Impact of Heat Shock Protein 90 Inhibition on the Proteomic Profile of Lung Adenocarcinoma as Measured by Two-Dimensional Electrophoresis Coupled with Mass Spectrometry

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. siRNA Transfection
2.3. Drug Treatment
2.4. Immunoblotting
2.5. Proteomic Sample Preparation
2.6. Two-Dimensional Electrophoresis and Gel Image Analysis
2.7. MALDI-TOF/TOF Mass Spectrometry Analysis
2.8. Protein Identification
2.9. Protein Functional Analysis
3. Results
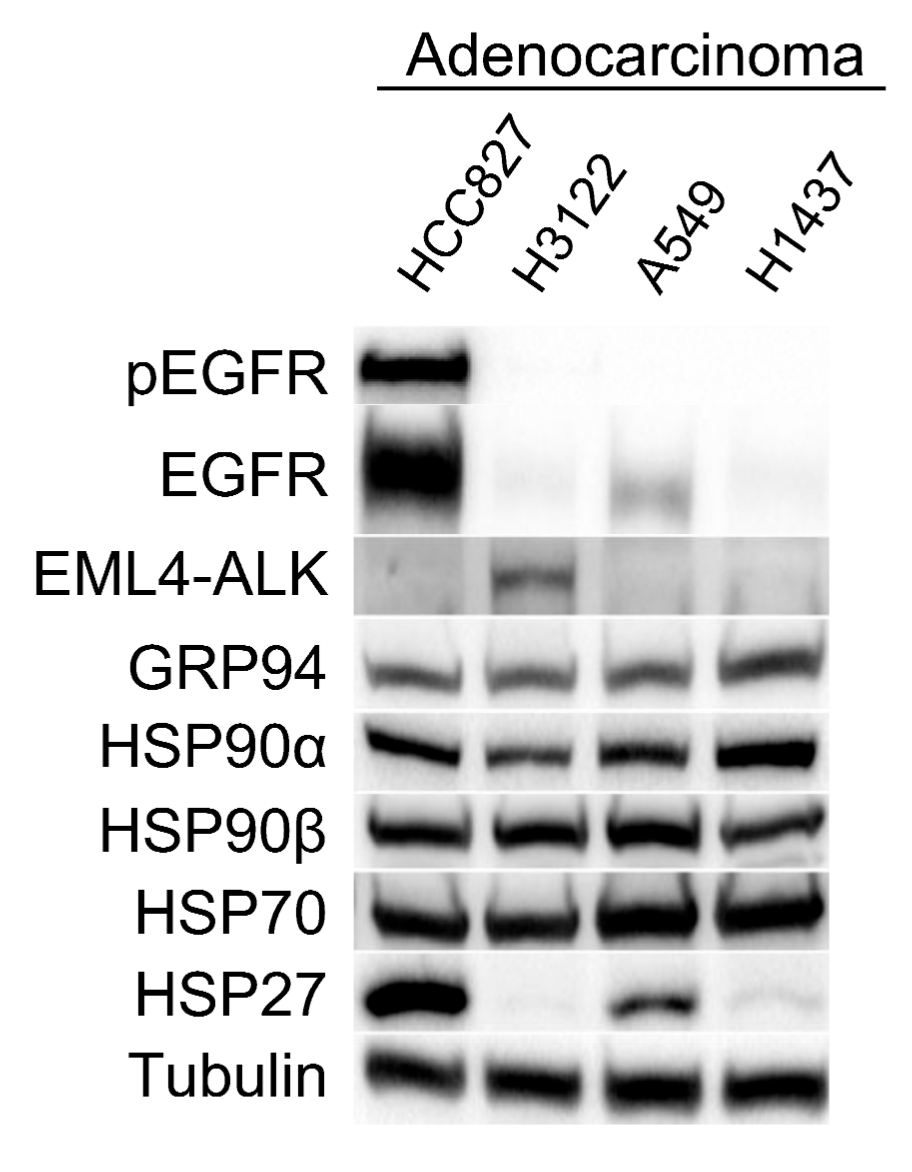
3.1. Characterization of Lung Adenocarcinoma Cell Lines
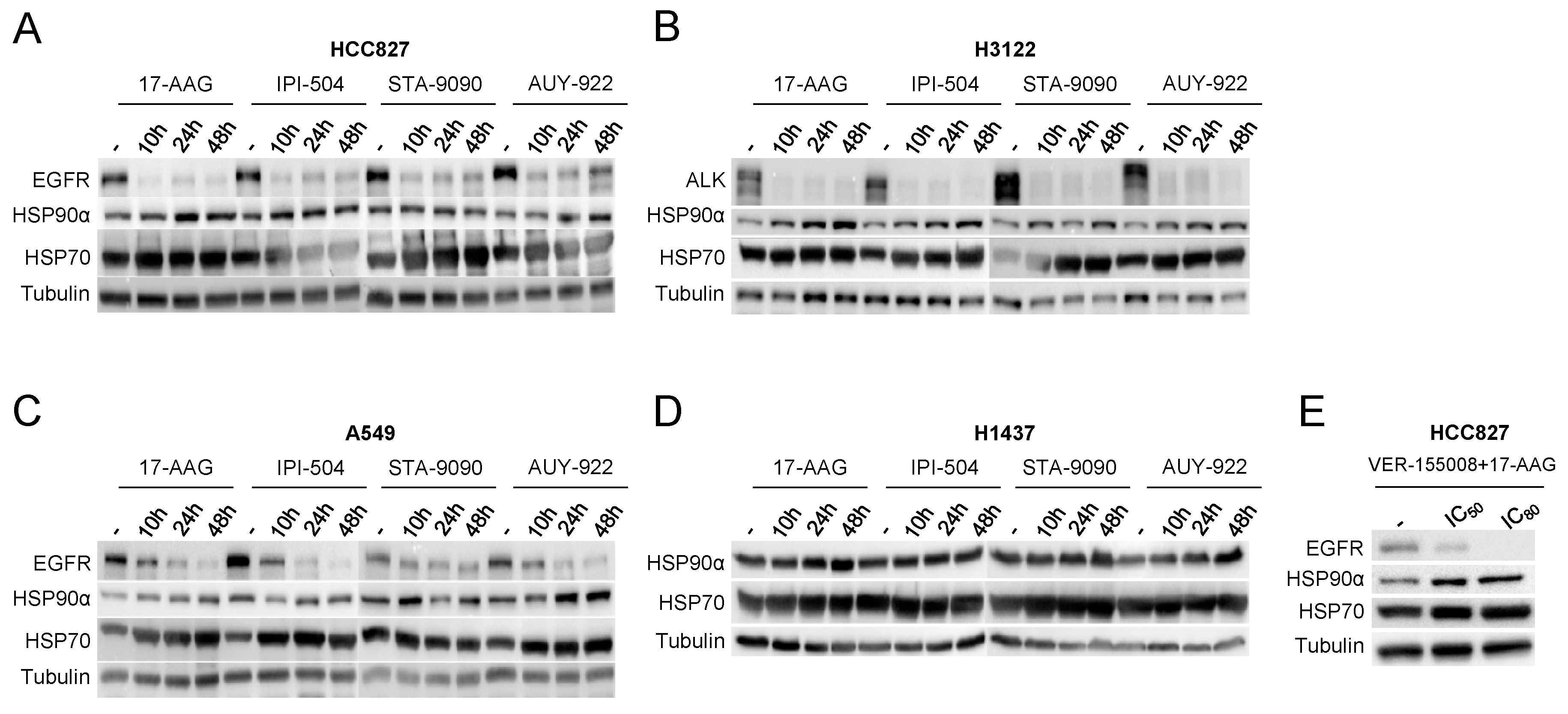
3.2. Effectiveness of HSP90 Inhibitors in Lung Adenocarcinoma Cell Lines
3.3. Effects of HSP90 Inhibition on Protein Expression Patterns in Adenocarcinoma Cell Lines
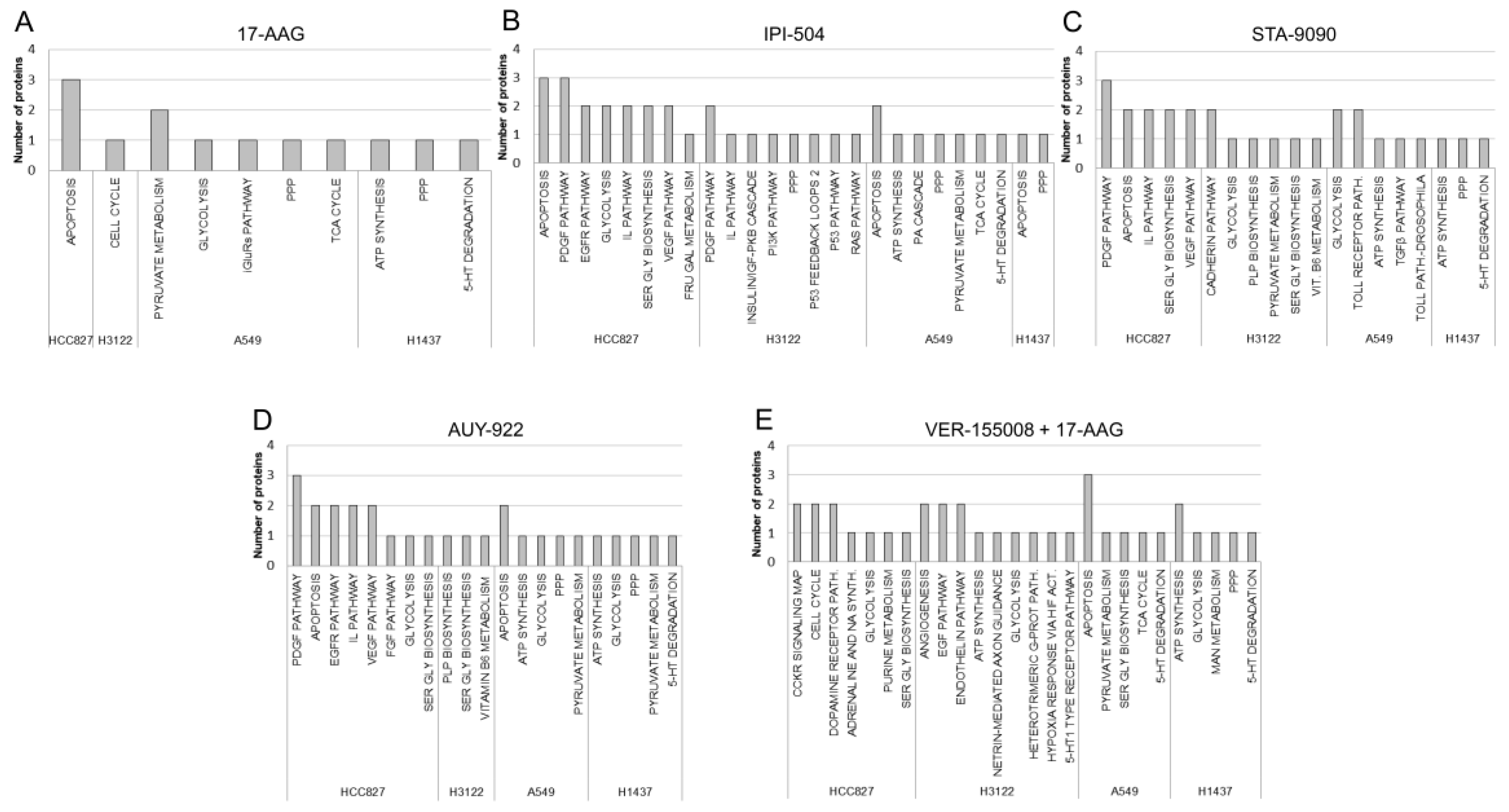
3.4. Functional Annotation Analysis of Differentially Expressed Proteins
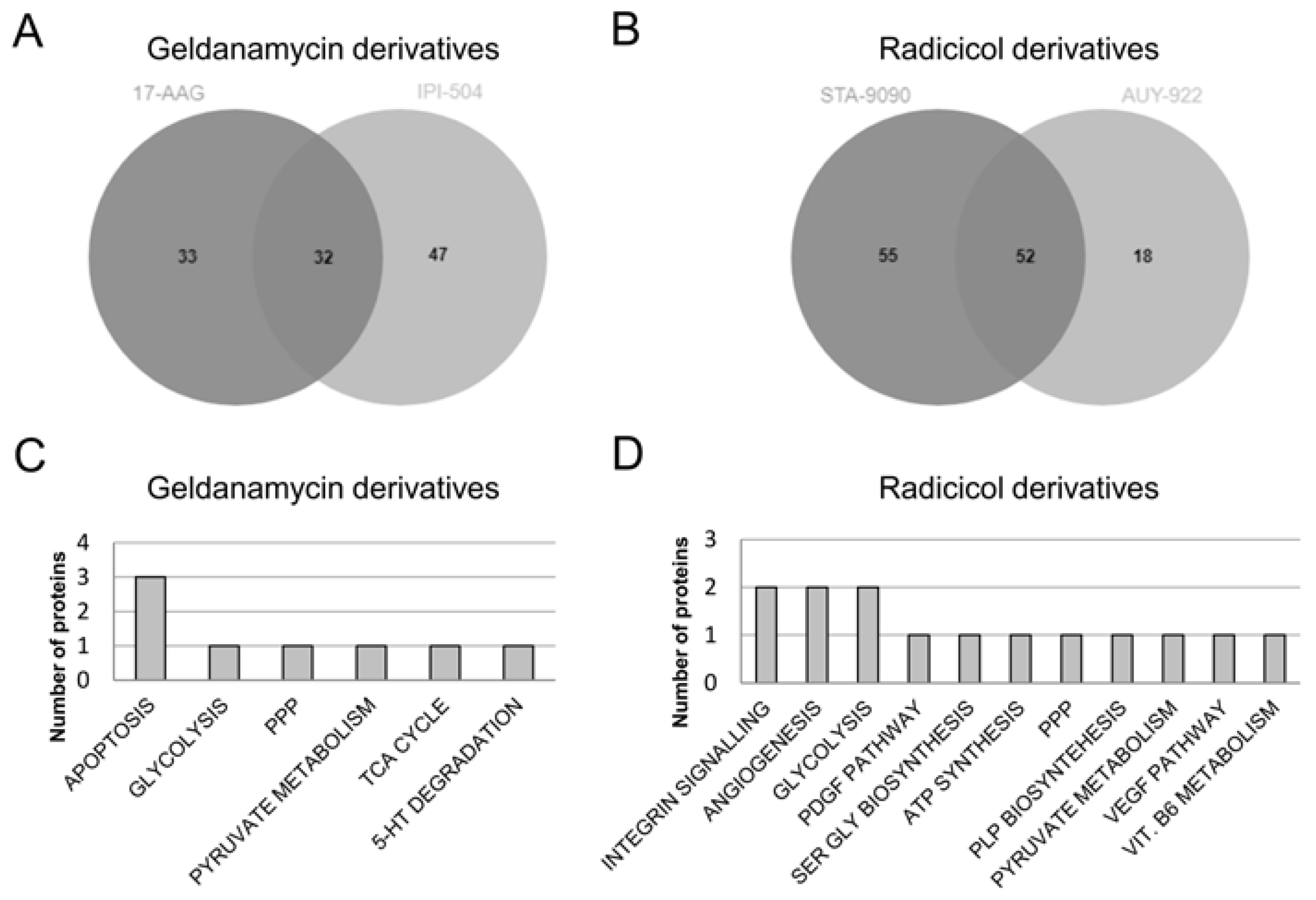
3.5. Protein Expression Profiles and Signalling Pathways Associated with the HSP90 Inhibitor Family in Lung Adenocarcinoma Cell Lines
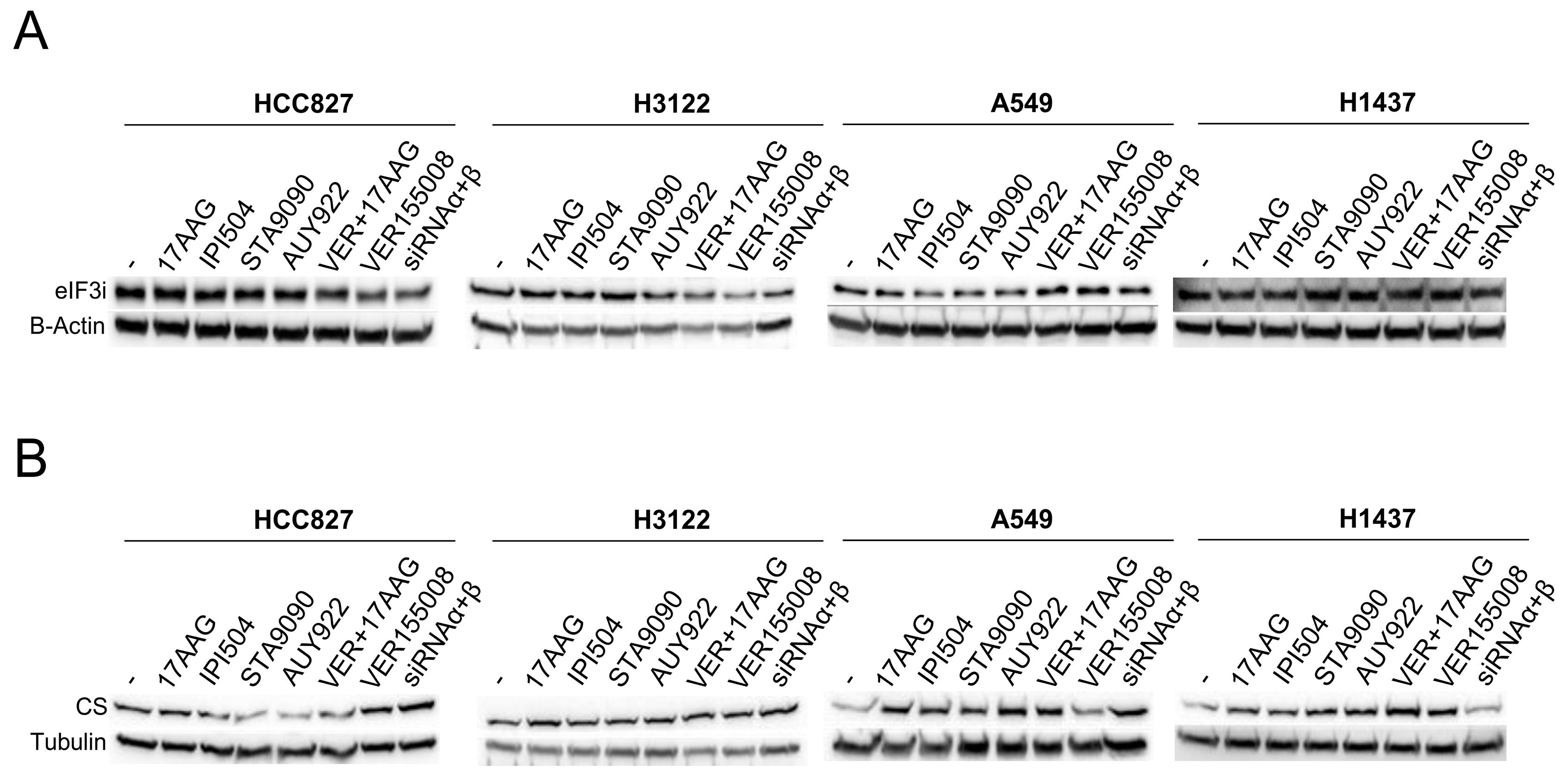
3.6. Validation of the Differential Expression of Proteins Involved in the Response to Chaperone Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Molina-Pinelo, S.; Lopez-Rios, F.; Paz-Ares, L. Biological therapies in nonsmall cell lung cancer. Eur. Respir. J. 2017, 49, 1601520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, A.; A Socinski, M.; Burns, T.F. Personalized treatment of EGFR mutant and ALK-positive patients in NSCLC. Expert Opin. Pharmacother. 2014, 15, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bhattacharya, S.; A Socinski, M.; Burns, T.F. HSP90 inhibitors in lung cancer: Promise still unfulfilled. Clin. Adv. Hematol. Oncol. 2016, 14, 346–356. [Google Scholar] [PubMed]
- Thunnissen, E.; Van Der Oord, K.; Bakker, M.D. Prognostic and predictive biomarkers in lung cancer. A review. Virchows Arch. 2014, 464, 347–358. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar]
- Sequist, L.V.; Gettinger, S.; Senzer, N.N.; Martins, R.G.; Janne, P.A.; Lilenbaum, R.; Gray, J.E.; Iafrate, A.J.; Katayama, R.; Hafeez, N.; et al. Activity of IPI-504, a Novel Heat-Shock Protein 90 Inhibitor, in Patients With Molecularly Defined Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 4953–4960. [Google Scholar] [CrossRef]
- Ruiz, M.I.G.; Floor, K.; Roepman, P.; Rodriguez, J.A.; Meijer, G.A.; Mooi, W.J.; Jassem, E.; Nikliński, J.; Muley, T.; Van Zandwijk, N.; et al. Integration of Gene Dosage and Gene Expression in Non-Small Cell Lung Cancer, Identification of HSP90 as Potential Target. PLoS ONE 2008, 3, e0001722. [Google Scholar]
- Liu, W.; Wu, Y.; Wang, L.; Gao, L.; Wang, Y.; Liu, X.; Zhang, K.; Song, J.; Wang, H.; A Bayer, T.; et al. Protein signature for non-small cell lung cancer prognosis. Am. J. Cancer Res. 2014, 4, 256–269. [Google Scholar]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef]
- Park, K.-S.; Oh, B.; Lee, M.-H.; Nam, K.-Y.; Jin, H.R.; Yang, H.; Choi, J.; Kim, S.-W.; Lee, D.H. The HSP90 inhibitor, NVP-AUY922, sensitizes KRAS-mutant non-small cell lung cancer with intrinsic resistance to MEK inhibitor, trametinib. Cancer Lett. 2016, 372, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Toyooka, S.; Soh, J.; Yamamoto, H.; Dote, H.; Kawasaki, K.; Otani, H.; Kubo, T.; Jida, M.; Ueno, T.; et al. