Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects

1
Department of Neurosciences, University of Montreal and CHU Sainte Justine Research Center, Montreal, QC H3T1C5, Canada
2
UOSD Laboratorio Neurogenetica e Neuroscienze, IRCCS Istituto Giannina Gaslini, 16147 Genoa, Italy
3
UOC Neurochirurgia, IRCCS Istituto Giannina Gaslini, 16147 Genoa, Italy
*
Author to whom correspondence should be addressed.
Cells 2019, 8(10), 1198; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101198
Submission received: 3 September 2019
/
Revised: 26 September 2019
/
Accepted: 1 October 2019
/
Published: 4 October 2019
(This article belongs to the Special Issue Wnt Signaling in Health and Diseases)
Abstract
:Neural tube defects (NTDs), including spina bifida and anencephaly, represent the most severe and common malformations of the central nervous system affecting 0.7–3 per 1000 live births. They result from the failure of neural tube closure during the first few weeks of pregnancy. They have a complex etiology that implicate a large number of genetic and environmental factors that remain largely undetermined. Extensive studies in vertebrate models have strongly implicated the non-canonical Wnt/planar cell polarity (PCP) signaling pathway in the pathogenesis of NTDs. The defects in this pathway lead to a defective convergent extension that is a major morphogenetic process essential for neural tube elongation and subsequent closure. A large number of genetic studies in human NTDs have demonstrated an important role of PCP signaling in their etiology. However, the relative contribution of this pathway to this complex etiology awaits a better picture of the complete genetic architecture of these defects. The emergence of new genome technologies and bioinformatics pipelines, complemented with the powerful tool of animal models for variant interpretation as well as significant collaborative efforts, will help to dissect the complex genetics of NTDs. The ultimate goal is to develop better preventive and counseling strategies for families affected by these devastating conditions.
1. Introduction
Neural tube defects (NTDs) result from the failure of the neural tube closure during weeks 3–5 after fertilization [1,2]. Their clinical manifestation varies in type and severity depending on the site of the closure defect along the embryonic axis. They are classified into two major types: Open or closed NTDs, where the damaged nervous tissue is either exposed to the environment or covered by skin respectively. The two most common forms of open NTDs are anencephaly and myelomeningocele (MMC). The former results from the failure of the neural tube closure at the cranial region, while the latter at the spinal region. Anencephaly is characterized by an absent cranium and is invariably lethal, whereas many infants with MMC now survive but with severe and lifelong physical and developmental disabilities. A number of skin-covered (closed) NTDs are categorized clinically depending on the presence (lipomyeloschisis, lipomyelomeningocele, meningocele, and myelocystocele) or absence of a subcutaneous mass (including dermal sinus, caudal regression, and spinal segmental dysgenesis) [1].
NTDs represent the second most common group of human birth defects. Their incidence has been significantly reduced due to food fortification and maternal periconceptional supplementation of folic acid [3,4]. Their regional average prevalence per 1000 live births in 2015 varied between 0.7–1 in North America and Europe to 3.1 in Southern Asia [5]. Some NTDs are resistant to folic acid and thousands of families are still affected by these devastating conditions each year, necessitating a better understanding of their etiology and the development of novel preventive strategies [3,6]. More recently, a few pilot studies conducted in Europe have suggested a protective effect of inositol when taken periconceptionally in combination with folic acid [7,8]. Larger studies are needed to confirm this promising novel preventive strategy against NTDs.
Population and family studies indicate a complex etiology to NTDs involving environmental and genetic factors. Most NTD cases are sporadic [2,9,10]. Women who have one previous affected fetus have an empirical recurrence risk of 3% in any subsequent pregnancy and of 10% after conceiving a second NTD embryo [10]. A few families have been reported to be segregating NTDs but with a complex pattern of inheritance hindering the identification of the underlying genetic variants [9,11]. NTDs in humans are hypothesized to follow a multifactorial threshold model where many genetic variants, alone or in interaction with many environmental factors, modulate the incidence and severity of the disease. Heritability in NTDs is estimated to be 60% with multiple susceptibility genes involved [12]. However, the identity and relative contribution of such genes to NTDs remain unknown.
This article reviews the role of the non-canonical Wnt/planar cell polarity (PCP) genes in NTDs’ pathogenesis. The process of neural tube formation and how defects in this process lead to the various types of NTDs is first briefly described. Then, the components and readouts of PCP signaling during development are described. The focus is placed on the process of convergent extension that is mediated by PCP signaling and that is the major force underlying the elongation and narrowing of the neural tube. This is followed by a summary of the undebatable evidence of an important role of defective PCP signaling in the causation of NTDs in animal models and an update on the findings from genetic studies of this pathway in human NTDs. Finally, the contribution of this pathway is discussed in the larger context of the genetic architecture of NTDs and the challenges faced by researchers in their quest for dissecting the complex etiology of these debilitating birth defects and their ultimate prevention.
2. Neural Tube Formation: A Rapid, Multi-Step and Complex Process
The neural tube is the precursor of the brain and spinal cord. Its formation is a dynamic and rapid process that occurs in 2 phases: Primary neurulation that leads to formation of the brain and most of the spinal cord during weeks 3 and 4 of pregnancy; secondary neurulation that occurs in the lower sacral and coccygeal regions during week 5 of pregnancy [1].
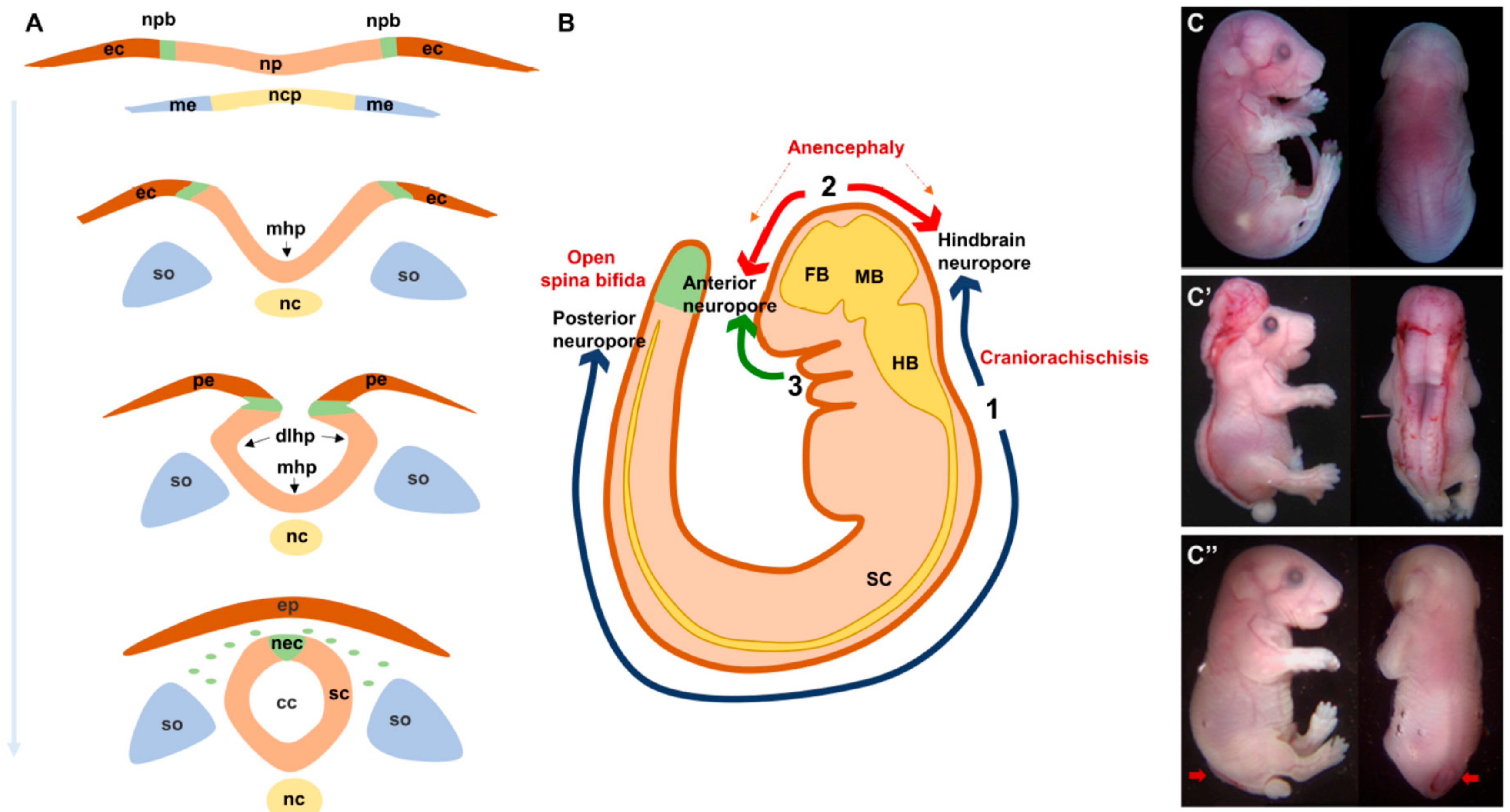
Primary neurulation starts with a flat sheet of specialized dorsal ectodermal cells, the neural plate, which first undergoes extensive shaping and then elevates to form bilateral neural folds that bends to face one another across the midline, fuse at their tips, and separate from the overlying non-neural ectoderm (Figure 1A). The dynamic development of the neural tube is also mediated by the signals and biomechanical forces from the surrounding tissues [13,14].
