ZAP-70 Regulates Autoimmune Arthritis via Alterations in T Cell Activation and Apoptosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
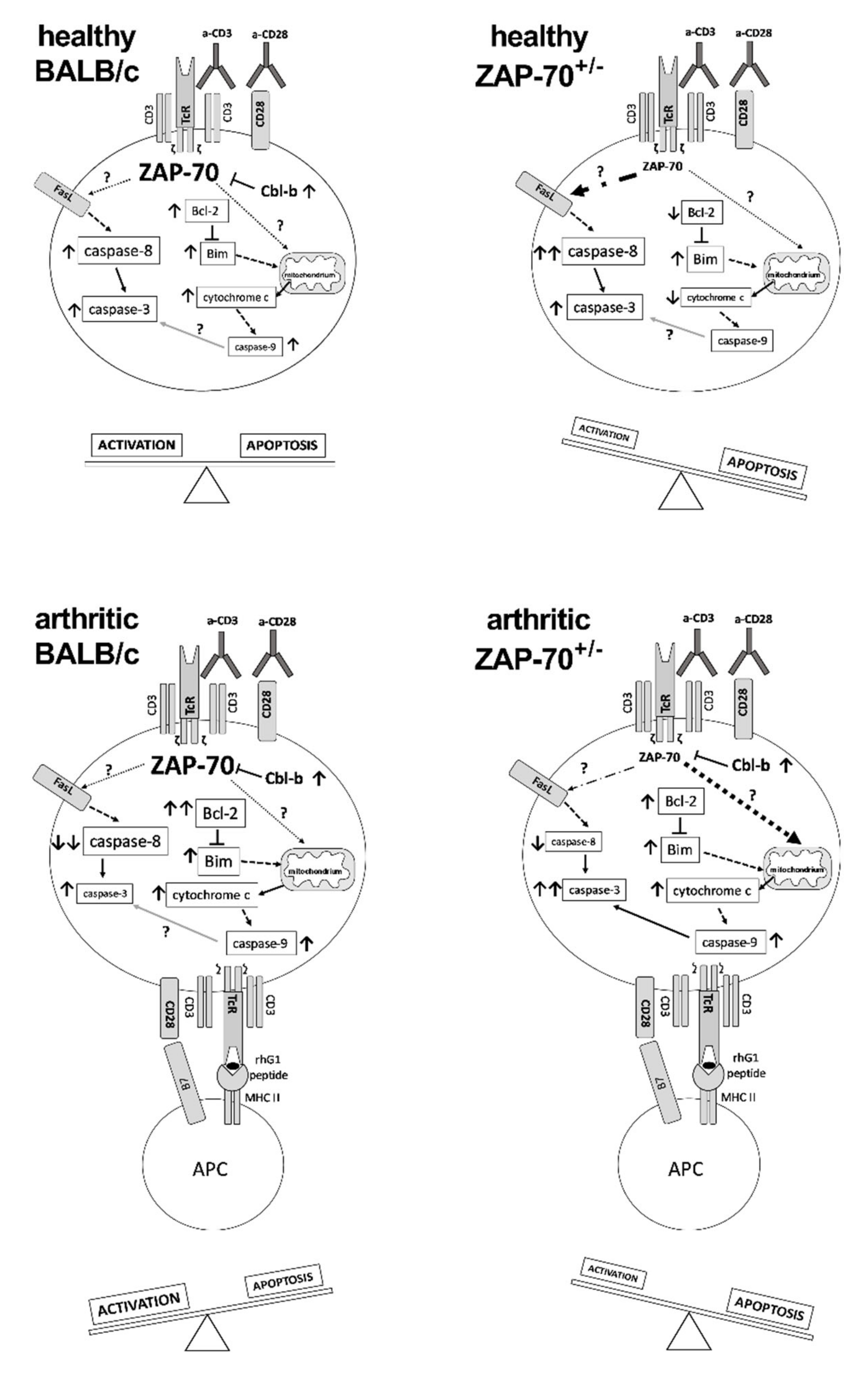
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Arthritis Induction
2.3. In Vivo Bioluminescent Imaging
2.4. Intracellular Cytokine Measurement
2.5. In Vitro Spleen Cell Culture
2.6. Antigen-Specific Proliferation
2.7. ELISA Measurements
2.8. T Cell Activation and Apoptosis
2.9. Statistical Methods
3. Results
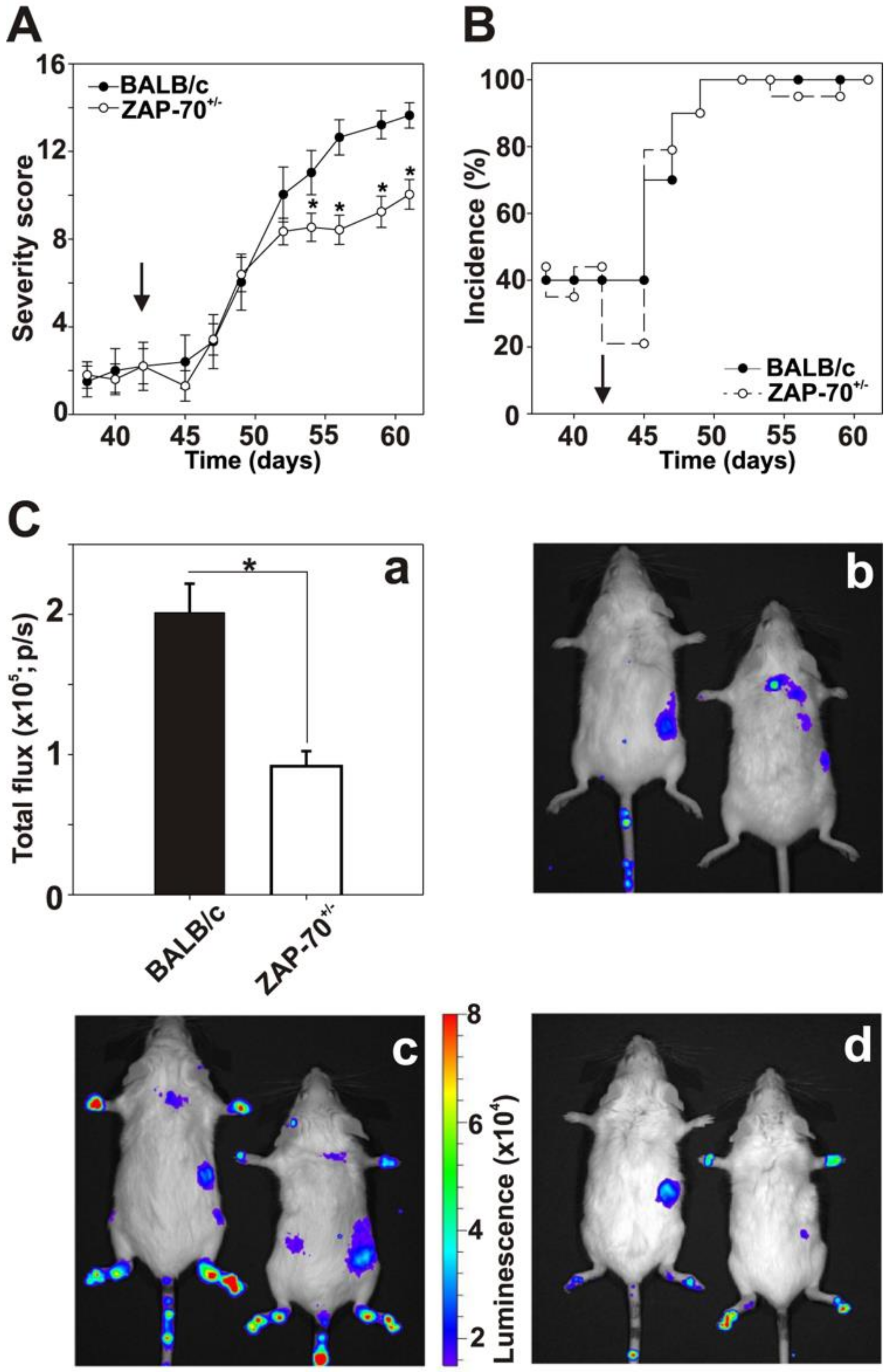
3.1. Partial Deficiency of the ZAP-70 Ameliorated the Clinical Picture of Autoimmune Arthritis
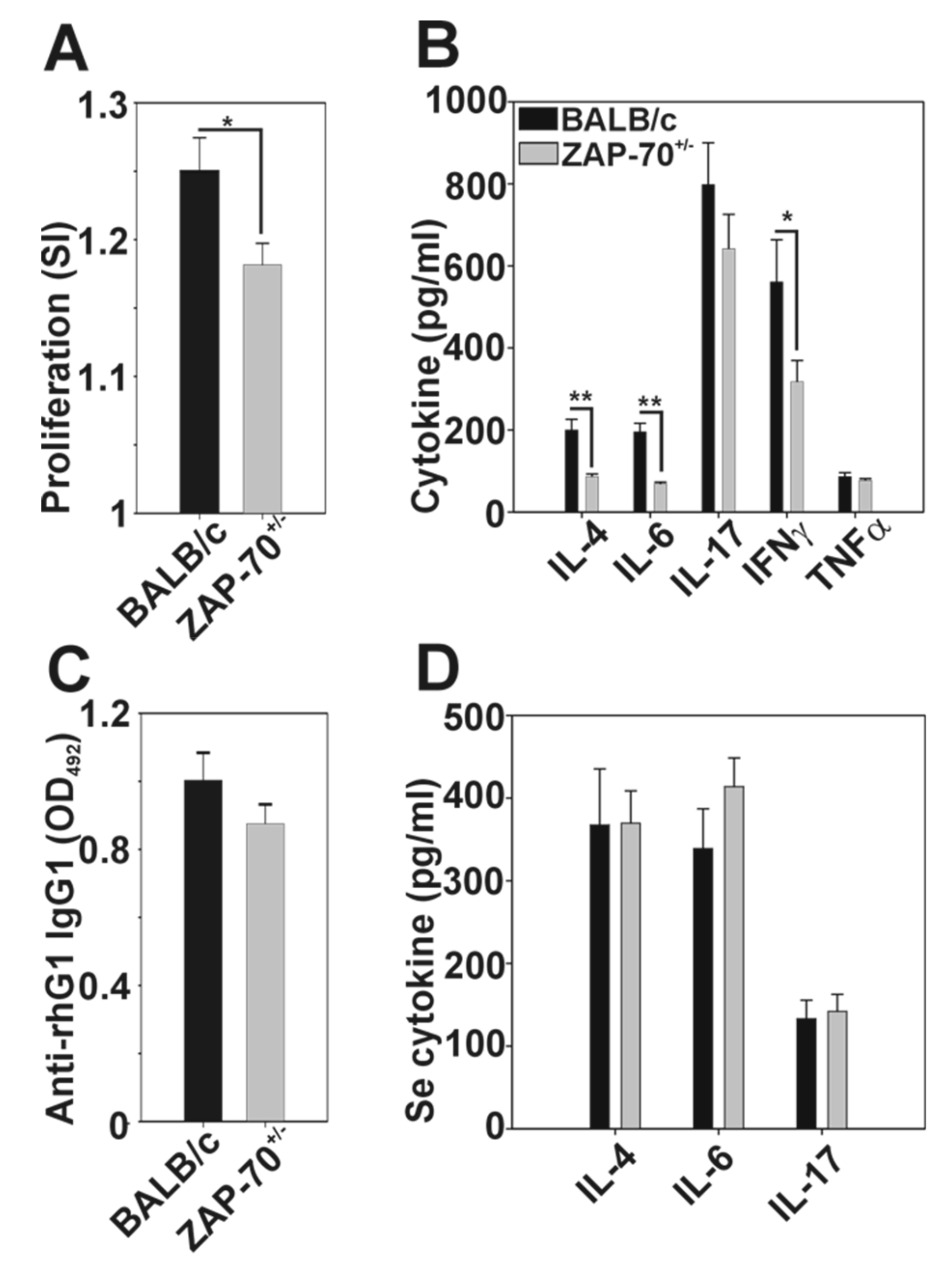
3.2. Comparison of the G1-Specific Immune Response between ZAP-70+/− and Control Mice
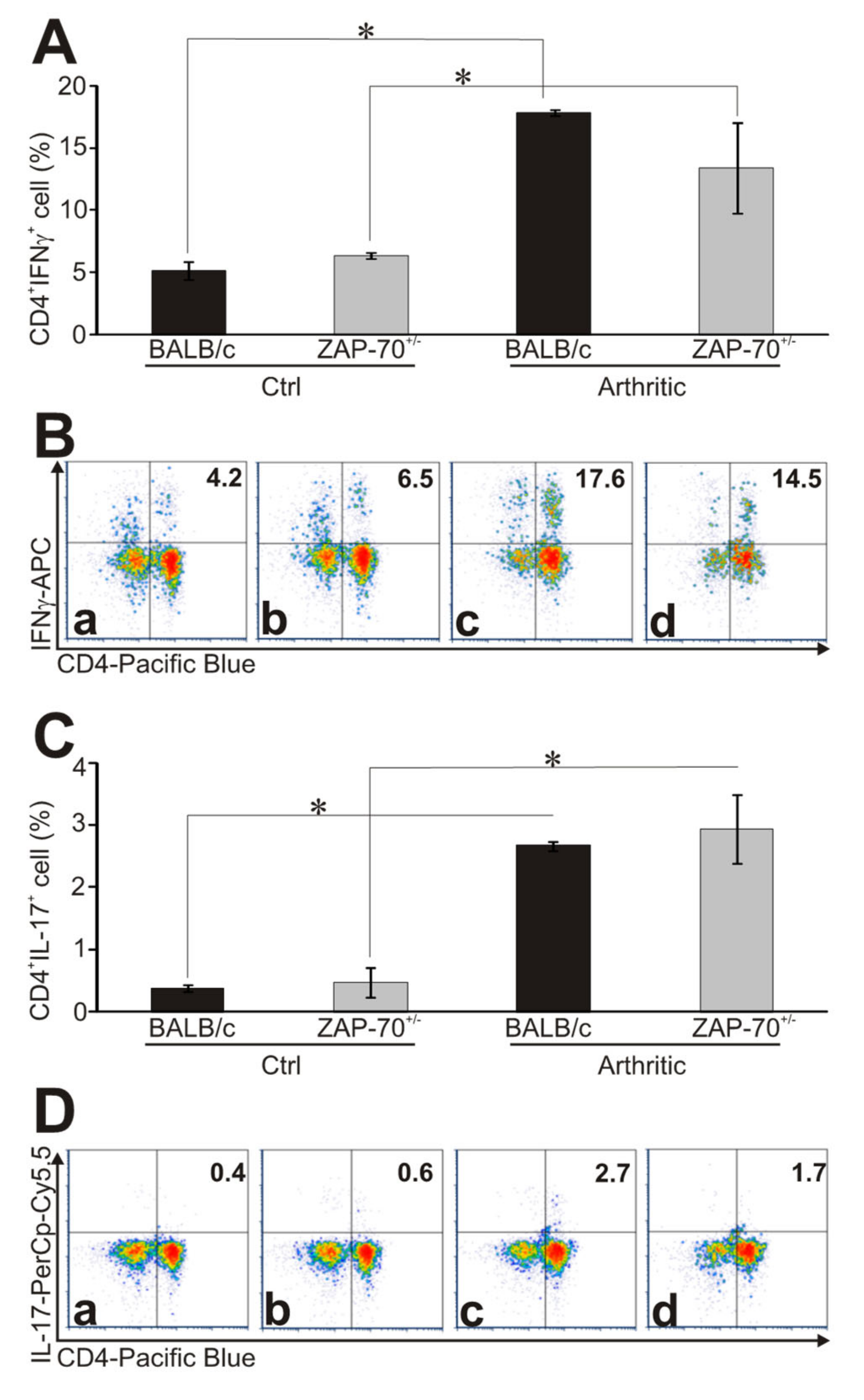
3.3. Altered Th1/Th17 Polarization in the Partially ZAP-70 Deficient Arthritic Mice
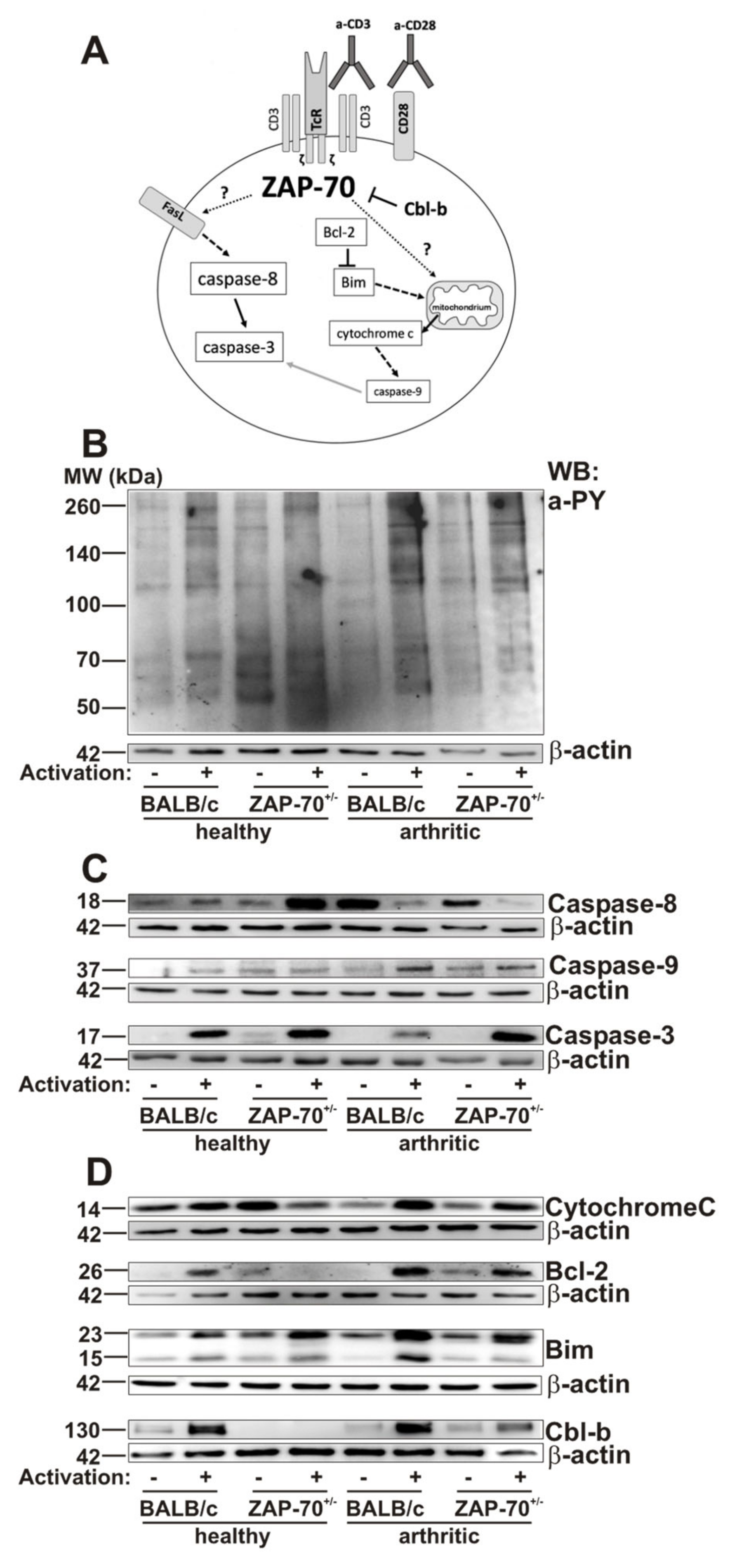
3.4. Alterations in T Cell Activation and Apoptosis in ZAP-7+/− Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, A.C.; Irving, B.A.; Fraser, J.D.; Weiss, A. The Zeta Chain Is Associated with a Tyrosine Kinase and upon T-Cell Antigen Receptor Stimulation Associates with ZAP-70, a 70-KDa Tyrosine Phosphoprotein. Proc. Natl. Acad. Sci. USA 1991, 88, 9166–9170. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Iwashima, M.; Turck, C.W.; Weiss, A. ZAP-70: A 70 Kd Protein-Tyrosine Kinase That Associates with the TCR Zeta Chain. Cell 1992, 71, 649–662. [Google Scholar] [CrossRef]
- Vivier, E.; da Silva, A.J.; Ackerly, M.; Levine, H.; Rudd, C.E.; Anderson, P. Association of a 70-KDa Tyrosine Phosphoprotein with the CD16: Zeta: Gamma Complex Expressed in Human Natural Killer Cells. Eur. J. Immunol. 1993, 23, 1872–1876. [Google Scholar] [CrossRef]
- Scielzo, C.; Camporeale, A.; Geuna, M.; Alessio, M.; Poggi, A.; Zocchi, M.R.; Chilosi, M.; Caligaris-Cappio, F.; Ghia, P. ZAP-70 Is Expressed by Normal and Malignant Human B-Cell Subsets of Different Maturational Stage. Leukemia 2006, 20, 689–695. [Google Scholar] [CrossRef]