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-DMAG, on non-small-cell lung cancers being resistant to EGFR tyrosine kinase inhibitor. Lung Cancer 2012, 75, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, H.; Yamada, T.; Takeuchi, S.; Nakagawa, T.; Kita, K.; Nakamura, T.; Matsumoto, K.; Suda, K.; Mitsudomi, T.; Yano, S. Hsp90 Inhibition Overcomes HGF-Triggering Resistance to EGFR-TKIs in EGFR-Mutant Lung Cancer by Decreasing Client Protein Expression and Angiogenesis. J. Thorac. Oncol. 2012, 7, 1078–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, H.; Fujisawa, H.; Masaoka, A.; Noguchi, M.; Hirayama, R.; Takahashi, M.; Fujimori, A.; Okayasu, R. The combination of Hsp90 inhibitor 17AAG and heavy-ion irradiation provides effective tumor control in human lung cancer cells. Cancer Med. 2015, 4, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.F.; Vos, M.H.; Lindquist, S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc. Nat. Acad. Sci. USA 1997, 94, 12949–12956. [Google Scholar] [CrossRef] [PubMed]
- Weidenauer, L.; Wang, T.; Joshi, S.; Chiosis, G.; Quadroni, M.R. Proteomic interrogation of HSP90 and insights for medical research. Expert Rev. Proteom. 2017, 14, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Erlejman, A.G.; Lagadari, M.; Toneatto, J.; Piwien-Pilipuk, G.; Galigniana, M.D. Regulatory role of the 90-kDa-heat-shock protein (Hsp90) and associated factors on gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Shimamura, T. Epidermal Growth Factor Receptors Harboring Kinase Domain Mutations Associate with the Heat Shock Protein 90 Chaperone and Are Destabilized following Exposure to Geldanamycins. Cancer Res. 2005, 65, 6401–6408. [Google Scholar] [CrossRef] [Green Version]
- Da Rocha Dias, S.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Soga, S.; Beebe, K.; Lee, M.-J.; Kim, Y.S.; Trepel, J.; Neckers, L. Sensitivity of epidermal growth factor receptor and ErbB2 exon 20 insertion mutants to Hsp90 inhibition. Br. J. Cancer 2007, 97, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pashtan, I.; Tsutsumi, S.; Xu, W.; Neckers, L. Cancer cells harboring MET gene amplification activate alternative signaling pathways to escape MET inhibition but remain sensitive to Hsp90 inhibitors. Cell Cycle 2009, 8, 2050–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Kim, S.Y.; So, K.S.; Baek, I.-J.; Kim, W.S.; Choi, S.H.; Lee, J.C.; Bivona, T.G.; Rho, J.K.; Choi, C.-M. AUY922 Effectively Overcomes MET- and AXL-Mediated Resistance to EGFR-TKI in Lung Cancer Cells. PLoS ONE 2015, 10, e0119832. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Tsukuda, K.; Toyooka, S.; Ando, M.; Takaoka, M.; Soh, J.; Asano, H.; Maki, Y.; Muraoka, T.; Tanaka, N.; et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer 2012, 76, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Lin, J.; Feng, T. Effects of 17-AAG on the proliferation and apoptosis of human lung cancer A549 and H446 cells. Chin. J. Tuberc. Resp. Dis. 2015, 38, 267–272. [Google Scholar]
- Normant, E.; Paez, G.; A West, K.; Lim, A.R.; Slocum, K.L.; Tunkey, C.; McDougall, J.; A Wylie, A.; Robison, K.; Caliri, K.; et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4–ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2011, 30, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Cohen, V. HSP90 as a novel molecular target in non-small-cell lung cancer. Lung Cancer 2016, 7, 11–17. [Google Scholar] [PubMed]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A Multicenter Phase II Study of Ganetespib Monotherapy in Patients with Genotypically Defined Advanced Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef]
