New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Glucose Tolerance Test (GTT)
2.3. In Vivo Hemodynamic Analysis
2.4. Biochemical Measurements
2.5. Liver Histology
2.6. Oil Red O Staining
2.7. Cell Death
2.8. Immunohistochemistry
2.9. Western Blotting
2.10. Transcriptome Profiling
2.11. Bioinformatic Data Analysis
2.12. Statistical Analysis
3. Results
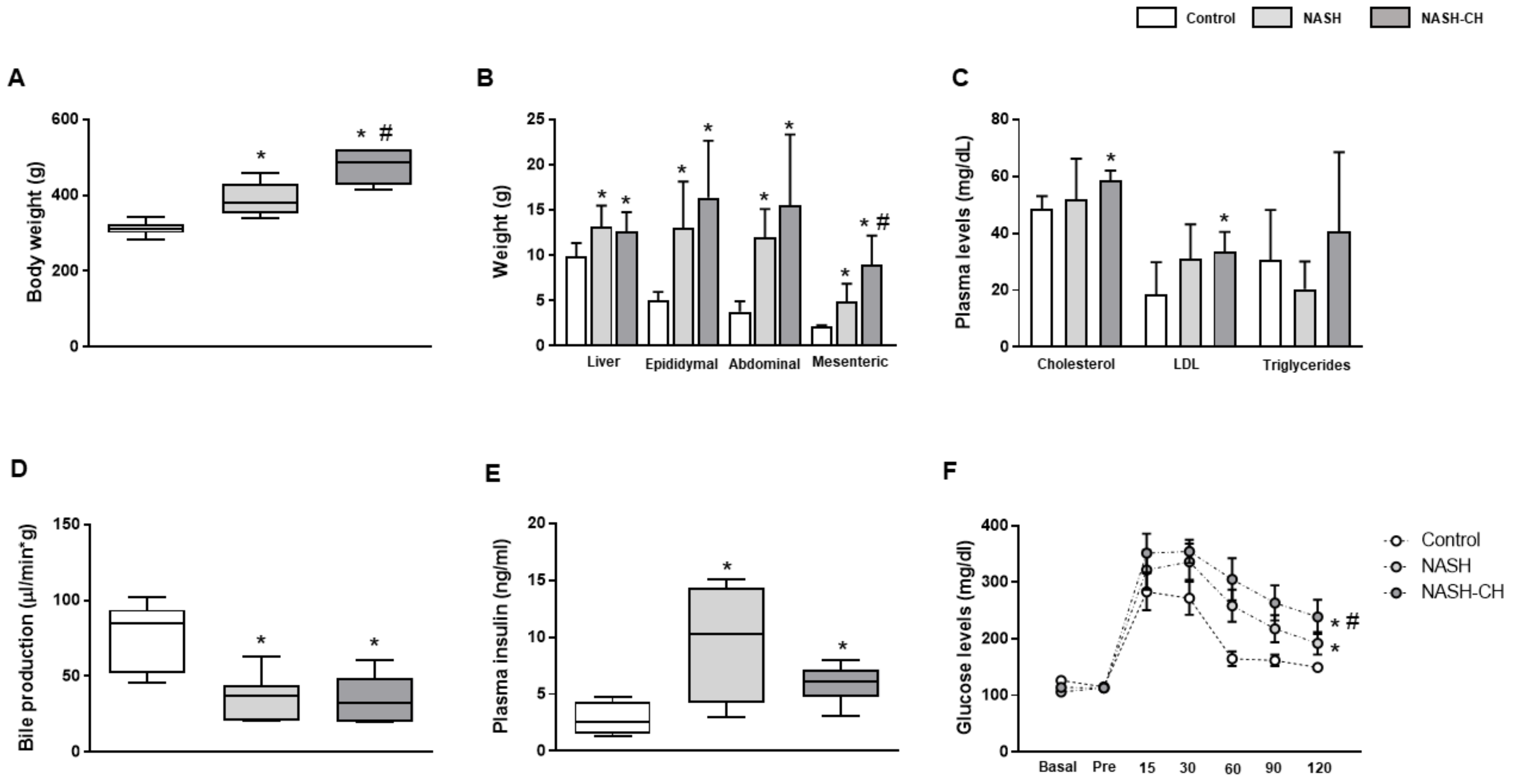
3.1. BarNa Rats Develop Obesity, Liver Disfunction, and Insulin Resistance
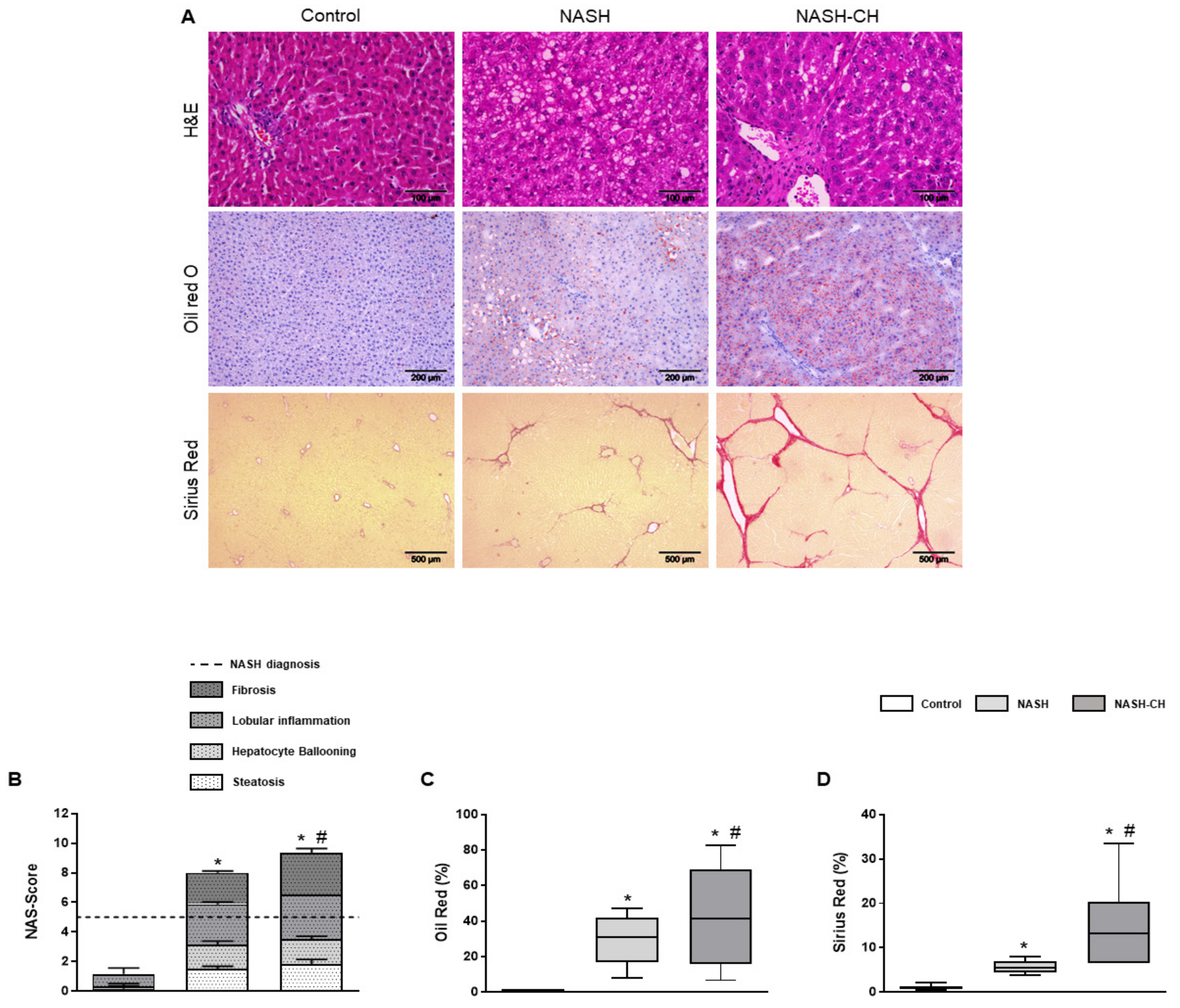
3.2. BarNa Rats Present Liver Histology Features Distinctive of Human NASH
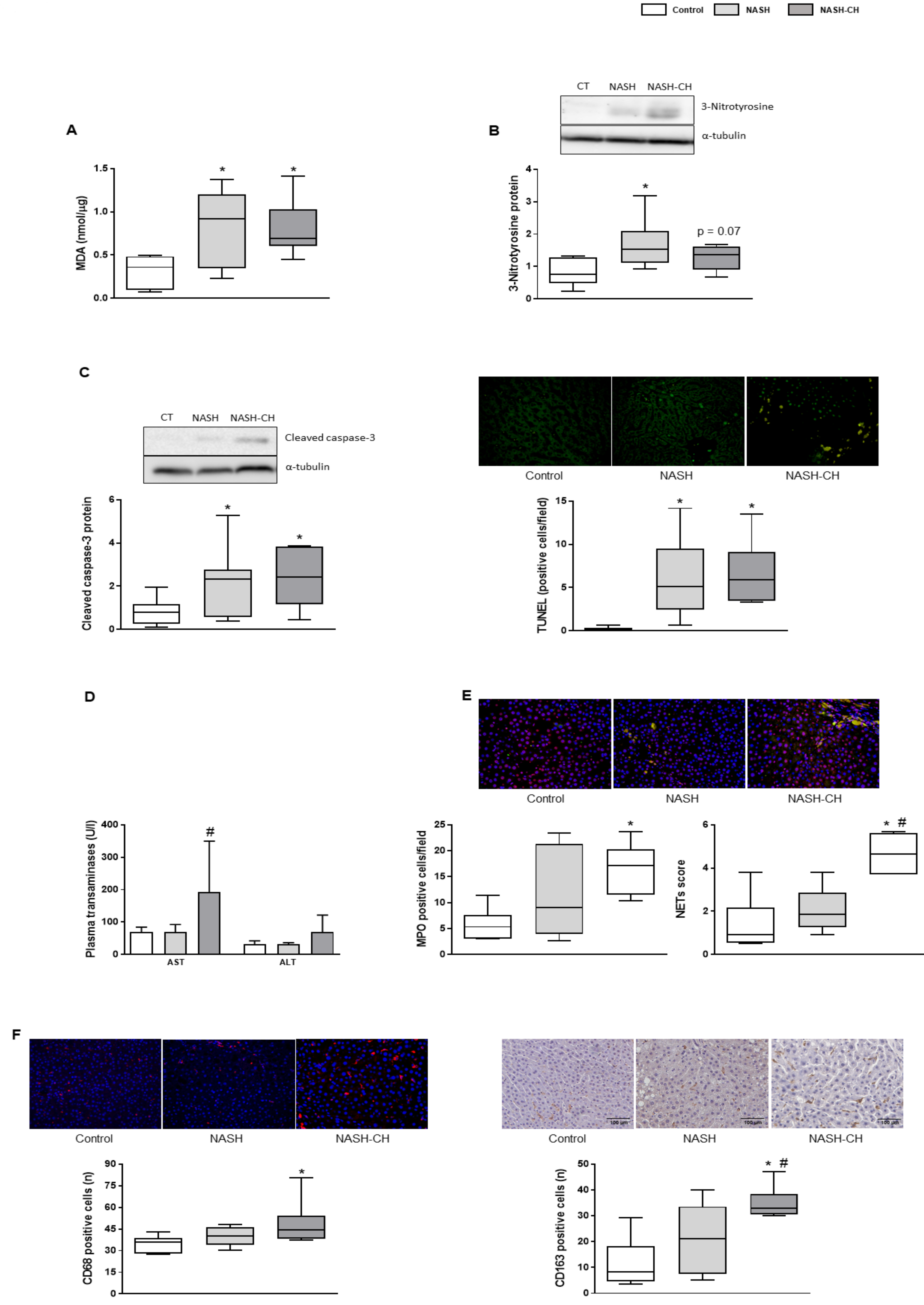
3.3. BarNa Rats Show Elevated Oxidative Stress Levels, Cell Death, and Inflammation
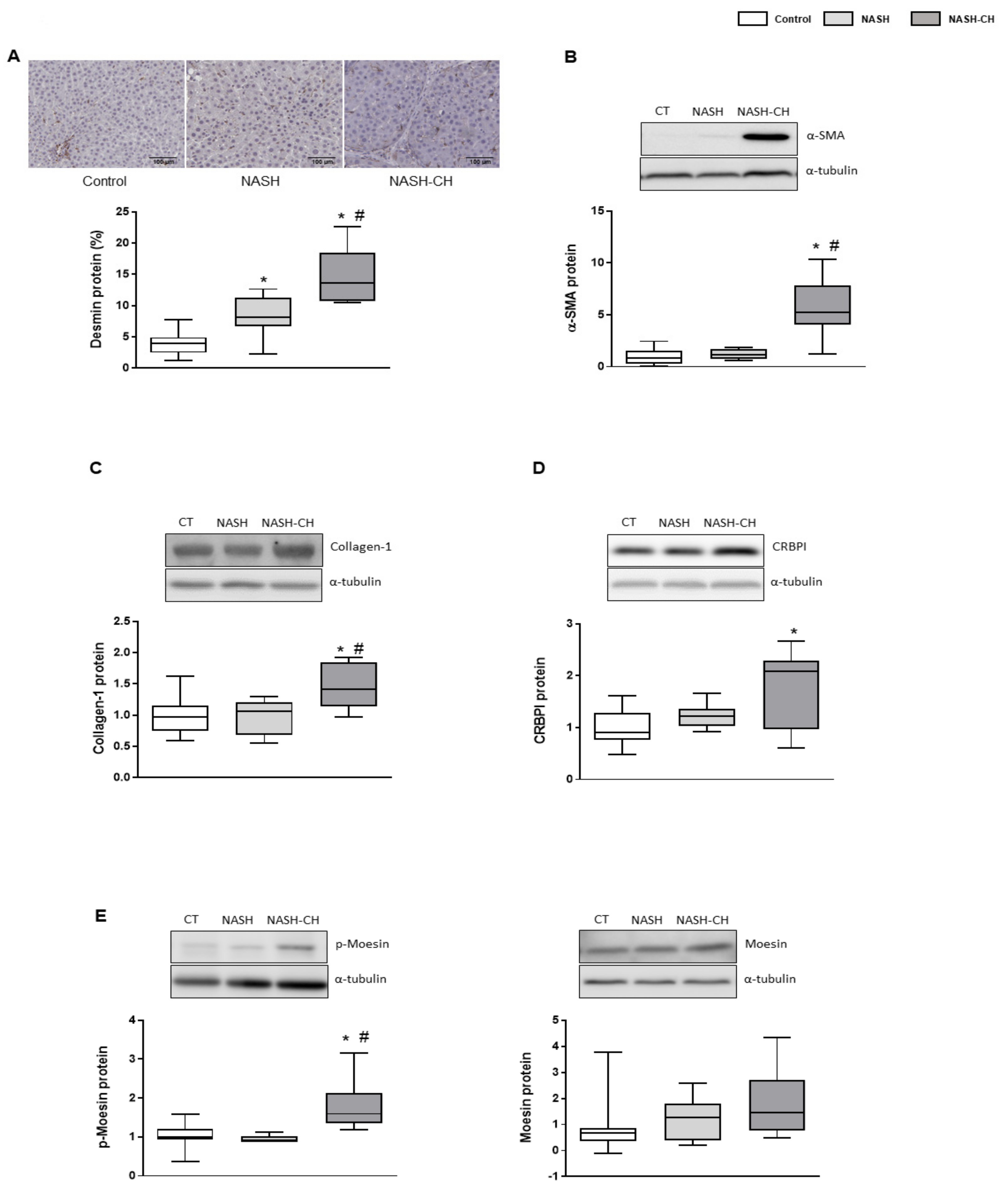
3.4. BarNa Model Promotes the Activation and Proliferation of HSCs
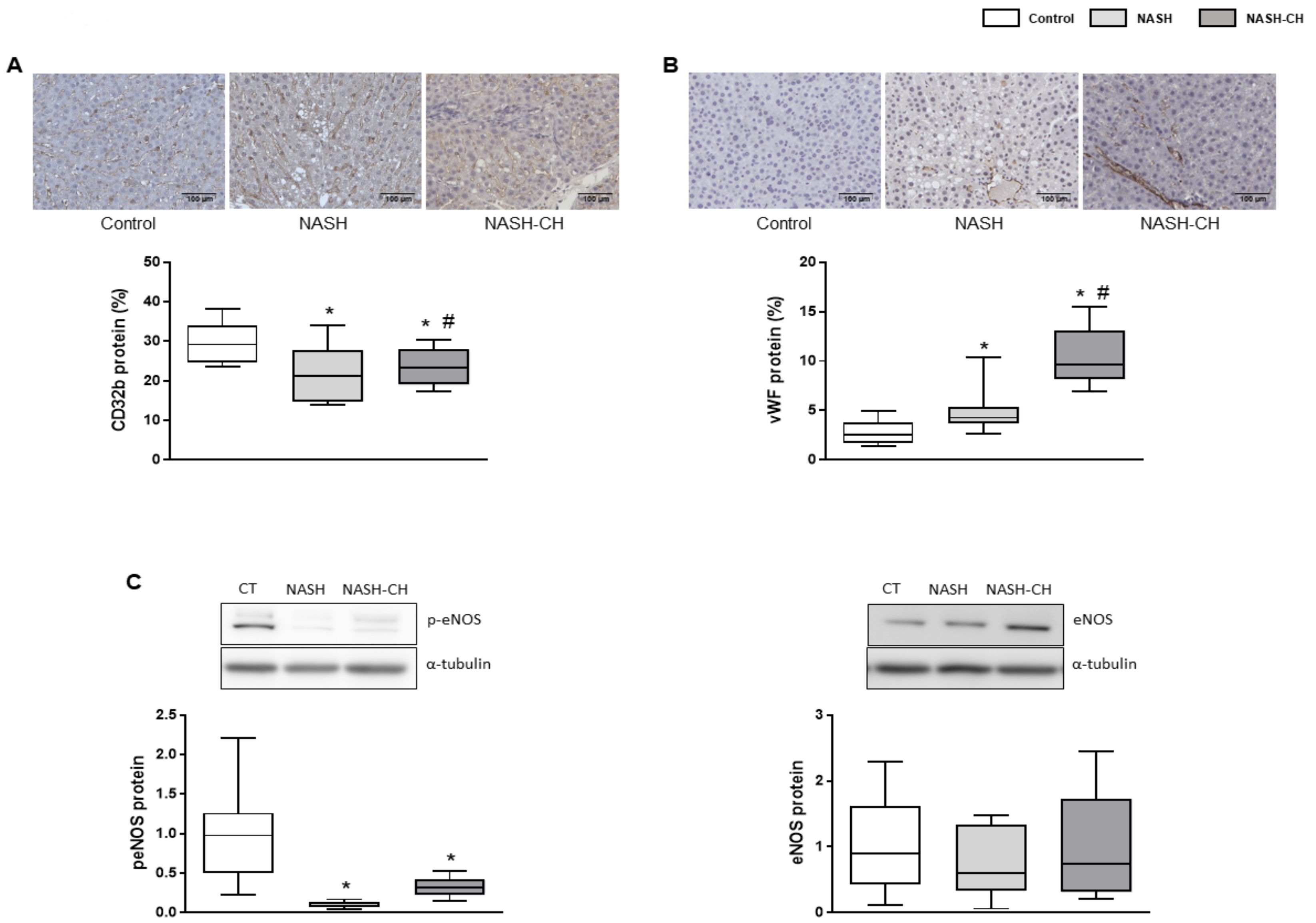
3.5. BarNa Rats Exhibit Hepatic Endothelial Dysfunction
3.6. BarNa Rats Show Significant Portal Hypertension
3.7. BarNa Rats Share De-Regulations in Main Pathways Involved in Human NAFLD and NASH Pathophysiology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of Nonalcoholic Fatty Liver Disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- Wegermann, K.; Diehl, A.M.; Moylan, C.A. Disease