- Chakupurakal, G.; Bell, A.; Griffiths, M.; Wandroo, F.; Moss, P. Analysis of ZAP70 Expression in Adult Acute Lymphoblastic Leukaemia by Real Time Quantitative PCR. Mol. Cytogenet. 2012, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Schweighoffer, E.; Vanes, L.; Mathiot, A.; Nakamura, T.; Tybulewicz, V.L. Unexpected Requirement for ZAP-70 in Pre-B Cell Development and Allelic Exclusion. Immunity 2003, 18, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Fallah-Arani, F.; Schweighoffer, E.; Vanes, L.; Tybulewicz, V.L.J. Redundant Role for Zap70 in B Cell Development and Activation. Eur. J. Immunol. 2008, 38, 1721–1733. [Google Scholar] [CrossRef]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.S.; Hsu, L.-Y.; Freedman, T.S.; Weiss, A. ZAP-70: An Essential Kinase in T-Cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [PubMed]
- Kadlecek, T.A.; van Oers, N.S.C.; Lefrancois, L.; Olson, S.; Finlay, D.; Chu, D.H.; Connolly, K.; Killeen, N.; Weiss, A. Differential Requirements for ZAP-70 in TCR Signaling and T Cell Development. J. Immunol. 1998, 161, 4688–4694. [Google Scholar] [PubMed]
- Walkovich, K.; Vander Lugt, M. ZAP70-Related Combined Immunodeficiency; Rockville Pike: Bethesda, MD, USA, 1993. [Google Scholar]
- Palacios, E.H.; Weiss, A. Distinct Roles for Syk and ZAP-70 during Early Thymocyte Development. J. Exp. Med. 2007, 204, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Negishi, I.; Motoyama, N.; Nakayama, K.; Senju, S.; Hatakeyama, S.; Zhang, Q.; Chan, A.C.; Loh, D.Y. Essential Role for ZAP-70 in Both Positive and Negative Selection of Thymocytes. Nature 1995, 376, 435–438. [Google Scholar] [CrossRef]
- Au-Yeung, B.B.; Levin, S.E.; Zhang, C.; Hsu, L.-Y.; Cheng, D.A.; Killeen, N.; Shokat, K.M.; Weiss, A. A Genetically Selective Inhibitor Demonstrates a Function for the Kinase Zap70 in Regulatory T Cells Independent of Its Catalytic Activity. Nat. Immunol. 2010, 11, 1085–1092. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid Arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid Arthritis. Nat. Rev. Dis. Prim. 2018, 4, 18001. [Google Scholar] [CrossRef]
- Boissier, M.-C.; Semerano, L.; Challal, S.; Saidenberg-Kermanac’h, N.; Falgarone, G. Rheumatoid Arthritis: From Autoimmunity to Synovitis and Joint Destruction. J. Autoimmun. 2012, 39, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.; Matthys, P. Collagen-Induced Arthritis and Related Animal Models: How Much of Their Pathogenesis Is Auto-Immune, How Much Is Auto-Inflammatory? Cytokine Growth Factor Rev. 2011, 22, 339–344. [Google Scholar] [CrossRef]
