. Cell Biol. 1998, 143, 469–486. [Google Scholar] [CrossRef]
- SundarRaj, N.; Rizzo, J.D.; Anderson, S.C.; Gesiotto, J.P. Expression of vimentin by rabbit corneal epithelial cells during wound repair. Cell Tissue Res. 1992, 267, 347–356. [Google Scholar] [CrossRef]
- Yoon, K.H.; Yoon, M.; Moir, R.D.; Khuon, S.; Flitney, F.W.; Goldman, R.D. Insights into the dynamic properties of keratin intermediate filaments in living epithelial cells. J. Cell Biol. 2001, 153, 503–516. [Google Scholar] [CrossRef]
- Klymkowsky, M.W. Vimentin and keratin intermediate filament systems in cultured PtK2 epithelial cells are interrelated. EMBO J. 1982, 1, 161–165. [Google Scholar] [CrossRef]
- Velez-delValle, C.; Marsch-Moreno, M.; Castro-Munozledo, F.; Galvan-Mendoza, I.J.; Kuri-Harcuch, W. Epithelial cell migration requires the interaction between the vimentin and keratin intermediate filaments. Sci. Rep. 2016, 6, 24389. [Google Scholar] [CrossRef]
- Lee, I.-C.; Choi, B.Y. Withaferin-A—A Natural Anticancer Agent with Pleitropic Mechanisms of Action. Int. J. Mol. Sci. 2016, 17, 290. [Google Scholar] [CrossRef]
- Nagalingam, A.; Kuppusamy, P.; Singh, S.V.; Sharma, D.; Saxena, N.K. Mechanistic elucidation of the antitumor properties of withaferin a in breast cancer. Cancer Res. 2014, 74, 2617–2629. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| IF Protein | Mouse Model | IF Gene Alteration | Outcome upon IF Gene Alteration | Ref. |
|---|---|---|---|---|
| K10 | Transgenic-Skin carcinoma | overexpression of K10 | Decreased tumor growth | [54] |
| bovineK5-hK10 injected | ||||
| K14 | Xenograft-Breast carcinoma | K14 shRNA | Decreased metastasis | [55] |
| MMTV-PyMT tumor cells injected | ||||
| K17 | Transgenic-Skin carcinoma | Krt17−/− | Decreased tumor growth | [50] |
| K5-Gli2tg | ||||
| K17 | Transgenic-Skin carcinoma | Krt17−/− | Decreased tumor growth | [51] |
| K14-HPV16tg | ||||
| K17 | Xenograft-Ewing Sarcoma | K17 shRNA | Decreased tumor growth | [52] |
| A673 and SK-N-MC cell lines injected | ||||
| K17 | Xenograft-Cervical carcinoma | K17 shRNA | Decreased tumor growth | [53] |
| SiHa and CaSki cell lines injected | ||||
| K19 | Xenograft-Breast carcinoma | K19 antibody injected to block K19 | Decreased tumor growth | [56] |
| KPL-4 cell line injected | ||||
| K19 | Xenograft-Breast carcinoma | K19 shRNA | Increased tumor growth | [57] |
| SKBR3 cell line injected | ||||
| K8 | Xenograft-Oral cavity squamous cell carcinoma | overexpression of KRT8 WT, S73A or S431A | K8 S73A and S431A increased tumor growth compared to WT | [58] |
| AW13516 cell line injected | ||||
| Vimentin | Transgenic-Lung adenocarcinoma | Vim−/− | Decreased metastasis | [59] |
| LSL-KrasG12D/Lkb1fl/fl | ||||
| Vimentin | Xenograft-Teratocarcinoma | Vim−/− | No change | [60] |
| Embryonic stem cells injected | ||||
| Vimentin | Xenograft-MCK–KR–hIGF-IR | Vimentin shRNA | Decreased metastasis | [61] |
| MVT-1 cell line injected | ||||
| Vimentin | Xenograft-Leiomyosarcoma | overexpression of VIM S39A or S29D | Vim S39D increased tumor growth and metastasis | [62] |
| SKLMS1 cell line injected | ||||
| Nestin | Xenograft-Nasopharyngeal carcinoma | Nestin shRNA | Decreased tumor growth | [63] |
| 5-8F cell line injected | ||||
| Nestin | Xenograft-Hepatocellular carcinoma | Nestin shRNA | Decreased tumor growth | [64] |
| Huh7 cell line injected | ||||
| Nestin | Transgenic-Hepatocellular carcinoma | Nestin shRNA | Decreased tumor growth | [64] |
| Transposons encoding YAP and p53 shRNA injected | ||||
| Nestin | Xenograft-Glioblastoma | Nestin shRNA | Decreased tumor growth | [65] |
| A172 cell line injected | ||||
| Nestin | Xenograft-Pancreatic carcinoma | Nestin shRNA | Decreased metastasis | [66] |
| PANC cell line injected | ||||
| Nestin | Xenograft-Pancreatic carcinoma | Nestin shRNA | Decreased tumor growth | [67] |
| KLM-1 cell line injected |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, P.; Alsharif, S.; Fallatah, A.; Chung, B.M. Intermediate Filaments as Effectors of Cancer Development and Metastasis: A Focus on Keratins, Vimentin, and Nestin. Cells 2019, 8, 497. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050497
Sharma P, Alsharif S, Fallatah A, Chung BM. Intermediate Filaments as Effectors of Cancer Development and Metastasis: A Focus on Keratins, Vimentin, and Nestin. Cells. 2019; 8(5):497. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050497
Chicago/Turabian StyleSharma, Pooja, Sarah Alsharif, Arwa Fallatah, and Byung Min Chung. 2019. "Intermediate Filaments as Effectors of Cancer Development and Metastasis: A Focus on Keratins, Vimentin, and Nestin" Cells 8, no. 5: 497. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050497