pathways and molecular mechanisms of nonalcoholic steatohepatitis. Clin. Liver Dis. 2018, 11, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Hirsova, P.; Malhi, H.; Gores, G.J. Animal Models of Nonalcoholic Steatohepatitis: Eat, Delete, and Inflame. Dig. Dis. Sci. 2016, 61, 1325–1336. [Google Scholar] [CrossRef]
- Romestaing, C.; Piquet, M.A.; Bedu, E.; Rouleau, V.; Dautresme, M.; Hourmand-Ollivier, I.; Filippi, C.; Duchamp, C.; Sibille, B. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr. Metab. 2007. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Blusztajn, J.K. Choline and Human Nutrition. Annu. Rev. Nutr. 1994, 14, 269–296. [Google Scholar] [CrossRef]
- Nakamoto, K.; Takayama, F.; Mankura, M.; Hidaka, Y.; Egashira, T.; Ogino, T.; Kawasaki, H.; Mori, A. Beneficial effects of fermented green tea extract in a rat model of non-alcoholic steatohepatitis. J. Clin. Biochem. Nutr. 2009, 44, 239–246. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Mendes, F.D.; Suzuki, A.; Sanderson, S.O.; Lindor, K.D.; Angulo, P. Prevalence and Indicators of Portal Hypertension in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2012, 10, 1028–1033. [Google Scholar] [CrossRef]