Shaping of the neural plate involves its convergence towards the midline and extension along the anterior-posterior axis. This morphogenetic process of convergent extension (CE) is discussed in details below. As CE is progressing, the neural plate elevates towards the dorsal midline leading to the formation of bilateral neural folds. This is followed by formation of 3 bending or hinge points that facilitate the apposition of the neural folds across the midline: one median hinge point (MHP) at the midline overlying the notochord and two dorsolateral hinge points (DLHP) [13,14] (Figure 1A). The pattern of HP formation along the neural tube is influenced by factors secreted by the notochord, mainly Sonic Hedgehog, and by the surface ectoderm, mainly Bone Morphogenic Proteins [15,16]. The major morphogenetic process that occurs during HP formation is apical constriction whereby cells convert their shape from cuboidal to wedge-like due to a decrease in their apical area and that is mediated mainly by an actin-binding protein called Shroom3 [17,18,19]. The last step of NT closure is fusion of the opposing tips of the neural folds and the separation of the closed NT from the overlying non-neural ectoderm which later develops into the epidermis (Figure 1A). This step involves significant tissue remodeling and dynamic behaviors including the formation of cellular bridges, ruffles, filopodia and lamellipodia [20]. The molecular mechanisms underlying this step remain largely unknown but a few studies implicated members of the protease activated receptors signaling (Par1 and Par2) [21], the grainyhead-like transcription factors (Grhl2) [22], the Rho family of GTPases (Rac1 and Cdc42) [23] and EphrinA-EphA interactions [20,24].
Neural tube closure is initiated at specific sites along the embryonic axis and proceeds in a zipper-like fashion rostrally and/or caudally (Figure 1B). These sites have been well defined in mouse models and are still poorly defined in humans. In mice, closure 1 occurs at the hindbrain/cervical brain boundary, closure 2 at the forebrain/midbrain boundary and closure 3 at the rostral end of the future forebrain. Closures 1 and 2 proceed bidirectionally while closure 3 proceeds only caudally. These multiple closure sites create three neuropores: The anterior and hindbrain neuropores in the cranial region and the posterior neuropore in the low spinal region [2,9]. In humans, gross and histological examination of fetuses support a multisite closure model. However, inconsistencies in the number of sites varying between 2 and 5 have been reported [25,26,27].
Secondary neurulation is a canalisation process where a group of self-renewing stem cells in the caudal tailbud condense and undergo canalisation, converting the solid neural precursor into a hollow secondary neural tube that becomes continuous with the primary NT [13,14]. The cellular and molecular mechanisms underlying secondary neurulation remain largely unknown. Fate mapping studies in chick embryos suggested similar cellular or molecular mechanisms to primary neurulation [28].
3. Neural Tube Defects: What Could Go Wrong?
Any lesion at any of the mechanisms described above could lead to NTDs. In mouse models, the mutations in at least 250 genes were found to be responsible for the failure of NT closure [29]. The majority of these mutations follow a simple Mendelian mode of inheritance with high penetrance, as opposed to the complex pattern inheritance observed in humans. The type and severity of the resulting NTD depend on which closure site was affected. In mouse models, the failure of closure 1 leads to a rare and very severe form of NTDs called craniorachischisis (CRS) characterized by an open neural tube throughout the spinal region. Incomplete closure 1 at the caudal side causes open spina bifida (Figure 1C). The failure of closure 2 leads to anencephaly and that of closure 3 causes a split face and usually a forebrain anencephaly. The defects in secondary neurulation are thought to cause the milder forms of closed NTDs and often involves tethering of the spinal cord along with lipoma [9,14].
4. PCP Signaling: A Non-Canonical Branch of Wnt Signaling with Unresolved Mysteries
4.1. Non-Canonical Wnt/PCP Signaling: From Flies to Vertebrates
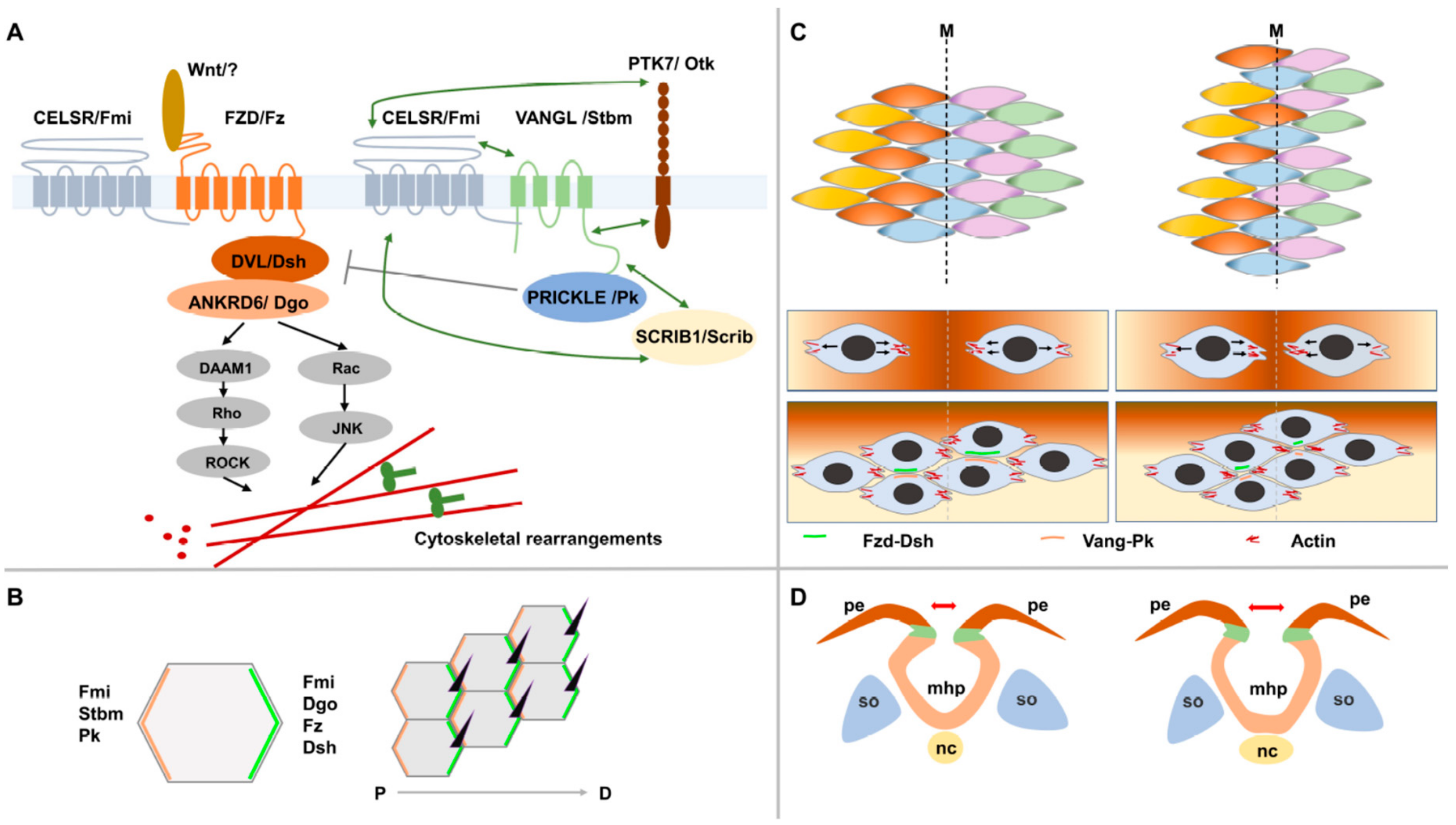
The first Wnt-1 gene was discovered in 1982 and since then, an explosion of studies has been conducted to dissect the role of this gene along with other Wnt members in signaling in development and disease [30,31,32]. This long-studied branch of Wnt signaling is referred to as canonical and is regulated by the intracellular levels of ß catenin. It has a crucial role in mediating the cell proliferation and cell fate during development and is deregulated in many types of cancer [32,33]. Canonical Wnt signaling shares upstream effectors with another less well-studied and more recently discovered branch called the non-canonical PCP pathway [34]. PCP is β-catenin independent and mediates tissue polarity that describes the collective cell polarity within the plane of an epithelium and that is usually perpendicular to the apicobasal polarity within the cell [34,35]. It was first described in the fly where it mediates the polarity of highly organized structures including the distal orientation of wing trichomes (hair cells) and the complex organization of the ommatidia (eye units) in the adult eye [36,37]. Genetic studies of mutants affecting these structures have identified a group of six core PCP genes: Vang gogh/Strabsimus (Vang/Stbm), Frizzled (Fz), Dishevelled (Dsh), Flamingo (Fmi), Prickle (Pk) and Diego (Dg) (Figure 2A). PCP in the developing wing is established through an asymmetric localization of two mutually exclusive signaling complexes along its proximal-distal axis (Figure 2B). Upon binding of PCP-specific Wnt ligands to the transmembrane protein Fz receptor, the latter recruits the cytosolic proteins Dsh and Dgo distally while the transmembrane protein Vang/Stbm recruits the cytosolic protein Pk and localizes it proximally. The atypical cadherin Fmi is localized to both proximal and distal membranes and promotes the assembly of these complexes and the propagation of PCP asymmetry throughout the tissue (Figure 2B). In the fly, it was shown that PCP signaling directly impacts the cytoskeleton by controlling the actomyosin machinery via Rho GTPases and Rho Kinase and thereby inducing the observed morphological and cell behaviour changes (Figure 2A) [35,36,37].