- McNamee, K.; Williams, R.; Seed, M. Animal Models of Rheumatoid Arthritis: How Informative Are They? Eur. J. Pharmacol. 2015, 759, 278–286. [Google Scholar] [CrossRef]
- Kobezda, T.; Ghassemi-Nejad, S.; Mikecz, K.; Glant, T.T.; Szekanecz, Z. Of Mice and Men: How Animal Models Advance Our Understanding of T-Cell Function in RA. Nat. Rev. Rheumatol. 2014, 10, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Bessis, N.; Decker, P.; Assier, E.; Semerano, L.; Boissier, M.-C. Arthritis Models: Usefulness and Interpretation. Semin. Immunopathol. 2017, 39, 469–486. [Google Scholar] [CrossRef]
- Glant, T.T.; Radacs, M.; Nagyeri, G.; Olasz, K.; Laszlo, A.; Boldizsar, F.; Hegyi, A.; Finnegan, A.; Mikecz, K. Proteoglycan-Induced Arthritis and Recombinant Human Proteoglycan Aggrecan G1 Domain-Induced Arthritis in BALB/c Mice Resembling Two Subtypes of Rheumatoid Arthritis. Arthritis Rheum. 2011, 63, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, A.; Mikecz, K.; Tao, P.; Glant, T.T. Proteoglycan (Aggrecan)-Induced Arthritis in BALB/c Mice Is a Th1-Type Disease Regulated by Th2 Cytokines. J. Immunol. 1999, 163, 5383–5390. [Google Scholar]
- Boldizsar, F.; Tarjanyi, O.; Nemeth, P.; Mikecz, K.; Glant, T.T. Th1/Th17 Polarization and Acquisition of an Arthritogenic Phenotype in Arthritis-Susceptible BALB/c, but Not in MHC-Matched, Arthritis-Resistant DBA/2 Mice. Int. Immunol. 2009, 21, 511–522. [Google Scholar] [CrossRef]
- Cope, A.P.; Schulze-Koops, H.; Aringer, M. The Central Role of T Cells in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2007, 25, S4–S11. [Google Scholar]
- Cope, A.P. Studies of T-Cell Activation in Chronic Inflammation. Arthritis Res. 2002, 4, S197. [Google Scholar] [CrossRef]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [PubMed]