- Johnson, M.L.; Yu, H.A.; Hart, E.M.; Weitner, B.B.; Rademaker, A.W.; Patel, J.D.; Kris, M.G.; Riely, G.J. Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer with Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J. Clin. Oncol. 2015, 33, 1666–1673. [Google Scholar] [CrossRef]
- Felip, E.; Barlesi, F.; Besse, B.; Chu, Q.; Gandhi, L.; Kim, S.-W.; Carcereny, E.; Sequist, L.V.; Brunsvig, P.; Chouaïd, C.; et al. Phase 2 Study of the HSP-90 Inhibitor AUY922 in Previously Treated and Molecularly Defined Patients with Advanced Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of Hsp90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Rong, B.; Yang, S. Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and developments (Review). Int. J. Oncol. 2018, 52, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Pastor, M.D.; Nogal, A.; Molina-Pinelo, S.; Carnero, A.; Paz-Ares, L. Proteomic biomarkers in lung cancer. Clin. Trans. Oncol. 2013, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C. Application of proteomics in non-small-cell lung cancer. Expert Rev. Proteom. 2016, 13, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Marrugal, Á.; Ojeda, L.; Paz-Ares, L.; Molina-Pinelo, S.; Ferrer, I. Proteomic-Based Approaches for the Study of Cytokines in Lung Cancer. Dis. Markers 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, Y.; Du, J.; Ma, L.; Jiang, H.; Jin, F.; Yang, S. Early Candidate Biomarkers of Non-Small Cell Lung Cancer Are Screened and Identified in Premalignant Lung Lesions. Technol. Cancer Res. Treat. 2017, 16, 66–74. [Google Scholar] [CrossRef]
- Pastor, M.; Nogal, A.; Molina-Pinelo, S.; Melendez, R.; Salinas, A.; De La Peña, M.G.; Martín-Juan, J.; Corral, J.; Garcia-Carbonero, R.; Carnero, A.; et al. Identification of proteomic signatures associated with lung cancer and COPD. J. Proteom. 2013, 89, 227–237. [Google Scholar] [CrossRef]
- Kang, S.-M.; Sung, H.-J.; Ahn, J.-M.; Park, C.-S.; Park, J.-Y.; Lee, S.-Y.; Cho, J.-Y. The Haptoglobin β chain as a supportive biomarker for human lung cancers. Mol. BioSyst. 2011, 7, 1167. [Google Scholar] [CrossRef]
- Chou, H.-C.; Lin, D.-Y.; Wen, Y.-F.; Lin, C.-C.; Lin, S.-H.; Chung, T.-W.; Liao, E.-C.; Chen, J.-Y.; Lin, C.-H.; Chen, Y.-J.; et al. Identification of Up- and Down-Regulated Proteins in Pemetrexed-Resistant Human Lung Adenocarcinoma: Flavin Reductase and Calreticulin Play Key Roles in the Development of Pemetrexed-Associated Resistance. J. Proteome Res. 2015, 14, 4907–4920. [Google Scholar] [CrossRef]
- Nagano, K.; Imai, S.; Zhao, X.; Yamashita, T.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; Kamada, H.; Nakagawa, S.; Tsutsumi, Y.; et al. Identification and evaluation of metastasis-related proteins, oxysterol binding protein-like 5 and calumenin, in lung tumors. Int. J. Oncol. 2015, 47, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Okano, T.; Seike, M.; Kuribayashi, H.; Soeno, C.; Ishii, T.; Kida, K.; Gemma, A. Identification of haptoglobin peptide as a novel serum biomarker for lung squamous cell carcinoma by serum proteome and peptidome profiling. Int. J. Oncol. 2016, 48, 945–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Niu, Y.; Li, M.; Fang, S.; Guo, L. Identification of DJ-1 as a contributor to multidrug resistance in human small-cell lung cancer using proteomic analysis. Int. J. Exp. Pathol. 2017, 98, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tang, M.; Liu, Y.; Wang, J.; Wu, Z. Comparative Proteomics of Chromium-Transformed Beas-2B Cells by 2D-DIGE and MALDI-TOF/TOF MS. Boil. Trace Element Res. 2018, 185, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Voruganti, S.; Kline, J.T.; Balch, M.J.; Rogers, J.; Matts, R.L.; Hartson, S.D. Proteomic Profiling of Hsp90 Inhibitors. Methods Mol. Biol. 2018, 1709, 139–162. [Google Scholar] [CrossRef]