The studies of vertebrate models have demonstrated a high level of evolutionary conservation at PCP core genes (Figure 2A) and have identified a new set of PCP genes including those coding for PCP vertebrate-specific Wnt ligands (Wnt 5A, 5B and 11) and Wnt co-receptors (Ryk, Ror2 and Ptk7) [37,38,39,40]. These studies also discovered a new role of the apicobasal polarity gene Scribble1 (Scrib1) in PCP signaling [41]. PCP signaling in vertebrates mediates planar polarity and collective cell movements in a large number of developmental processes and in some pathogenic states. Among these processes are the convergent extension during gastrulation and neural tube formation, axonal guidance, neuronal polarity, eyelid closure, hair bundle orientation in inner ear sensory cells, hair follicle orientation in the skin, epidermal wound repair, and branching morphogenesis in lung and kidney. The defects in PCP signaling in vertebrates result in a range of developmental anomalies and diseases including NTDs, polycystic kidneys, and congenital heart diseases [42,43,44,45,46]. Recent studies have discovered an important role of this pathway in promoting directed cell migration during the invasion and metastasis of malignant cells [47,48]. The underlying molecular mechanisms of PCP signaling in health and diseases remain a mystery despite extensive studies conducted in animal models.
4.2. PCP and CE during Neural Tube Formation: Strong Evidence from Animal Models
The first process that was shown to be mediated by the non-canonical Wnt/PCP signaling in vertebrates was the convergent extension during gastrulation and neurulation in Xenopus and zebrafish. CE involves two major types of cell movements: Collective cell migration followed by cell intercalations. Neuroepithelial cells produce mediolaterally-oriented protrusions that enable them to move directionally in a manner similar to the leading edge of a migrating cell. Next, mediolaterally-oriented junctions undergo actomyosin contractions, thereby driving cell intercalations (Figure 2C) [35,49,50,51]. Extensive studies of these models have established a similar molecular basis of PCP signaling to that described in Drosophila from the first step of the asymmetric localisation of the transmembrane complexes to the downstream effectors of actin regulators including RhoA, Rho kinase, the formin Daam1 and the Rho GTPase Cdc42 [35]. In vertebrate cells undergoing CE, it was shown that PK and VANGL localize anteriorly, whereas DVL localizes posteriorly [35] (Figure 2C). The knockdown or overexpression of PCP genes in Xenopus and zebrafish caused CE defects, manifested by a shortened body axis and an open neural tube (in Xenopus) [35,52,53,54]. In mammals, the defects in PCP signaling impair CE and neural tube formation [2,9,55]. In fact, the first mouse model to implicate PCP signaling in NTDs was the Looptail (Lp) mouse that suffers from the severe form of CRS. The traditional linkage and positional cloning studies have identified the core PCP gene Vangl2 as the gene defective in Lp [56,57]. It was demonstrated in this mutant that disrupting non-canonical Wnt/PCP core genes caused a defective CE of midline neuroepithelial cells that led to a widened neural plate and far apart neural folds that failed to fuse (Figure 2D) [58]. The subsequent studies of other single or double mouse mutants at other core PCP genes, Celsr1 (orthologue of Fmi) [59], Fz3/Fz6 [60] and Dvl1/Dvl2 [61], and at vertebrate PCP genes, Ptk7 [38] and Scrib1 [41] have further confirmed the role this pathway in NTDs’ pathogenesis. CRS is the hallmark phenotype of a defective PCP signaling in mice. However, it was demonstrated that double-mutant embryos at PCP genes (Vangl2, Scrib1, Celsr1 and Ptk7) generated a range of open NTDs including anencephaly and open spina bifida [38,62].
4.3. PCP: Molecular Crosstalks to Other Processes and Pathways during Neurulation
PCP signaling does not act alone during neurulation. It forms part of an intricate and finely regulated network of processes and signaling pathways that together ensure proper neurulation. Among these processes are apicobasal polarity (ABP) [41,63], apical constriction [64], oriented cell divisions [43,44], ciliogenesis [35,65], and the canonical Wnt signaling pathway [34]. The molecular cross-talks between PCP and each of ABP and the Wnt canonical signaling that have been associated with NTDs in mouse mutants and human cohorts are briefly discussed.
PCP and ABP: ABP is essential for normal epithelial cell shape and function and is generated and maintained by antagonistic interactions between the apical aPkc–Par6–Par3 and Crumbs–Pals1–Patj complexes and the basolateral proteins Scribble, Lgl and Dlg [66]. The defects in ABP affect tissue integrity and structure and have been linked to many types of cancer [67]. Scrib1 was the first gene to link ABP to PCP signaling. In mice, mutations in Scrib1 cause CRS and genetically interact with the core PCP gene Vangl2 to mediate the orientation of ear sensory cells, CE and neural tube formation [41,63]. Histological studies of the developing neural tube in these mutants and of cultured epithelial cells following partial knockdown of Scrib1 showed an abnormal localization of Vangl2 and of the apical protein Par-3 [68]. It was hypothesized that mutations in Scrib1 cause or predispose to NTDs through its effect on Par-3 and Vangl2 localization and most likely independently of ABP [68]. Additional studies have confirmed a role for Scrib during neural tube morphogenesis in zebrafish [69] and in Drosophila eye and wing development as part of PCP signaling [70]. The details of genetic studies of SCRIB1 in human NTDs are described below.
PCP and the canonical Wnt/β-catenin pathway: Several studies have identified a group of molecular switches that act on both Wnt/β-catenin and Wnt/PCP pathways simultaneously in the neural tube closure [71]. These mediators include Lrp6 and Ptk7. Lrp6 acts as a Wnt co-receptor and is crucial for activation of Wnt/β-catenin canonical pathway while simultaneously inhibiting the non-canonical pathway [72,73]. Lrp6 downregulation in Xenopus embryos disrupts CE during gastrulation [74] and knockout in mice causes exencephaly, spina bifida, absent tail, and limb deformities [75,76]. Interestingly, PCP defects in both Xenopus and mouse embryos lacking Lrp6 function were rescued by the deletion of the non-canonical ligand Wnt5a, indicating that these phenotypes resulted from non-canonical Wnt gain-of-function [72,73,74]. Ptk7 activates the Wnt/PCP pathway while simultaneously inhibiting the Wnt/β-catenin canonical pathway [77]. Ptk7 mouse mutants suffer from CRS and genetically interact with Vangl2 [38]. In frog and zebrafish models, morpholino oligonucleotides (MO) induced knockdown of Ptk7 as well as its overexpression lead to similar CE defects manifested mainly by a shortened body axis and widened somites [77,78]. Depending on the context, Ptk7 is also able to positively regulate Wnt signaling in the posterior neural plate in Xenopus through the interaction with ß catenin. The depletion of Ptk7 in these cells inhibited the induction of direct targets of canonical Wnt signaling resulting in a loss of posterior neural cell fates and preventing later neural CE [79,80]. The details of genetic studies of LRP6 and PTK7 in human NTDs as mediators of PCP signaling are described below.
5. PCP Signaling Genes in Human NTDs: What do We Know So Far?
5.1. Genetic Studies of PCP Signaling in Human NTDs
The strong candidacy of PCP genes in NTDs’ pathogenesis as deduced from animal models has provided a solid rationale for investigating their orthologues in human NTDs. The first genetic study of PCP signaling in humans NTDs implicated orthologues of Vangl2 mutated in Looptail. In that study, the authors analyzed VANGL2 and its paralogue VANGL1 in a cohort of 137 Italian patients with nonsyndromic spinal dysraphisms and 7 fetuses with CRS [81]. Surprisingly, they did not detect any nonsynonymous variant in VANGL2. On the other hand, they identified in VANGL1 three variants detected in a heterozygous state, p.Val239Ile, p.Arg274Gln, in two familial NTD cases and p.Met328Thr in one sporadic case. The p.Val239Ile was a de novo mutation that abrogated the interaction between VANGL1 and all three Dvl proteins [81]. However, this study was published more than a decade ago at a time where the majority of researchers in this field adopted a candidate gene approach to dissect the complex genetics of NTDs. It had a high impact in the field of NTD genetics as it was the first genetic and functional study that demonstrated a role of PCP signaling in human NTDs [81]. Since then, a large number of studies have been conducted on the role of PCP core genes and mediators in NTDs’ pathogenesis making it the most extensively studied pathway in human NTDs [81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102]. The majority of these studies included genetic as well as functional data in cell cultures and zebrafish models. Table 1 summarizes the major positive findings from these studies. It lists rare PCP variants that were associated with NTDs and that were predicted to be pathogenic and/or functionally validated. This table is an underestimate of the contribution of PCP signaling to NTDs. It does not include rare variants that were predicted to be benign and/or were not functionally validated and/or synonymous that could affect gene splicing and/or regulatory that could represent novel PCP mutations in human NTDs. Most of these studies focused on coding rare variants present in patients and might have missed other rare damaging variants because of the strategy of excluding variants present in as few as one control. Only a few studies did not identify NTD-associated variants of each of DVL3, FZD3, PTK7, VANGL1, VANGL2 and PK1, in the cohorts analyzed and were not included in this table [82,89,90,91,102]. Only a few studies assessed the role of common variants of PCP genes (MAF > 1%) in NTDs and the majority did not find a significant association between these variants and the NTDs’ phenotype [82,83,86,87]. Only one study has detected a significant association between the common SNP rs4839469 in VANGL1 and NTDs [103].