- Königshofer, P.; Brusilovskaya, K.; Schwabl, P.; Reiberger, T. Animal models of portal hypertension. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1565, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Kado, S.; Kano, M.; Masuoka, N.; Nagata, Y.; Kobayashi, T.; Miyazaki, K.; Ishikawa, F. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin. Exp. Pharmacol. Physiol. 2013, 40, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, P.M.; Jacob, H.J. Rats! Dis. Model. Mech. 2009, 2, 206–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, T.F.; Cheeseman, K.H.; Ingold, K.U. Carbon tetrachloride toxicity as a model for studying free-radical mediated liver injury. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1985, 311, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Pasarín, M.; La Mura, V.; Gracia-Sancho, J.; García-Calderó, H.; Rodríguez-Vilarrupla, A.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; Brandes, R.P.; Fernández, M.; Bosch, J.; García-Pagán, J.C. Evidence Against a Role for NADPH Oxidase Modulating Hepatic Vascular Tone in Cirrhosis. Gastroenterology 2007, 133, 959–966. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Manicardi, N.; Ortega-Ribera, M.; Maeso-Díaz, R.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Hide, D.; García-Calderó, H.; Boyer-Díaz, Z.; Contreras, P.C.; et al. Emricasan ameliorates portal hypertension and liver fibrosis in cirrhotic rats through a hepatocyte-mediated paracrine mechanism. Hepatol. Commun. 2019, 3, 987–1000. [Google Scholar] [CrossRef]
- Maeso-Díaz, R.; Ortega-Ribera, M.; Lafoz, E.; Lozano, J.J.; Baiges, A.; Cogger, V.C.; Gracia-Sancho, J. Aging Influences Hepatic Microvascular Biology and Liver Fibrosis in Advanced Chronic Liver Disease. Aging Dis. 2019, 10, 684–698. [Google Scholar] [CrossRef] [Green Version]
- Maeso-Díaz, R.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Hide, D.; Muñoz, L.; Hessheimer, A.J.; Vila, S.; Francés, R.; Fondevila, C.; Albillos, A.; et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 2018. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; Van Den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, 1–17. [Google Scholar] [CrossRef]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.M.; Vilaseca, M.; Lafoz, E.; Garcia-Caldero, H.; Haute, G.V.; Fernández-Iglesias, A.; de Oliveira, J.R.; Garcia-Pagan, J.C.; Bosch, J.; Gracia-Sancho, J. Simvastatin Prevents Progression of Acute on Chronic Liver Failure in Rats With Cirrhosis and Portal Hypertension. Gastroenterology 2018, 155, 1564–1577. [Google Scholar] [CrossRef] [Green Version]