- Walter, R.; Pan, K.T.; Doebele, C.; Comoglio, F.; Tomska, K.; Bohnenberger, H.; Young, R.M.; Jacobs, L.; Keller, U.; Bonig, H.; et al. HSP90 promotes Burkitt lymphoma cell survival by maintaining tonic B-cell receptor signaling. Blood 2017, 129, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, E.V.; Centenera, M.M.; Moldovan, M.; Das, R.; Irani, S.; Vincent, A.D.; Chan, H.; Horvath, L.G.; Lynn, D.J.; Daly, R.J.; et al. Identification of Novel Response and Predictive Biomarkers to Hsp90 Inhibitors Through Proteomic Profiling of Patient-derived Prostate Tumor Explants. Mol. Cell. Proteom. 2018, 17, 1470–1486. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef]
- Condé, R.; Belak, Z.R.; Nair, M.; O’Carroll, R.F.; Ovsenek, N. Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochem. Cell Boil. 2009, 87, 845–851. [Google Scholar] [CrossRef]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef] [PubMed]
- Grem, J.L. Phase I and Pharmacologic Study of 17-(Allylamino)-17-Demethoxygeldanamycin in Adult Patients with Solid Tumors. J. Clin. Oncol. 2005, 23, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Loo, A.; Jaeger, S.; Bagdasarian, L.; Yu, J.; Chung, F.; Korn, J.; Ruddy, D.; Guo, R.; et al. Targeting HSF1 sensitizes cancer cells to HSP90 inhibition. Oncotarget 2013, 4, 816–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, A.; Barone, G.; Lauria, A.; Martorana, A.; Piccionello, A.P.; Pierro, P.; Terenzi, A.; Almerico, A.M.; Buscemi, S.; Campanella, C.; et al. Hsp60, a novel target for antitumor therapy: structure-function features and prospective drugs design. Curr. Pharm. Des. 2013, 19, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Minegishi, H. HSP60 as a drug target. Curr. Pharm. Des. 2013, 19, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, W.; Shao, W.; Yin, W.; Chen, H.; Qiu, Y.; Mo, M.; Zhao, J.; Deng, Q.; He, J. Heat shock protein-60 expression was significantly correlated with the prognosis of lung adenocarcinoma. J. Surg. Oncol. 2011, 104, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Ağababaoğlu, İ.; Önen, A.; Demir, A.B.; Aktaş, S.; Altun, Z.; Ersöz, H.; Şanl, A.; Özdemir, N.; Akkoçlu, A. Chaperonin (HSP60) and annexin-2 are candidate biomarkers for non-small cell lung carcinoma. Medicine 2017, 96, e5903. [Google Scholar] [CrossRef]
- Korfanty, J.; Stokowy, T.; Widlak, P.; Gogler-Piglowska, A.; Handschuh, L.; Podkowiński, J.; Vydra, N.; Naumowicz, A.; Toma-Jonik, A.; Widlak, W. Crosstalk between HSF1 and HSF2 during the heat shock response in mouse testes. Int. J. Biochem. Cell Biol. 2014, 57, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Stanhill, A.; Levin, V.; Hendel, A.; Shachar, I.; Kazanov, D.; Arber, N.; Kaminski, N.; Engelberg, D. Ha-ras(val12) induces HSP70b transcription via the HSE/HSF1 system, but HSP70b expression is suppressed in Ha-ras(val12)-transformed cells. Oncogene 2006, 25, 1485–1495. [Google Scholar] [CrossRef]
- Ramirez, V.P.; Stamatis, M.; Shmukler, A.; Aneskievich, B.J. Basal and stress-inducible expression of HSPA6 in human keratinocytes is regulated by negative and positive promoter regions. Cell Stress Chaperones 2015, 20, 95–107. [Google Scholar] [CrossRef]