In mice, single mutants of PCP genes suffer mainly from the severe CRS and rarely exhibit open spina bifida [38,41,56,57,59,60,61]. CRS is an extremely rare type of NTDs in humans hindering the availability of large cohorts for genetic studies. Assuming no overlap in the cohorts included in Table 1, 8 PCP genes (VANGL1-2, CELSR1-3, DVL3, SCRIB1 and PTK7) have been re-sequenced in a couple of hundred of CRS cases and no obvious loss of function (LoF) variant was identified in these genes in the majority of CRS cases analyzed. Interestingly, the double mutants of PCP genes in mice exhibit a wide range of open NTDs including anencephaly and open spina bifida, providing a strong rationale for a comprehensive analysis of this pathway in other types of open NTDs in humans [38,62]. A large number of studies have been conducted on the role of PCP signaling in open NTDs with a focus on the most common type that is MMC. As shown in Table 1, potentially pathogenic rare variants were identified in each of the 14 PCP genes in a small fraction of open NTD patients. As in the case for CRS, the majority of these variants were missense with no obvious LoF effect. Further shown in Table 1, a few PCP studies have also included closed NTDs and obtained similar types of genetic variants, suggesting a shared role for this pathway in the pathogenesis of open and closed forms of NTDs.
The majority of NTD cases are sporadic. A few families of Italian origin with multiple individuals affected by open or closed NTDs were available for genetic studies of PCP. The potentially damaging PCP variants identified in these families did not segregate with the NTDs’ phenotype, consistent with a complex pattern of inheritance for NTDs [68,83,85]. In both sporadic and familial cases of NTDs and when parents’ DNA was available, the majority of PCP variants were inherited from apparently healthy parents indicating low penetrance. Many PCP variants initially reported to be found only in a single family appeared later in the ExAC and gnomAD databases at a very low frequency. This should not be surprising as the vast majority of unaffected individuals are expected to carry one or more risk alleles and even large effect alleles are subject to background modification and incomplete penetrance.
All variants identified in PCP genes in NTDs were heterozygous in contrast to the recessive mode of action of mutations identified in their orthologues in mouse mutants. The most plausible interpretation for this finding is that these variants need to interact with other genetic variants and possibly environmental factors to modulate the incidence and severity of the NTD phenotype. Hence, NTDs might be explained by the inheritance of different combinations of mutant alleles in different individuals. Researchers have tried to investigate this hypothesis by the identification of NTD patients who are double or compound heterozygotes for damaging variants of PCP genes and by assessing the genetic burden of PCP in human NTDs. NTD patients who were double heterozygous for potentially pathogenic PCP mutations were identified in many studies but were not further investigated for a combined functional effect on PCP signaling [87,91,95,97,98,101]. One recent study used targeted next generation sequencing (NGS) of 30 PCP genes in 184 Chinese NTD cases followed by a replication of findings in CELSR1 in an independent cohort of 292 American patients [100]. This study identified double damaging variants in CELSR genes and other PCP genes in 3.3% of their cohort and demonstrated a combined effect of two heterozygous variants of CELSR2 and DVL3 in down regulation of PCP signaling in vitro. The same study demonstrated a significant enrichment of LoF variants in CELSR1 in NTDs when compared to the frequency reported in the ExAC database. Other studies have demonstrated a significant enrichment of predicted damaging variants in NTDs compared to controls in 3 other PCP genes: VANGL1 [97], FZD6 [89] and PTK7 [102].
The functional validation of missense variants identified in NTD patients in core PCP genes focused mainly on protein-protein interactions and on protein localization that are essential for proper PCP signaling. For example, variants of VANGL genes affected the interactions with DVL members and vice versa [81,84,100] and variants of CELSR1 affected its membrane localization and its recruitment of Vangl2 to the cell membrane [90,94]. Other validation assays included the zebrafish model where abnormal PCP signaling led to a defective CE manifested by a shortened body axis and widened somites. This easily measured phenotype was used as a readout for testing any potential pathogenic effect of PCP variants on the process of CE. For example, two variants of VANGL1 failed to induce CE defects when overexpressed and to rescue MO-induced CE defects and they were hypothesized to be hypomorphic or LoF [104]. Three variants of PK1 were hypothesized to be hypermorphic in an overexpression assay and one of these variants antagonized the CE phenotype induced by the overexpression of the wild-type PK1 in a dominant fashion [85]. On the other hand, the functional validation of variants detected in PCP mediators including LRP6 and ANKRD6 (that codes for DIVERSIN) mainly focused on their abilities to act antagonistically on both Wnt pathways. Interestingly, the variants of each of these 2 genes were shown to lose this antagonistic effect indicating the importance of a tightly regulated balance between Wnt pathways during neural tube formation [93,95,96]. The variants identified in SCRIB1 followed an unexpected finding of its effect on Par3 and Vangl2 localization in the neural tube in mouse mutants. Two variants of SCRIB1 failed to rescue the localization defect of Par-3 and Vangl1 caused by knockdown of SCRIB1 in MDCK cells [68].
5.2. Are PCP Genes Major Culprits and/or Accomplices in the Complex Etiology of Human NTDs?
Following the summarized findings of genetic studies of PCP signaling in human NTDs described above, can the relative contribution of PCP genes to NTD pathogenesis be predicted with any accuracy? Our current state of knowledge of the genetic architecture of these defects, similar to many other medically important complex diseases, is still too limited to answer this question. As mentioned above, NTDs have long been hypothesized to follow a multifactorial threshold model where a combination of a large number of genetic variants and environmental factors are needed to reach this threshold and develop a phenotype. The genetic component remains poorly defined. As severe forms of NTDs are deleterious to fitness, it is reasonable to hypothesize that predisposing genetic variants should be selected against and should therefore be rare. However, is genetic liability to severe NTDs attributed to an accumulation of rare alleles with large effects and/or rare alleles that all have minor effects? One WES study in 43 sporadic cases affected with MMC or anencephaly demonstrated an important role of LoF de novo mutations (DNMs) in the development of these severe forms of NTDs [106]. DNMs are more deleterious, on average, than inherited mutations and are represented as “single mutations with large effects” that play a major role in many rare and common forms of developmental diseases [107,108]. Genetic studies of PCP signaling described above demonstrated an enrichment of damaging variants in PCP genes in severe NTDs. While most of these mutations were missense, heterozygous and incompletely penetrant, it is still possible that they can exert a large effect in NTDs’ pathogenesis. PCP signaling is extremely important for tissue formation and therefore it is reasonable to suggest that obvious LoF variants of this pathway are not compatible with life and can escape detection. On the other hand, a recent whole genome sequencing study conducted in three different NTD cohorts (Han Chinese, Caucasian USA, and Middle Eastern/Qatar) have proposed an omnigenic model for genetic liability to NTDs that is determined by an accumulation of singleton LoF variants of many genes, all with minor effects. They did not detect any significant enrichment of these variants in any biological pathway. Interestingly, they showed that the accumulation of nine of these variants represents the threshold above which an individual develops NTDs [109]. This study did not include analyses of regulatory variants that have been hypothesized to play an important role in the etiology of complex traits. It is of course possible that both models, rare alleles with large effects and rare alleles with minor effects, exist for severe NTDs. The omnigenic model was proposed to explain the missing heritability of complex traits. It assumes that any given disease phenotype can be directly affected by a modest number of core pathways and genes with specific roles in disease pathogenesis. However, a large number of interconnected regulatory networks and genes that are outside core pathways can affect the function of these core pathways. Since core genes only constitute a tiny fraction of all genes, most heritability comes from minor and indirect effects of hundreds or even thousands of genes expressed in the disease relevant cell [110].
When discussing the genetic components of NTDs, it is important to differentiate between severe NTDs from the milder closed forms that do not affect reproductive fitness. Gene burden studies in PCP signaling were either conducted in a cohort of open and closed NTDs without stratification by clinical type or only in open NTDs. The only two NGS studies of NTDs were conducted on the two severe open forms of MMC and anencephaly. While this review cannot speculate on the genetic architecture of milder forms of NTDs without more focused studies of larger cohorts, the fact that they do not affect reproductive fitness argues against a major role of rare alleles with large effects in their etiology.