- Takemura, S.; Klimiuk, P.A.; Braun, A.; Goronzy, J.J.; Weyand, C.M. T Cell Activation in Rheumatoid Synovium Is B Cell Dependent. J. Immunol. 2001, 167, 4710–4718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalheiro, H.; Duarte, C.; Silva-Cardoso, S.; da Silva, J.A.P.; Souto-Carneiro, M.M. CD8+ T Cell Profiles in Patients with Rheumatoid Arthritis and Their Relationship to Disease Activity. Arthritis Rheumatol. 2015, 67, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Shoda, H.; Fujio, K.; Sakurai, K.; Ishigaki, K.; Nagafuchi, Y.; Shibuya, M.; Sumitomo, S.; Okamura, T.; Yamamoto, K. Autoantigen BiP-Derived HLA-DR4 Epitopes Differentially Recognized by Effector and Regulatory T Cells in Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1171–1181. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sakoda, H.; Nakajima, T.; Kato, T.; Okubo, M.; Dohi, M.; Mizushima, Y.; Ito, K.; Nishioka, K. Accumulation of Multiple T Cell Clonotypes in the Synovial Lesions of Patients with Rheumatoid Arthritis Revealed by a Novel Clonality Analysis. Int. Immunol. 1992, 4, 1219–1223. [Google Scholar] [CrossRef]
- Cutolo, M.; Sulli, A.; Paolino, S.; Pizzorni, C. CTLA-4 Blockade in the Treatment of Rheumatoid Arthritis: An Update. Expert Rev. Clin. Immunol. 2016, 12, 417–425. [Google Scholar] [CrossRef]
- Venerito, V.; Lopalco, G.; Cacciapaglia, F.; Fornaro, M.; Iannone, F. A Bayesian Mixed Treatment Comparison of Efficacy of Biologics and Small Molecules in Early Rheumatoid Arthritis. Clin. Rheumatol. 2019, 38, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Iannone, F.; Lopalco, G.; Cantarini, L.; Galeazzi, M.; Lapadula, G. Efficacy and Safety of Combination Therapy for Preventing Bone Damage in Rheumatoid Arthritis. Clin. Rheumatol. 2016, 35, 19–23. [Google Scholar] [CrossRef]
- Malemud, C.; Malemud, J.C. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells 2018, 7, 223. [Google Scholar] [CrossRef]
- Scatizzi, J.C.; Hutcheson, J.; Bickel, E.; Haines, G.K.; Perlman, H. Pro-Apoptotic Bid Is Required for the Resolution of the Effector Phase of Inflammatory Arthritis. Arthritis Res. Ther. 2007, 9, R49. [Google Scholar] [CrossRef] [PubMed]
- Hanyecz, A.; Olasz, K.; Tarjanyi, O.; Nemeth, P.; Mikecz, K.; Glant, T.T.; Boldizsar, F. Proteoglycan Aggrecan Conducting T Cell Activation and Apoptosis in a Murine Model of Rheumatoid Arthritis. Biomed Res. Int. 2014, 2014, 942148. [Google Scholar] [CrossRef]