- Khalouei, S.; Chow, A.M.; Brown, I.R. Stress-induced localization of HSPA6 (HSP70B’) and HSPA1A (HSP70-1) proteins to centrioles in human neuronal cells. Cell Stress Chaperones 2014, 19, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-S.; Song, J.-H.; Hwang, B.; Noh, D.-H.; Park, S.L.; Kim, W.T.; Park, S.-S.; Kim, W.-J.; Moon, S.-K. HSPA6 augments garlic extract-induced inhibition of proliferation, migration, and invasion of bladder cancer EJ cells; Implication for cell cycle dysregulation, signaling pathway alteration, and transcription factor-associated MMP-9 regulation. PLoS ONE 2017, 12, e0171860. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.; Kaul, S.C.; Ryu, J.; Lee, J.-S.; Ahn, H.M.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.-O.; Wadhwa, R. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-O.; Bhargava, P.; Na, Y.; Lee, J.-S.; Ryu, J.; Kaul, S.C.; Wadhwa, R. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci. Rep. 2017, 7, 42016. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Che, S.-L.; Piao, J.-J.; Xu, M.; Chen, L.-Y.; Lin, Z.-H. Mortalin overexpression predicts poor prognosis in early stage of non–small cell lung cancer. Tumor Boil. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Grills, C.; Jithesh, P.V.; Blayney, J.; Zhang, S.-D.; Fennell, D.A. Gene Expression Meta-Analysis Identifies VDAC1 as a Predictor of Poor Outcome in Early Stage Non-Small Cell Lung Cancer. PLoS ONE 2011, 6, e14635. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.P.; Wadhwa, R.; Luk, J.M. Mortalin–p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yan, X.; Zhang, G.; Zhao, W.; Jiao, S. The predictive value of ERCC1 and p53 for the effect of panobinostat and cisplatin combination treatment in NSCLC. Oncotarget 2015, 6, 18997–19005. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, M.; Gurtner, K.; Can, Y.; Brammer, I.; Rieckmann, T.; Oertel, R.; Wysocki, M.; Dorniok, F.; Gal, A.; Grob, T.J.; et al. Radiosensitization of NSCLC cells by EGFR inhibition is the result of an enhanced p53-dependent G1 arrest. Radiother. Oncol. 2015, 115, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Rockliffe, N.; Johnson, G.G.; Sherrington, P.D.; Pettitt, A.R. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene 2008, 27, 2445–2455. [Google Scholar] [CrossRef] [PubMed]
- Chai, K.; Ning, X.; Nguyễn, T.T.T.; Zhong, B.; Morinaga, T.; Li, Z.; Shingyoji, M.; Tada, Y.; Tatsumi, K.; Shimada, H.; et al. Heat shock protein 90 inhibitors augment endogenous wild-type p53 expression but down-regulate the adenovirally-induced expression by inhibiting a proteasome activity. Oncotarget 2018, 9, 26130–26143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.-L.; Hwang, J.; Perng, R.-P. p53 gene status modulates the chemosensitivity of non-small cell lung cancer cells. J. Biomed. Sci. 2000, 7, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Duarte, A.; Lacerda, R.; Menezes, J.; Romão, L. eIF3: A factor for human health and disease. RNA Biol. 2018, 15, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Spilka, R.; Ernst, C.; Mehta, A.K.; Haybaeck, J.; Seeböck, R. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013, 340, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Hershey, J.W. Regulation of protein synthesis and the role of eIF3 in cancer. Braz. J. Med Boil. Res. 2010, 43, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershey, J.W. The role of eIF3 and its individual subunits in cancer. Biochim. Biophys. Acta 2015, 1849, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Katsumata, R.; Okuda, T.; Yamamoto, T.; Miyazaki, K.; Senga, T.; Machida, K.; A Thant, A.; Nakatsugawa, S.; Hamaguchi, M. Molecular cloning and characterization of human MAWD, a novel protein containing WD-40 repeats frequently overexpressed in breast cancer. Cancer Res. 2000, 60, 13–17. [Google Scholar] [PubMed]