- Guixé-Muntet, S.; de Mesquita, F.C.; Vila, S.; Hernández-Gea, V.; Peralta, C.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. J. Hepatol. 2017, 66, 86–94. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. Bioinforma. Impact Accurate Quantif. Proteomic Genet. Anal. Res. 2014. [Google Scholar] [CrossRef]
- Curwen, V.; Eyras, E.; Andrews, T.D.; Clarke, L.; Mongin, E.; Searle, S.M.J.; Clamp, M. The Ensembl automatic gene annotation system. Genome Res. 2004, 14, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.; Shi, W.; Smyth, G. limma powers di erential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Després, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meyer, C.; Xu, C.; Weng, H.; Hellerbrand, C.; ten Dijke, P.; Dooley, S. Animal models of chronic liver diseases. Am. J. Physiol. Liver Physiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Venu, M.; Martin, E.; Saeian, K.; Gawrieh, S. High prevalence of vitamin A deficiency and vitamin D deficiency in patients evaluated for liver transplantation. Liver Transplant. 2013, 19, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herández-Gea, V.; Friedman, S.L. Autophagy fuels tissue fibrogenesis. Autophagy 2012, 8, 849–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed vitamin A metabolism in non-alcoholic fatty liver disease (NAFLD). Nutrients 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Ashla, A.A.; Hoshikawa, Y.; Tsuchiya, H.; Hashiguchi, K.; Enjoji, M.; Nakamuta, M.; Taketomi, A.; Maehara, Y.; Shomori, K.; Kurimasa, A.; et al. Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease. Hepatol. Res. 2010, 40, 594–604. [Google Scholar] [CrossRef]
- Villaça Chaves, G.; Pereira, S.E.; Saboya, C.J.; Ramalho, A. Non-alcoholic fatty liver disease and its relationship with the nutritional status of vitamin A in individuals with class III obesity. Obes. Surg. 2008, 18, 378–385. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Persico, M.; Masarone, M.; Damato, A.; Ambrosio, M.; Federico, A.; Rosato, V.; Bucci, T.; Carrizzo, A.; Vecchione, C. Non alcoholic fatty liver disease and eNOS dysfunction in humans. BMC Gastroenterol. 2017. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Schall, R.A.; Jia, C.; McColgan, B.; et al. The Natural History of Advanced Fibrosis due to Nonalcoholic Steatohepatitis: Data from the Simtuzumab Trials. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | NASH | p-Value vs. Control | NASH-CH | p-Value vs. Control | |