- Szodoray, P.; Jellestad, S.; Nakken, B.; Brun, J.G.; Jonsson, R. Programmed Cell Death in Rheumatoid Arthritis Peripheral Blood T-Cell Subpopulations Determined by Laser Scanning Cytometry. Lab. Invest. 2003, 83, 1839–1848. [Google Scholar] [CrossRef] [Green Version]
- Lundy, S.K.; Sarkar, S.; Tesmer, L.A.; Fox, D.A. Cells of the Synovium in Rheumatoid Arthritis. T Lymphocytes. Arthritis Res. Ther. 2007, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One Year in Review 2018: Pathogenesis of Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar]
- Sakaguchi, N.; Takahashi, T.; Hata, H.; Nomura, T.; Tagami, T.; Yamazaki, S.; Sakihama, T.; Matsutani, T.; Negishi, I.; Nakatsuru, S.; et al. Altered Thymic T-Cell Selection Due to a Mutation of the ZAP-70 Gene Causes Autoimmune Arthritis in Mice. Nature 2003, 426, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Hashimoto, M.; Yoshitomi, H.; Tanaka, S.; Nomura, T.; Yamaguchi, T.; Iwakura, Y.; Sakaguchi, N.; Sakaguchi, S. T Cell Self-Reactivity Forms a Cytokine Milieu for Spontaneous Development of IL-17+ Th Cells That Cause Autoimmune Arthritis. J. Exp. Med. 2007, 204, 41–47. [Google Scholar] [CrossRef]
- Thomas, R.; Turner, M.; Cope, A.P. High Avidity Autoreactive T Cells with a Low Signalling Capacity through the T-Cell Receptor: Central to Rheumatoid Arthritis Pathogenesis? Arthritis Res. Ther. 2008, 10, 210. [Google Scholar] [CrossRef]
- Gross, S.; Gammon, S.T.; Moss, B.L.; Rauch, D.; Harding, J.; Heinecke, J.W.; Ratner, L.; Piwnica-Worms, D. Bioluminescence Imaging of Myeloperoxidase Activity in Vivo. Nat. Med. 2009, 15, 455–461. [Google Scholar] [CrossRef]
- Tarjanyi, O.; Boldizsar, F.; Nemeth, P.; Mikecz, K.; Glant, T.T. Age-Related Changes in Arthritis Susceptibility and Severity in a Murine Model of Rheumatoid Arthritis. Immun. Ageing 2009, 6, 8. [Google Scholar] [CrossRef]
- Kugyelka, R.; Kohl, Z.; Olasz, K.; Mikecz, K.; Rauch, T.A.; Glant, T.T.; Boldizsar, F. Enigma of IL-17 and Th17 Cells in Rheumatoid Arthritis and in Autoimmune Animal Models of Arthritis. Mediators Inflamm. 2016, 2016, 6145810. [Google Scholar] [CrossRef]
- Brenner, D.; Krammer, P.H.; Arnold, R. Concepts of Activated T Cell Death. Crit. Rev. Oncol. Hematol. 2008, 66, 52–64. [Google Scholar] [CrossRef]