To add complexity to the etiology of NTDs, a possible role that should not be excluded is for common variants of small effect size that could still contribute to the background liability, genotype-genotype interactions, genotype-environment interactions, and epigenetic effects. NTDs might indeed belong to a broad sense heritability model that takes into accounts all these genetic, environmental, and epigenetic factors [111].
6. Conclusions and Challenges
The evidence for an important role of PCP signaling in NTDs’ pathogenesis in vertebrate models is overwhelming. These models remain crucial for dissecting the molecular and cellular mechanisms by which PCP signaling mediates the process of CE and interacts with other developmental processes and pathways during neural tube formation. These models are also essential for the interpretation of the role of genetic variants identified in human studies and for the investigation of the role of gene-gene and gene-environment interactions in the NTDs’ etiology. In humans, the evidence has accumulated for an important contribution of PCP genes to NTDs’ pathogenesis. The majority of these variants were heterozygous, missense and incompletely penetrant, consistent with a complex etiology that implicates a large number of factors that together increase the risk of NTDs. As PCP mutations are not completely penetrant, it would still not be possible to offer an accurate recurrence risk after identifying a mutation in a PCP gene.
Despite intensive efforts, the etiology of NTDs remain a challenge and thousands of families are still affected resulting in a significant clinical, emotional and financial burden to the society. As genes and variants that are responsible for these devastating conditions are identified through NGS strategies, large collaborative efforts are needed to dissect this etiology and only then a better picture of the relative contribution of PCP signaling as well as other disease-relevant signaling pathways will emerge. In the more distant future, advances in genome technologies and in bioinformatics pipelines as well as the devotion of a large community of NTD researchers will provide affected families and clinicians with the tools for a personalized medicine in addition to better estimates of risk factors and appropriate prevention and counseling strategies.
Author Contributions
All authors contributed to writing the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rossi, A.; Biancheri, R.; Cama, A.; Piatelli, G.; Ravegnani, M.; Tortori-Donati, P. Imaging in spine and spinal cord malformations. Eur. J. Radiol. 2004, 50, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Stanier, P.; Greene, N.D.E. Neural tube defects: Recent advances, unsolved questions, and controversies. Lancet Neurol. 2013, 12, 799–810. [Google Scholar] [CrossRef]
- Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet 1991, 338, 131–137. [CrossRef]
- Blom, H.J.; Shaw, G.M.; den Heijer, M.; Finnell, R.H. Neural tube defects and folate: Case far from closed. Nat. Rev. Neurosci. 2006, 7, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, H.; Kancherla, V.; Moorthie, S.; Darlison, M.W.; Modell, B. Estimates of global and regional prevalence of neural tube defects for 2015: A systematic analysis. Ann. N. Y. Acad. Sci. 2018, 1414, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Mosley, B.S.; Cleves, M.A.; Siega-Riz, A.M.; Shaw, G.M.; Canfield, M.A.; Waller, D.K.; Werler, M.M.; Hobbs, C.A. National Birth Defects Prevention Study Neural tube defects and maternal folate intake among pregnancies conceived after folic acid fortification in the United States. Am. J. Epidemiol. 2009, 169, 9–17. [Google Scholar] [CrossRef]
- Greene, N.D.E.; Leung, K.-Y.; Gay, V.; Burren, K.; Mills, K.; Chitty, L.S.; Copp, A.J. Inositol for the prevention of neural tube defects: A pilot randomised controlled trial. Br. J. Nutr. 2016, 115, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.E.; Leung, K.-Y.; Copp, A.J. Inositol, neural tube closure and the prevention of neural tube defects. Birth Defects Res. 2017, 109, 68–80. [Google Scholar] [CrossRef]
- Bassuk, A.G.; Kibar, Z. Genetic basis of neural tube defects. Semin. Pediatr. Neurol. 2009, 16, 101–110. [Google Scholar] [CrossRef]
- Avagliano, L.; Massa, V.; George, T.M.; Qureshy, S.; Bulfamante, G.P.; Finnell, R.H. Overview on neural tube defects: From development to physical characteristics. Birth Defects Res. 2018. [Google Scholar] [CrossRef]
- Detrait, E.R.; George, T.M.; Etchevers, H.C.; Gilbert, J.R.; Vekemans, M.; Speer, M.C. Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicol. Teratol. 2005, 27, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.A. Non-multifactorial neural tube defects. Am. J. Med. Genet. C Semin Med. Genet. 2005, 135C, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Massarwa, R.; Ray, H.J.; Niswander, L. Morphogenetic movements in the neural plate and neural tube: Mouse. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulou, E.; Galea, G.L.; Rolo, A.; Greene, N.D.E.; Copp, A.J. Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development 2017, 144, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Ybot-Gonzalez, P.; Cogram, P.; Gerrelli, D.; Copp, A.J. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development 2002, 129, 2507–2517. [Google Scholar] [PubMed]
- Ybot-Gonzalez, P.; Gaston-Massuet, C.; Girdler, G.; Klingensmith, J.; Arkell, R.; Greene, N.D.E.; Copp, A.J. Neural plate morphogenesis during mouse neurulation is regulated by antagonism of Bmp signalling. Development 2007, 134, 3203–3211. [Google Scholar] [CrossRef] [Green Version]
- Haigo, S.L.; Hildebrand, J.D.; Harland, R.M.; Wallingford, J.B. Shroom induces apical constriction and is required for hingepoint formation during neural tube closure. Curr. Biol. 2003, 13, 2125–2137. [Google Scholar] [CrossRef]
- Martin, P. Morphogenesis: Shroom in to close the neural tube. Curr. Biol. 2004, 14, 150–151. [Google Scholar] [CrossRef]
- Sawyer, J.M.; Harrell, J.R.; Shemer, G.; Sullivan-Brown, J.; Roh-Johnson, M.; Goldstein, B. Apical constriction: A cell shape change that can drive morphogenesis. Dev. Biol. 2010, 341, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Pai, Y.-J.; Abdullah, N.L.; Mohd-Zin, S.W.; Mohammed, R.S.; Rolo, A.; Greene, N.D.E.; Abdul-Aziz, N.M.; Copp, A.J. Epithelial fusion during neural tube morphogenesis. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Camerer, E.; Barker, A.; Duong, D.N.; Ganesan, R.; Kataoka, H.; Cornelissen, I.; Darragh, M.R.; Hussain, A.; Zheng, Y.-W.; Srinivasan, Y.; et al. Local protease signaling contributes to neural tube closure in the mouse embryo. Dev. Cell 2010, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Pyrgaki, C.; Liu, A.; Niswander, L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev. Biol. 2011, 353, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolo, A.; Savery, D.; Escuin, S.; de Castro, S.C.; Armer, H.E.J.; Munro, P.M.G.; Molè, M.A.; Greene, N.D.E.; Copp, A.J. Regulation of cell protrusions by small GTPases during fusion of the neural folds. Elife 2016. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, N.M.; Turmaine, M.; Greene, N.D.E.; Copp, A.J. EphrinA-EphA receptor interactions in mouse spinal neurulation: Implications for neural fold fusion. Int. J. Dev. Biol. 2009, 53, 559–568. [Google Scholar] [CrossRef]
- Van Allen, M.I.; Kalousek, D.K.; Chernoff, G.F.; Juriloff, D.; Harris, M.; McGillivray, B.C.; Yong, S.L.; Langlois, S.; MacLeod, P.M.; Chitayat, D. Evidence for multi-site closure of the neural tube in humans. Am. J. Med. Genet. 1993, 47, 723–743. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, M.I. Multisite neural tube closure in humans. Birth Defects Orig. Artic. Ser. 1996, 30, 203–225. [Google Scholar] [PubMed]
- De Bakker, B.S.; Driessen, S.; Boukens, B.J.D.; van den Hoff, M.J.B.; Oostra, R.-J. Single-site neural tube closure in human embryos revisited. Clin. Anat. 2017, 30, 988–999. [Google Scholar] [CrossRef]
- Catala, M. Genetic control of caudal development. Clin. Genet. 2002, 61, 89–96. [Google Scholar] [CrossRef]
- Harris, M.J.; Juriloff, D.M. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. Part. A Clin. Mol. Teratol. 2010, 88, 653–669. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.T.; Wallingford, J.B. Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Maung, S.M.T.W.; Jenny, A. Planar cell polarity in Drosophila. Organogenesis 2011, 7, 165–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Borchers, A.G.M.; Jolicoeur, C.; Rayburn, H.; Baker, J.C.; Tessier-Lavigne, M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 2004, 430, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Oishi, I.; Endo, M.; Nishita, M. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: Their implications in developmental morphogenesis and human diseases. Dev. Dyn. 2010, 239, 1–15. [Google Scholar] [CrossRef]
- Macheda, M.L.; Sun, W.W.; Kugathasan, K.; Hogan, B.M.; Bower, N.I.; Halford, M.M.; Zhang, Y.F.; Jacques, B.E.; Lieschke, G.J.; Dabdoub, A.; et al. The Wnt receptor Ryk plays a role in mammalian planar cell polarity signaling. J. Biol. Chem. 2012, 287, 29312–29323. [Google Scholar] [CrossRef]