- Huang, J.-S.; Chao, C.-C.; Su, T.-L.; Yeh, S.-H.; Chen, D.-S.; Chen, C.-T.; Chen, P.-J.; Jou, Y.-S. Diverse cellular transformation capability of overexpressed genes in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2004, 315, 950–958. [Google Scholar] [CrossRef]
- Ahlemann, M.; Zeidler, R.; Lang, S.; Mack, B.; Münz, M.; Gires, O. Carcinoma-associated eIF3i overexpression facilitates mTOR-dependent growth transformation. Mol. Carcinog. 2006, 45, 957–967. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Lin, K.-T.; Chen, S.-C.; Gu, D.-L.; Chen, C.-F.; Tu, P.-H.; Jou, Y.-S. Overexpressed-eIF3I interacted and activated oncogenic Akt1 is a theranostic target in human hepatocellular carcinoma. Hepatology 2013, 58, 239–250. [Google Scholar] [CrossRef]
- Qi, J.; Dong, Z.; Liu, J.; Zhang, J.T. EIF3i promotes colon oncogenesis by regulating COX-2 protein synthesis and beta-catenin activation. Oncogene 2014, 33, 4156–4163. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.X.; Joseph, P.; Ong, T.M. Antisense inhibition of translation initiation factor 3 reverses its oncogenic potential. Teratog. Carcinog. Mutagen 2002, 22, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Lei, Y.-X.; Ong, T.-M. Up-regulation of expression of translation factors—A novel molecular mechanism for cadmium carcinogenesis. Mol. Cell. Biochem. 2004, 255, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, Y.; Yao, S.; Shi, H.; Huang, X.; Li, Y.; Wei, Y.; Lin, S. The Translation Initiation Factor eIF3i Up-regulates Vascular Endothelial Growth Factor A, Accelerates Cell Proliferation, and Promotes Angiogenesis in Embryonic Development and Tumorigenesis. J. Boil. Chem. 2014, 289, 28310–28323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, P.; Zhang, Q.; Yao, X.; Zhao, L.; Liu, Y.; Liu, X.; Tao, R.; Yu, C.; Li, Y.; et al. eIF3i activity is critical for endothelial cells in tumor induced angiogenesis through regulating VEGFR and ERK translation. Oncotarget 2017, 8, 19968–19979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucci, C.; Chiariello, M. Signal transduction gRABs attention. Cell. Signal. 2006, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Boil. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Masuda, E.S.; Luo, Y.; Young, C.; Shen, M.; Rossi, A.B.; Huang, B.C.; Yu, S.; Bennett, M.K.; Payan, D.G.; Scheller, R.H. Rab37 is a novel mast cell specific GTPase localized to secretory granules. FEBS Lett. 2000, 470, 61–64. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Tseng, R.-C.; Hsu, H.-S.; Wang, Y.-C.; Hsu, M.-T. Frequent down-regulation of hRAB37 in metastatic tumor by genetic and epigenetic mechanisms in lung cancer. Lung Cancer 2009, 63, 360–367. [Google Scholar] [CrossRef]
- Tzeng, H.-T.; Li, T.-H.; Tang, Y.-A.; Tsai, C.-H.; Lu, P.-J.F.; Lai, W.-W.; Chiang, C.-W.; Wang, Y.-C. Phosphorylation of Rab37 by protein kinase C alpha inhibits the exocytosis function and metastasis suppression activity of Rab37. Oncotarget 2017, 8, 108556–108570. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Cheng, H.-C.; Wang, Y.-S.; Lin, P.; Jen, J.; Kuo, I.-Y.; Chang, Y.-H.; Liao, P.-C.; Chen, R.-H.; Yuan, W.-C.; et al. Small GTPase Rab37 targets tissue inhibitor of metalloproteinase 1 for exocytosis and thus suppresses tumour metastasis. Nat. Commun. 2014, 5, 4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA mismatch repair gene homologue hMLH 1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, B.; Yuan, Y.; Fei, J.; Wang, J. MutL homolog 1 expression in thyroid carcinoma and its clinical significance. J. Cancer Res. Ther. 2016, 12, 281. [Google Scholar] [CrossRef]