- Arakaki, R.; Yamada, A.; Kudo, Y.; Hayashi, Y.; Ishimaru, N. Mechanism of Activation-Induced Cell Death of T Cells and Regulation of FasL Expression. Crit. Rev. Immunol. 2014, 34, 301–314. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and Cell Death: Outer Membrane Permeabilization and Beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Czabotar, P.E. The BCL-2 Family of Proteins and Mitochondrial Outer Membrane Permeabilisation. Semin. Cell Dev. Biol. 2017, 72, 152–162. [Google Scholar] [CrossRef]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef]
- Shamim, M.; Nanjappa, S.G.; Singh, A.; Plisch, E.H.; LeBlanc, S.E.; Walent, J.; Svaren, J.; Seroogy, C.; Suresh, M. Cbl-b Regulates Antigen-Induced TCR down-Regulation and IFN-Gamma Production by Effector CD8 T Cells without Affecting Functional Avidity. J. Immunol. 2007, 179, 7233–7243. [Google Scholar] [CrossRef] [PubMed]
- Olasz, K.; Boldizsar, F.; Kis-Toth, K.; Tarjanyi, O.; Hegyi, A.; van Eden, W.; Rauch, T.A.; Mikecz, K.; Glant, T.T. T Cell Receptor (TCR) Signal Strength Controls Arthritis Severity in Proteoglycan-Specific TCR Transgenic Mice. Clin. Exp. Immunol. 2012, 167, 346–355. [Google Scholar] [CrossRef]
- Farber, D.L.; Hussain, S.F.; Farber, D.L. Differential TCR Signaling and the Generation of Memory T Cells. J. Immunol. 1998, 160, 535–539. [Google Scholar]
- Qingjun, L.; Zhou, H.; Langdon, W.; Zhang, J. E3 Ubiquitin Ligase Cbl-b in Innate and Adaptive Immunity. Cell Cycle 2014, 13, 1875–1884. [Google Scholar] [CrossRef]
- Zhang, Z.; Elly, C.; Qiu, L.; Altman, A.; Liu, Y.C. A Direct Interaction between the Adaptor Protein Cbl-b and the Kinase Zap-70 Induces a Positive Signal in T Cells. Curr. Biol. 1999, 9, 203–206. [Google Scholar] [CrossRef]
- Jeon, M.-S.; Atfield, A.; Venuprasad, K.; Krawczyk, C.; Sarao, R.; Elly, C.; Yang, C.; Arya, S.; Bachmaier, K.; Su, L.; et al. Essential Role of the E3 Ubiquitin Ligase Cbl-b in T Cell Anergy Induction. Immunity 2004, 21, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Liu, Y.; Koretzky, G.A.; Durden, D.L. SLP-76-Cbl-Grb2-Shc Interactions in FcgammaRI Signaling. Blood 1998, 92, 1697–1706. [Google Scholar]