- Murdoch, J.N.; Henderson, D.J.; Doudney, K.; Gaston-Massuet, C.; Phillips, H.M.; Paternotte, C.; Arkell, R.; Stanier, P.; Copp, A.J. Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Hum. Mol. Genet. 2003, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Barrow, J.R. Wnt/PCP signaling: A veritable polar star in establishing patterns of polarity in embryonic tissues. Semin. Cell Dev. Biol. 2006, 17, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Vladar, E.K.; Antic, D.; Axelrod, J.D. Planar cell polarity signaling: The developing cell’s compass. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- McNeill, H. Planar cell polarity: Keeping hairs straight is not so simple. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Sebbagh, M.; Borg, J.-P. Insight into planar cell polarity. Exp. Cell Res. 2014, 328, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.J.; Long, D.A.; Dean, C.H. Planar cell polarity in organ formation. Curr. Opin. Cell Biol. 2018, 55, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, A.C.; Mlodzik, M. From instruction to output: Wnt/PCP signaling in development and cancer. Curr. Opin. Cell Biol. 2018, 51, 110–116. [Google Scholar] [CrossRef] [PubMed]
- VanderVorst, K.; Dreyer, C.A.; Konopelski, S.E.; Lee, H.; Ho, H.-Y.H.; Carraway, K.L. Wnt/PCP Signaling Contribution to Carcinoma Collective Cell Migration and Metastasis. Cancer Res. 2019, 79, 1719–1729. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nathans, J. Tissue/planar cell polarity in vertebrates: New insights and new questions. Development 2007, 134, 647–658. [Google Scholar] [CrossRef]
- Roszko, I.; Sawada, A.; Solnica-Krezel, L. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin. Cell Dev. Biol. 2009, 20, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Huebner, R.J.; Wallingford, J.B. Coming to Consensus: A Unifying Model Emerges for Convergent Extension. Dev. Cell 2018, 46, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallingford, J.B. Neural tube closure and neural tube defects: Studies in animal models reveal known knowns and known unknowns. Am. J. Med. Genet. C Semin Med. Genet. 2005, 135, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Heisenberg, C.-P. Convergent extension: Using collective cell migration and cell intercalation to shape embryos. Development 2012, 139, 3897–3904. [Google Scholar] [CrossRef] [PubMed]
- Shindo, A. Models of convergent extension during morphogenesis. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Vogan, K.J.; Groulx, N.; Justice, M.J.; Underhill, D.A.; Gros, P. Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat. Genet. 2001, 28, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Doudney, K.; Paternotte, C.; Copp, A.J.; Stanier, P. Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Hum. Mol. Genet. 2001, 10, 2593–2601. [Google Scholar] [CrossRef]
- Ybot-Gonzalez, P.; Savery, D.; Gerrelli, D.; Signore, M.; Mitchell, C.E.; Faux, C.H.; Greene, N.D.E.; Copp, A.J. Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development 2007, 134, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.A.; Quint, E.; Tsipouri, V.; Arkell, R.M.; Cattanach, B.; Copp, A.J.; Henderson, D.J.; Spurr, N.; Stanier, P.; Fisher, E.M.; et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003, 13, 1129–1133. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, N.; Nathans, J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 2006, 26, 2147–2156. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Ray, S.; Li, S.; Hamblet, N.S.; Lijam, N.; Tsang, M.; Greer, J.; Kardos, N.; Wang, J.; Sussman, D.J.; et al. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008, 4, e1000259. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Damrau, C.; Paudyal, A.; Bogani, D.; Wells, S.; Greene, N.D.E.; Stanier, P.; Copp, A.J. Genetic interactions between planar cell polarity genes cause diverse neural tube defects in mice. Dis. Model. Mech. 2014, 7, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Montcouquiol, M.; Rachel, R.A.; Lanford, P.J.; Copeland, N.G.; Jenkins, N.A.; Kelley, M.W. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 2003, 423, 173–177. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, E.M.; Vijayraghavan, D.; Davidson, L.A.; Hildebrand, J.D. Shroom3 functions downstream of planar cell polarity to regulate myosin II distribution and cellular organization during neural tube closure. Biol. Open 2015, 4, 186–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallingford, J.B.; Mitchell, B. Strange as it may seem: The many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011, 25, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Assémat, E.; Bazellières, E.; Pallesi-Pocachard, E.; Le Bivic, A.; Massey-Harroche, D. Polarity complex proteins. Biochim. Biophys. Acta 2008, 1778, 614–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halaoui, R.; McCaffrey, L. Rewiring cell polarity signaling in cancer. Oncogene 2015, 34, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Kharfallah, F.; Guyot, M.C.; El Hassan, A.R.; Allache, R.; Merello, E.; De Marco, P.; Di Cristo, G.; Capra, V.; Kibar, Z. Scribble1 plays an important role in the pathogenesis of neural tube defects through its mediating effect of Par-3 and Vangl1/2 localization. Hum. Mol. Genet. 2017, 26, 2307–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žigman, M.; Trinh, L.A.; Fraser, S.E.; Moens, C.B. Zebrafish Neural Tube Morphogenesis Requires Scribble-Dependent Oriented Cell Divisions. Curr. Biol. 2011, 21, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milgrom-Hoffman, M.; Humbert, P.O. Regulation of cellular and PCP signalling by the Scribble polarity module. Semin. Cell Dev. Biol. 2018, 81, 33–45. [Google Scholar] [CrossRef]
- Weidinger, G.; Moon, R.T. When Wnts antagonize Wnts. J. Cell Biol. 2003, 162, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Andersson, E.R.; Schambony, A.; Esner, M.; Bryjová, L.; Biris, K.K.; Hall, A.C.; Kraft, B.; Cajanek, L.; Yamaguchi, T.P.; et al. The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol. Biol. Cell 2009, 20, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Bryjova, L.; Biris, K.; Yamaguchi, T.P.; Arenas, E.; Bryja, V. Genetic interaction between Lrp6 and Wnt5a during mouse development. Dev. Dyn. 2010, 239, 237–245. [Google Scholar]
- Tahinci, E.; Thorne, C.A.; Franklin, J.L.; Salic, A.; Christian, K.M.; Lee, L.A.; Coffey, R.J.; Lee, E. Lrp6 is required for convergent extension during Xenopus gastrulation. Development 2007, 134, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef]
- Carter, M.; Chen, X.; Slowinska, B.; Minnerath, S.; Glickstein, S.; Shi, L.; Campagne, F.; Weinstein, H.; Ross, M.E. Crooked tail (Cd) model of human folate-responsive neural tube defects is mutated in Wnt coreceptor lipoprotein receptor-related protein 6. Proc. Natl. Acad. Sci. USA 2005, 102, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.; Naito, M.; Daulat, A.; Angers, S.; Ciruna, B. Ptk7 promotes non-canonical Wnt/PCP-mediated morphogenesis and inhibits Wnt/β-catenin-dependent cell fate decisions during vertebrate development. Development 2013, 140, 1807–1818. [Google Scholar] [CrossRef]
- Peradziryi, H.; Kaplan, N.A.; Podleschny, M.; Liu, X.; Wehner, P.; Borchers, A.; Tolwinski, N.S. PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J. 2011, 30, 3729–3740. [Google Scholar] [CrossRef] [Green Version]
- Puppo, F.; Thomé, V.; Lhoumeau, A.-C.; Cibois, M.; Gangar, A.; Lembo, F.; Belotti, E.; Marchetto, S.; Lécine, P.; Prébet, T.; et al. Protein tyrosine kinase 7 has a conserved role in Wnt/β-catenin canonical signalling. EMBO Rep. 2011, 12, 43–49. [Google Scholar] [CrossRef]
- Bin-Nun, N.; Lichtig, H.; Malyarova, A.; Levy, M.; Elias, S.; Frank, D. PTK7 modulates Wnt signaling activity via LRP6. Development 2014, 141, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Kibar, Z.; Torban, E.; McDearmid, J.R.; Reynolds, A.; Berghout, J.; Mathieu, M.; Kirillova, I.; De Marco, P.; Merello, E.; Hayes, J.M.; et al. Mutations in VANGL1 associated with neural-tube defects. N. Engl. J. Med. 2007, 356, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Doudney, K.; Ybot-Gonzalez, P.; Paternotte, C.; Stevenson, R.E.; Greene, N.D.E.; Moore, G.E.; Copp, A.J.; Stanier, P. Analysis of the planar cell polarity gene Vangl2 and its co-expressed paralogue Vangl1 in neural tube defect patients. Am. J. Med. Genet. A 2005, 136, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Bosoi, C.M.; Kooistra, M.; Salem, S.; Finnell, R.H.; De Marco, P.; Merello, E.; Bassuk, A.G.; Capra, V.; Gros, P. Novel mutations in VANGL1 in neural tube defects. Hum. Mutat. 2009, 30, E706–E715. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.-P.; Zhang, T.; Li, H.; Wu, B.-L.; Jin, L.; Wang, H.-Y. VANGL2 mutations in human cranial neural-tube defects. N. Engl. J. Med. 2010, 362, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.M.; Capra, V.; Allache, R.; Trinh, V.Q.-H.; De Marco, P.; Merello, E.; Drapeau, P.; Bassuk, A.G.; Kibar, Z. Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum. Mutat. 2011, 32, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Salem, S.; Bosoi, C.M.; Pauwels, E.; De Marco, P.; Merello, E.; Bassuk, A.G.; Capra, V.; Gros, P. Contribution of VANGL2 mutations to isolated neural tube defects. Clin. Genet. 2011, 80, 76–82. [Google Scholar] [CrossRef]