- Hu, H.; Chen, X.; Zhou, C.; Li, B.; Yang, Y.; Ying, X.; Mao, Y.; Zhang, Y.; Zhong, J.; Dai, J.; et al. Aberrant methylation of mutL homolog 1 is associated with increased risk of non-small cell lung cancer. J. Clin. Lab. Anal. 2018, 32, e22370. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Li, S.; Huang, C. Clinical significance of chromodomain helicase/ATPase DNA binding protein 1-like and human mutL homolog 1 gene expression in cholangiocarcinoma. Oncol. Lett. 2018, 16, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Shi, B.; Chu, J.; Tian, T. Promoter methylation of human mutL homolog 1 and colorectal cancer risk: A meta-analysis. J. Cancer Res. Ther. 2018, 14, 851. [Google Scholar] [CrossRef]
- Hashimoto, T.; Kurokawa, Y.; Takahashi, T.; Miyazaki, Y.; Tanaka, K.; Makino, T.; Yamasaki, M.; Nakajima, K.; Ikeda, J.-I.; Mori, M.; et al. Predictive value of MLH1 and PD-L1 expression for prognosis and response to preoperative chemotherapy in gastric cancer. Gastric Cancer 2019, 22, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweitzig, D.R.; Smirnov, D.A.; Connelly, M.C.; Terstappen, L.W.M.M.; O’Hara, S.M.; Moran, E. Physiological stress induces the metastasis marker AGR2 in breast cancer cells. Mol. Cell. Biochem. 2007, 306, 255–260. [Google Scholar] [CrossRef]
- Di Maro, G.; Salerno, P.; Unger, K.; Orlandella, F.M.; Monaco, M.; Chiappetta, G.; Thomas, G.; Oczko-Wojciechowska, M.; Masullo, M.; Jarząb, B.; et al. Anterior gradient protein 2 promotes survival, migration and invasion of papillary thyroid carcinoma cells. Mol. Cancer 2014, 13, 160. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, D.H.; Kang, D.; Kim, J.H. Expression of anterior gradient 2 is decreased with the progression of human biliary tract cancer. Tohoku J. Exp. Med. 2014, 234, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Maresh, E.L.; Mah, V.; Alavi, M.; Horvath, S.; Bagryanova, L.; Liebeskind, E.S.; A Knutzen, L.; Zhou, Y.; Chia, D.; Liu, A.Y.; et al. Differential expression of anterior gradient gene AGR2 in prostate cancer. BMC Cancer 2010, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.; Villalobos, V.M.; Gevaert, O.; Abramovitz, M.; Williams, C.; Sikic, B.I.; Leyland-Jones, B. Single Gene Prognostic Biomarkers in Ovarian Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0149183. [Google Scholar] [CrossRef]
- Riener, M.O.; Thiesler, T.; Hellerbrand, C.; Amann, T.; Cathomas, G.; Fritzsche, F.R.; Dahl, E.; Bahra, M.; Weichert, W.; Terracciano, L.; et al. Loss of anterior gradient-2 expression is an independent prognostic factor in colorectal carcinomas. Eur. J. Cancer 2014, 50, 1722–1730. [Google Scholar] [CrossRef]