|---|---|---|---|---|---|
| PP (mmHg) | 8.77 ± 0.48 | 12.14 ± 0.85 | <0.01 | 13.26 ± 0.82 | <0.001 |
| HVR (mmHg × min/mL × g) | 5.14 ± 0.79 | 13.14 ± 2.77 | <0.05 | 12.77 ± 2.38 | <0.05 |
| PBF (mL/min × g) | 1.9 ± 0.25 | 1.24 ± 0.25 | >0.2 | 1.19 ± 0.22 | >0.2 |
| MAP (mmHg) | 110→6 | 110→7 | >0.2 | 112 ± 10 | >0.2 |
| HR (BPM) | 382 ± 16 | 365 ± 17 | >0.2 | 347 ± 19 | >0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeso-Díaz, R.; Boyer-Diaz, Z.; Lozano, J.J.; Ortega-Ribera, M.; Peralta, C.; Bosch, J.; Gracia-Sancho, J. New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease. Cells 2019, 8, 1062. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8091062
Maeso-Díaz R, Boyer-Diaz Z, Lozano JJ, Ortega-Ribera M, Peralta C, Bosch J, Gracia-Sancho J. New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease. Cells. 2019; 8(9):1062. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8091062
Chicago/Turabian StyleMaeso-Díaz, Raquel, Zoe Boyer-Diaz, Juan José Lozano, Martí Ortega-Ribera, Carmen Peralta, Jaime Bosch, and Jordi Gracia-Sancho. 2019. "New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease" Cells 8, no. 9: 1062. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8091062