- Bubeck Wardenburg, J.; Fu, C.; Jackman, J.K.; Flotow, H.; Wilkinson, S.E.; Williams, D.H.; Johnson, R.; Kong, G.; Chan, A.C.; Findell, P.R. Phosphorylation of SLP-76 by the ZAP-70 Protein-Tyrosine Kinase Is Required for T-Cell Receptor Function. J. Biol. Chem. 1996, 271, 19641–19644. [Google Scholar] [CrossRef]
- Doníz-Padilla, L.; Martínez-Jiménez, V.; Niño-Moreno, P.; Abud-Mendoza, C.; Hernández-Castro, B.; González-Amaro, R.; Layseca-Espinosa, E.; Baranda-Cándido, L. Expression and Function of Cbl-b in T Cells from Patients with Systemic Lupus Erythematosus, and Detection of the 2126 A/G Cblb Gene Polymorphism in the Mexican Mestizo Population. Lupus 2011, 20, 628–635. [Google Scholar] [CrossRef]
- Korb, A.; Pavenstädt, H.; Pap, T. Cell Death in Rheumatoid Arthritis. Apoptosis 2009, 14, 447–454. [Google Scholar] [CrossRef]
- Van Loosdregt, J.; Rossetti, M.; Spreafico, R.; Moshref, M.; Olmer, M.; Williams, G.W.; Kumar, P.; Copeland, D.; Pischel, K.; Lotz, M.; et al. Increased Autophagy in CD4 + T Cells of Rheumatoid Arthritis Patients Results in T-Cell Hyperactivation and Apoptosis Resistance. Eur. J. Immunol. 2016, 46, 2862–2870. [Google Scholar] [CrossRef]
- Malemud, C.J. Chondrocyte Apoptosis in Rheumatoid Arthritis: Is Preventive Therapy Possible? Immunotherapy 2015, 1, 102. [Google Scholar] [CrossRef]
- Green, D.R.; Droin, N.; Pinkoski, M. Activation-Induced Cell Death in T Cells. Immunol. Rev. 2003, 193, 70–81. [Google Scholar] [CrossRef]
- Arnold, R.; Brenner, D.; Becker, M.; Frey, C.R.; Krammer, P.H. How T Lymphocytes Switch between Life and Death. Eur. J. Immunol. 2006, 36, 1654–1658. [Google Scholar] [CrossRef]
- Zhang, J.; Bárdos, T.; Mikecz, K.; Finnegan, A.; Glant, T.T. Impaired Fas Signaling Pathway Is Involved in Defective T Cell Apoptosis in Autoimmune Murine Arthritis. J. Immunol. 2001, 166, 4981–4986. [Google Scholar] [CrossRef]
- Zhang, N.; Hopkins, K.; He, Y.-W. C-FLIP Protects Mature T Lymphocytes from TCR-Mediated Killing. J. Immunol. 2008, 181, 5368–5373. [Google Scholar] [CrossRef]
- Eischen, C.M.; Williams, B.L.; Zhang, W.; Samelson, L.E.; Lynch, D.H.; Abraham, R.T.; Leibson, P.J. ZAP-70 Tyrosine Kinase Is Required for the up-Regulation of Fas Ligand in Activation-Induced T Cell Apoptosis. J. Immunol. 1997, 159, 1135–1139. [Google Scholar]