- Allache, R.; De Marco, P.; Merello, E.; Capra, V.; Kibar, Z. Role of the planar cell polarity gene CELSR1 in neural tube defects and caudal agenesis. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, O.; Kirmes, I.; Thiede, A.; Lechno, S.; Gocan, H.; Florian, I.S.; Haaf, T.; Zechner, U.; Sabova, L.; Horn, F. Novel VANGL1 Gene Mutations in 144 Slovakian, Romanian and German Patients with Neural Tube Defects. Mol. Syndromol. 2012, 3, 76–81. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Merello, E.; Rossi, A.; Piatelli, G.; Cama, A.; Kibar, Z.; Capra, V. FZD6 is a novel gene for human neural tube defects. Hum. Mutat. 2012, 33, 384–390. [Google Scholar] [CrossRef]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.E.; Copp, A.J.; Stanier, P. Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar] [CrossRef]
- De Marco, P.; Merello, E.; Consales, A.; Piatelli, G.; Cama, A.; Kibar, Z.; Capra, V. Genetic analysis of disheveled 2 and disheveled 3 in human neural tube defects. J. Mol. Neurosci. 2013, 49, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhu, H.; Duhon, C.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Mutations in planar cell polarity gene SCRIB are associated with spina bifida. PLoS ONE 2013, 8, e69262. [Google Scholar] [CrossRef] [PubMed]
- Allache, R.; Lachance, S.; Guyot, M.C.; De Marco, P.; Merello, E.; Justice, M.J.; Capra, V.; Kibar, Z. Novel mutations in Lrp6 orthologs in mouse and human neural tube defects affect a highly dosage-sensitive Wnt non-canonical planar cell polarity pathway. Hum. Mol. Genet. 2014, 23, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhu, H.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Identification of novel CELSR1 mutations in spina bifida. PLoS ONE 2014, 9, e92207. [Google Scholar] [CrossRef] [PubMed]
- Allache, R.; Wang, M.; De Marco, P.; Merello, E.; Capra, V.; Kibar, Z. Genetic studies of ANKRD6 as a molecular switch between Wnt signaling pathways in human neural tube defects. Birth Defects Res. Part. A Clin. Mol. Teratol. 2015, 103, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Fathe, K.; McCartney, D.; Zhu, H.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Rare LRP6 variants identified in spina bifida patients. Hum. Mutat. 2015, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Merello, E.; Mascelli, S.; Raso, A.; Piatelli, G.; Consales, A.; Cama, A.; Kibar, Z.; Capra, V.; Marco, P.D. Expanding the mutational spectrum associated to neural tube defects: Literature revision and description of novel VANGL1 mutations. Birth Defects Res. Part. A Clin. Mol. Teratol. 2015, 103, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; De Marco, P.; Merello, E.; Drapeau, P.; Capra, V.; Kibar, Z. Role of the planar cell polarity gene Protein tyrosine kinase 7 in neural tube defects in humans. Birth Defects Res. Part. A Clin. Mol. Teratol. 2015, 103, 1021–1027. [Google Scholar] [CrossRef]
- Qiao, X.; Liu, Y.; Li, P.; Chen, Z.; Li, H.; Yang, X.; Finnell, R.H.; Yang, Z.; Zhang, T.; Qiao, B.; et al. Genetic analysis of rare coding mutations in CELSR1-3 in Chinese Congenital Heart and Neural Tube Defects. Clin. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lei, Y.; Cao, X.; Zheng, Y.; Wang, F.; Bao, Y.; Peng, R.; Finnell, R.H.; Zhang, T.; Wang, H. Genetic analysis of Wnt/PCP genes in neural tube defects. BMC Med. Genomics 2018, 11, 38. [Google Scholar] [CrossRef]
- Wang, L.; Xiao, Y.; Tian, T.; Jin, L.; Lei, Y.; Finnell, R.H.; Ren, A. Digenic variants of planar cell polarity genes in human neural tube defect patients. Mol. Genet. Metab. 2018, 124, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Kim, S.-E.; Chen, Z.; Cao, X.; Zhu, H.; Yang, W.; Shaw, G.M.; Zheng, Y.; Zhang, T.; Wang, H.-Y.; et al. Variants identified in PTK7 associated with neural tube defects. Mol. Genet. Genomic. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Shi, O.; Wang, B.; Chang, B.; Yang, R.; Wang, Y.; Wang, F.; Shen, C. Association between VANGL1 gene polymorphisms and neural tube defects. Neuropediatrics 2014, 45, 234–239. [Google Scholar] [PubMed]
- Reynolds, A.; McDearmid, J.R.; Lachance, S.; De Marco, P.; Merello, E.; Capra, V.; Gros, P.; Drapeau, P.; Kibar, Z. VANGL1 rare variants associated with neural tube defects affect convergent extension in zebrafish. Mech. Dev. 2010, 127, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, M.; Gao, X.; Yu, L.X.; Paria, N.; Henkelman, R.M.; Wise, C.A.; Ciruna, B. ptk7 mutant zebrafish models of congenital and idiopathic scoliosis implicate dysregulated Wnt signalling in disease. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lemay, P.; Guyot, M.-C.; Tremblay, É.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, É.; Diallo, O.; De Marco, P.; Merello, E.; Massicotte, C.; et al. Loss-of-function de novo mutations play an important role in severe human neural tube defects. J. Med. Genet. 2015, 52, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Wang, W.; Corominas, R.; Lin, G.N. De novo Mutations from Whole Exome Sequencing in Neurodevelopmental and Psychiatric Disorders: From Discovery to Application. Front. Genet. 2019. [Google Scholar] [CrossRef]
- Chen, Z.; Lei, Y.; Zheng, Y.; Aguiar-Pulido, V.; Ross, M.E.; Peng, R.; Jin, L.; Zhang, T.; Finnell, R.H.; Wang, H. Threshold for neural tube defect risk by accumulated singleton loss-of-function variants. Cell Res. 2018, 28, 1039–1041. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Normal and abnormal neural tube formation. (A) Major steps of neural tube formation. The neural plate overlying the notochord elevates to form the neural folds around the midline, bends at the midline (mhp) and dorsolateral sites (dlhp) then fuses at the opposing tips of the neural folds and separates from the overlying epidermis. (B). Initiation sites of neural tube closure 1–3 in a mouse embryo. Defects in closure 1 and 2 lead to craniorachischisis and anencephaly respectively. Defects in closure at the caudal end of site 1 lead to open spina bifida. (C). Lateral and dorsal views of E18.5 embryos showing craniorachischisis (C′) and open spina bifida (C″, indicated by red arrows) as compared to wild type (C). cc, central canal; dlhp, dorsolateral hinge point; ec, ectoderm; ep, epidermis; fb, forebrain; hb, hindbrain; mb, midbrain; mhp, median hinge point; me, mesoderm; nc, notochord; ncp, notochordal plate; nec, neural crest; np, neural plate; npb, neural plate border; pe, presumptive epidermis; sc, spinal cord; so, somites.
Figure 1.
Normal and abnormal neural tube formation. (A) Major steps of neural tube formation. The neural plate overlying the notochord elevates to form the neural folds around the midline, bends at the midline (mhp) and dorsolateral sites (dlhp) then fuses at the opposing tips of the neural folds and separates from the overlying epidermis. (B). Initiation sites of neural tube closure 1–3 in a mouse embryo. Defects in closure 1 and 2 lead to craniorachischisis and anencephaly respectively. Defects in closure at the caudal end of site 1 lead to open spina bifida. (C). Lateral and dorsal views of E18.5 embryos showing craniorachischisis (C′) and open spina bifida (C″, indicated by red arrows) as compared to wild type (C). cc, central canal; dlhp, dorsolateral hinge point; ec, ectoderm; ep, epidermis; fb, forebrain; hb, hindbrain; mb, midbrain; mhp, median hinge point; me, mesoderm; nc, notochord; ncp, notochordal plate; nec, neural crest; np, neural plate; npb, neural plate border; pe, presumptive epidermis; sc, spinal cord; so, somites.

Figure 2.
The non-canonical Wnt/planar cell polarity signaling pathway (PCP). (A) A simplified diagram of the PCP signaling pathway showing its core components, its downstream effectors and two of its mediators Ptk7/Otk and Scribble1/Scrib. Nomenclature for core proteins is indicated in vertebrates and Drosophila (separated by/). Genetic interactions are indicated by double arrows. (B) An example of a PCP-regulated process in the wing blade of Drosophila. Asymmetric localization of PCP protein complexes at the proximal (Vang-Pk) and distal (Fz-Dsh) sides of adjacent cells mediates the distal positioning and orientation of a single trichome in each cell of the wing epithelium. (C) The process of convergent extension. Cells elongate mediolaterally, move and intercalate with neighboring cells resulting in convergence toward the midline and extension along the anteroposterior axis. During this process, PK and VANGL localize anteriorly whereas FZD and DVL localize posteriorly. (D) A simplified diagram showing a widened neural plate because of a defective convergent extension (CE) of midline neuroepithelial cells. The neural folds will be far apart and will fail to fuse leading to a neural tube defect.