- Alves, M.R.; E Melo, N.C.; Barros-Filho, M.C.; do Amaral, N.S.; Silva, F.I.B.; Baiocchi Neto, G.; Soares, F.A.; de Brot Andrade, L.; Rocha, R.M. Downregulation of AGR2, p21, and cyclin D and alterations in p53 function were associated with tumor progression and chemotherapy resistance in epithelial ovarian carcinoma. Cancer Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Millares, L.; Barreiro, E.; Cortés, R.; Martinez-Romero, A.; Balcells, C.; Cascante, M.; Enguita, A.B.; Alvarez, C.; Rami-Porta, R.; De Cos, J.S.; et al. Tumor-associated metabolic and inflammatory responses in early stage non-small cell lung cancer: Local patterns and prognostic significance. Lung Cancer 2018, 122, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-B.; Gu, J.-D.; Zhou, Q.-H. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Arelaki, S.; Koukourakis, M.I. Expression of enzymes related to glucose metabolism in non-small cell lung cancer and prognosis. Exp. Lung Res. 2017, 43, 1–8. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate Synthase Expression Affects Tumor Phenotype and Drug Resistance in Human Ovarian Carcinoma. PLoS ONE 2014, 9, e115708. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzola’, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonov, A.; Agostini, M.; Morello, M.; Minieri, M.; Melino, G.; Amelio, I. Bioinformatics analysis of the serine and glycine pathway in cancer cells. Oncotarget 2014, 5, 11004–11013. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | HSP90 Inhibitors | HSP70 Inhibitor | EGFR | ALK | KRAS | |||
|---|---|---|---|---|---|---|---|---|
| IC50 17-AAG nM | IC50 IPI-504 nM | IC50 STA-9090 nM | IC50 AUY-922 nM | IC50 VER-155008 µM | ||||
| HCC827 | 26.255 | 17.145 | 5.138 | 4.167 | 2.081 | E746-A750 del | WT | WT |
| H3122 | 26.165 | 28.371 | 7.991 | 9.11 | 33.898 | WT | EML4-ALK v1 | WT |
| A549 | 16.296 | 19.492 | 6.31 | 30.733 | 24.487 | WT | WT | p.G12S |
| H1437 | 3.708 | 3.473 | 6.794 | 2.814 | 24.811 | WT | WT | WT |
| Condition | Cell Line | |||
|---|---|---|---|---|
| HCC827 | H3122 | A549 | H1437 | |
| Control | 492 | 199 | 263 | 253 |
| 17-AAG | 488 | 200 | 259 | 264 |
| IPI-504 | 480 | 202 | 266 | 262 |
| STA-9090 | 499 | 200 | 235 | 265 |
| AUY-922 | 507 | 197 | 259 | 261 |
| VER-155008+17AAG | 374 | 209 | 257 | 275 |
| Treatment | Protein Deregulation | Cell Line | |||
|---|---|---|---|---|---|
| HCC827 | H3122 | A549 | H1437 | ||
| 17-AAG | Up | 23 | 4 | 9 | 10 |
| Down | 15 | 3 | 2 | 3 | |
| IPI-504 | Up | 26 | 3 | 11 | 18 |
| Down | 16 | 2 | 6 | 3 | |
| STA-9090 | Up | 25 | 14 | 6 | 9 |
| Down | 18 | 4 | 32 | 3 | |
| AUY-922 | Up | 23 | 5 | 6 | 9 |
| Down | 8 | 4 | 11 | 5 | |
| VER-155008 + 17AAG | Up | 30 | 10 | 12 | 15 |
| Down | 61 | 6 | 15 | 3 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrugal, Á.; Ferrer, I.; Pastor, M.D.; Ojeda, L.; Quintanal-Villalonga, Á.; Carnero, A.; Molina-Pinelo, S.; Paz-Ares, L. Impact of Heat Shock Protein 90 Inhibition on the Proteomic Profile of Lung Adenocarcinoma as Measured by Two-Dimensional Electrophoresis Coupled with Mass Spectrometry. Cells 2019, 8, 806. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080806
Marrugal Á, Ferrer I, Pastor MD, Ojeda L, Quintanal-Villalonga Á, Carnero A, Molina-Pinelo S, Paz-Ares L. Impact of Heat Shock Protein 90 Inhibition on the Proteomic Profile of Lung Adenocarcinoma as Measured by Two-Dimensional Electrophoresis Coupled with Mass Spectrometry. Cells. 2019; 8(8):806. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080806
Chicago/Turabian StyleMarrugal, Ángela, Irene Ferrer, Maria Dolores Pastor, Laura Ojeda, Álvaro Quintanal-Villalonga, Amancio Carnero, Sonia Molina-Pinelo, and Luis Paz-Ares. 2019. "Impact of Heat Shock Protein 90 Inhibition on the Proteomic Profile of Lung Adenocarcinoma as Measured by Two-Dimensional Electrophoresis Coupled with Mass Spectrometry" Cells 8, no. 8: 806. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080806