- Zhong, L.; Wu, C.-H.; Lee, W.-H.; Liu, C.-P. Zeta-Associated Protein of 70 KDa (ZAP-70), but Not Syk, Tyrosine Kinase Can Mediate Apoptosis of T Cells through the Fas/Fas Ligand, Caspase-8 and Caspase-3 Pathways. J. Immunol. 2004, 172, 1472–1482. [Google Scholar] [CrossRef]
- Zhan, Y.; Carrington, E.M.; Zhang, Y.; Heinzel, S.; Lew, A.M. Life and Death of Activated T Cells: How Are They Different from Naïve T Cells? Front. Immunol. 2017. [Google Scholar] [CrossRef]
- Sandalova, E.; Wei, C.-H.; Masucci, M.G.; Levitsky, V. Regulation of Expression of Bcl-2 Protein Family Member Bim by T Cell Receptor Triggering. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef]
- Hildeman, D.A.; Zhu, Y.; Mitchell, T.C.; Bouillet, P.; Strasser, A.; Kappler, J.; Marrack, P. Activated T Cell Death In vivo Mediated by Proapoptotic Bcl-2 Family Member Bim. Immunity 2002. [Google Scholar] [CrossRef]
- Zhu, Y.; Swanson, B.J.; Wang, M.; Hildeman, D.A.; Schaefer, B.C.; Liu, X.; Suzuki, H.; Mihara, K.; Kappler, J.; Marrack, P. Constitutive Association of the Proapoptotic Protein Bim with Bcl-2-Related Proteins on Mitochondria in T Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7681–7686. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, X. Cytochrome C-Mediated Apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kugyelka, R.; Prenek, L.; Olasz, K.; Kohl, Z.; Botz, B.; Glant, T.T.; Berki, T.; Boldizsár, F. ZAP-70 Regulates Autoimmune Arthritis via Alterations in T Cell Activation and Apoptosis. Cells 2019, 8, 504. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050504
Kugyelka R, Prenek L, Olasz K, Kohl Z, Botz B, Glant TT, Berki T, Boldizsár F. ZAP-70 Regulates Autoimmune Arthritis via Alterations in T Cell Activation and Apoptosis. Cells. 2019; 8(5):504. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050504
Chicago/Turabian StyleKugyelka, Réka, Lilla Prenek, Katalin Olasz, Zoltán Kohl, Bálint Botz, Tibor T. Glant, Timea Berki, and Ferenc Boldizsár. 2019. "ZAP-70 Regulates Autoimmune Arthritis via Alterations in T Cell Activation and Apoptosis" Cells 8, no. 5: 504. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050504