Figure 2.
The non-canonical Wnt/planar cell polarity signaling pathway (PCP). (A) A simplified diagram of the PCP signaling pathway showing its core components, its downstream effectors and two of its mediators Ptk7/Otk and Scribble1/Scrib. Nomenclature for core proteins is indicated in vertebrates and Drosophila (separated by/). Genetic interactions are indicated by double arrows. (B) An example of a PCP-regulated process in the wing blade of Drosophila. Asymmetric localization of PCP protein complexes at the proximal (Vang-Pk) and distal (Fz-Dsh) sides of adjacent cells mediates the distal positioning and orientation of a single trichome in each cell of the wing epithelium. (C) The process of convergent extension. Cells elongate mediolaterally, move and intercalate with neighboring cells resulting in convergence toward the midline and extension along the anteroposterior axis. During this process, PK and VANGL localize anteriorly whereas FZD and DVL localize posteriorly. (D) A simplified diagram showing a widened neural plate because of a defective convergent extension (CE) of midline neuroepithelial cells. The neural folds will be far apart and will fail to fuse leading to a neural tube defect.


{kind=link}
{kind=link}
Table 1.
List of rare coding PCP variants identified in neural tube defects (NTDs) and predicted to be damaging and/or affect protein function in NTDs.
Table 1.
List of rare coding PCP variants identified in neural tube defects (NTDs) and predicted to be damaging and/or affect protein function in NTDs.
| Gene | NTD Cohort | Variant | Patient Description | Functional Validation | Reference |
|---|---|---|---|---|---|
| PCP core genes | |||||
| ANKRD6 | 391 Italians and 82 French Canadians (14 cranial, 246 MMC, and 213 closed spinal NTDs) | p.Pro548Leu p.Arg587Gln p.Arg632His | MC LipoMS and MC CA | p.Pro548Leu and p.Arg632His were proven to be partially loss of function in activation of PCP signaling and inhibition of canonical Wnt pathway | [95] |
| CELSR1 | 36 fetuses with CRS from US, France and England | p.Ala773Val p.Arg2438Gln p.Ser2964Leu p.Pro2983Ala | 1 CRS 1 CRS 2 CRS 2 CRS | All 4 variants significantly affected frequency of membrane localisation | [90] |
| 391 Italians and 82 French Canadians (same as above) | p.Arg541Trp p.Val551Met p.Gln834X p.Arg.836Cys p.Val1008Leu p.Asp1401Gly p.Thr1443Pro p.Arg1456Gln p.Arg1526Trp p.Arg1835Cys p.Arg2121Cys p.Ser2190Leu p.Ala2228Val p.Arg2359Cys p.Ser2963Thr2966del | MMC LMC MMC Lipoma TFT MMC LMMC CA MMC MMC LMMC Lipoma Lipoma LMC CA | Not conducted | [87] | |
| 192 American SB infants | p.Ala1023Gly p.Ile1124Met p.Thr1362Met p.Gly1410Arg Truncated protein (c.5050_5051 ins GT) p.Gly1825Ser Truncated protein (c.5719_5720 del TG) p.Gly2062Ser p.Arg2354Cys p.Arg2497Cys | All SB | Two truncated mutations disrupted CELSR1 membrane localization and its recruitment of Vangl2 to cell membrane. | [94] | |
| 352 Chinese patients (26 CRS, 73EC, 64AN, 3Ex, 255 SB, 1 unknown) | p.Pro870Leu | CRS | p.Pro870Leu was a gain-of-function variant in zebrafish overexpression and rescue experiments. It increased both PCP and canonical Wnt signalling | [99] | |
| 184 Chinese patients (36 AN,12 CRS, 32 EC, 2 Ex, 91 SB, 11 unknown) and 292 American patients (100% SB) | p.Arg714His p.Pro870Leu p.Thr875Ile p.Ala1019Ser p.Thr1086Met p.Arg1194His p.Gln1473Ter | AN, SB SB SB | Not conducted | [100] | |
| 510 Chinese patients (125 AN, 232 SB, 46 EC, 79 AN & SB, 1 AN & EC, 15 SB & EC, 4 AN & SB & EC, 8 unknown) | p. Arg769Trp p.Arg1057Cys p.Gly1122Ser p.Gln2924His | SB SB AN SB | Not conducted | [101] | |
| CELSR2 | 352 Chinese patients (same as above) | p.Ser628Gly p.Thr2026Met p.Arg2153Gly p.Arg2480Cys p.Arg2015Gly fs*22 p.Phe2397Lys fs*584 | AN, SB SB EC SB CRS SB | p.Thr2026Met downregulated PCP signaling in a luciferase assay | [99,100] |
| 184 Chinese patients (same as above) | p.Ser628Gly p.Arg1990His p.Arg2015Gly fs*23 p.Thr2026His p.Arg2153Gly p.Arg2480Cys p.Arg2626Cys | Not conducted | [100] | ||
| CELSR3 | 352 Chinese patients (same as above) | p.Gly194Val p.Val446Met p.Gly1754Asp | 1 AN, 1 SB CRS CRS | Not conducted | [99] |
| 184 Chinese patients (same as above) | p.Val446Met p.Ile1102Leu p.Arg1194His p.Arg1453Cys p.Gly1754Asp p.Gly1754Ser p.Val2584Gly p.Met2630Ile | Not conducted | [100] | ||
| DVL2 | 473 Italian and French Canadian patients (same as above) | p.Ala53Val p.Glu620X (c.1801_1802ins) p.Tyr667Cys | Lipoma CA and TC MMC | Not conducted | [91] |
| DVL3 | 184 Chinese patients (same as above) | p.Asp403Asn | SB | p.Asp403Asn disrupted DVL3 interaction with VANGL2, upregulated canonical Wnt signaling and down regulated non-PCP signaling | [100] |
| 510 Chinese patients (same as above) | p.Arg148Gln | AN | Not conducted | [101] | |
| FZD6 | 473 Italian and French Canadian patients (same as above) | p.Cys615Ter p.Arg405Gln p.Arg511Cys p.Arg511His | MC MMC MMC CA | Not conducted | [89] |
| PK1 | 810 patients: 421 Italian (11 cranial, 211 open spinal including 208 MMC, 199 closed spinal) and 389 Americans (4 cranial, 325 MMC, 60 closed spinal) | p.Ile69Thr p.Asn81His p.Thr275Met p.Val550Met p.Arg682Cys p.Ser739Phe p.Asp771Asn | DM MMC MMC MMC MMC MMC CA | p.Ile69Thr, p.Asn81His, p.Thr275Met and p.Arg682Cys acted as hypermorphic alleles in inducing CE defects in an overexpression assay in zebrafish; p.Arg682Cys antagonized the CE phenotype induced by the wild-type zpk1a in a dominant fashion | [85] |
| VANGL1 | 137 Italian patients (80 MMC, 57 closed spinal) 7 French fetuses with CRS | p.Val239Ile p.Arg274Gln p.Met328Thr | CA MMC MMC, TC | p.Val239Ile abrogated interaction between VANGL1 and all three Dvl proteins; p.Val239Ile and p.Met328Thr failed to induce CE defects in overexpression assays and to rescue MO-induced CE defects in zebrafish | [81,104] |
| 673 patients: 284 Italians (11 cranial, 131 open spinal including 128 MMC, 142 closed spinal) and 389 Americans (4 cranial, 325 MMC, 60 closed spinal) | p.Ser83Ile p.Phe153Ser p.Arg181Gln p.Leu202Phe p.Ala404Ser | 3 TFT and TC TFT and TC MMC MMC CA | Not conducted | [83] | |
| 144 patients with open or closed NTDs from Slovakia, Romania and Germany | p.Gly205Arg p.Arg186His p.Arg173His | MMC TC and lipoma unknown | Not conducted | [88] | |
| 53 Italian patients (9 MMC, 44 closed spinal) | p.Alal187Val p.Arg517His p.His350 His | LipoMS MMC Cephalocele | Not conducted | [97] | |
| VANGL2 | 66 English patients (21 CRS, 24 SB, 21 AN) | A 7 bp duplication detected 30 nucleotides into intron six (IVS6+30) | CRS | Not conducted | [82] |
| 163 Han Chinese fetuses (16 AN, 63 CRS, 8 EC, 4 HPS, 14 iniencephaly, 58 SB) | p.Ser84Phe p.Arg353Cys p.Phe437Ser | HPS AN with SB AN | p.Phe437Ser completely abrogated interaction with Dvl; p.Arg353Cys diminished but did not abolish this interaction | [84] | |
| 673 Italian and American patients (same as above) | p.Arg135Trp p.Arg177His p.Leu242Val p.Thr247Met p.Arg270His p.Arg482His | MMC DM 1MCS, 1MMC Lipoma Fibrolipoma CA and TC | Not conducted | [86] | |
| PCP mediators | |||||
| LRP6 | 285 Italian patients (6 closed cranial, 153 MMC, 126 closed spinal) | p.Tyr306His p.Thr373Cys p.Val1386Leu p.Thr1541Cys | MMC MMC EC CA | p.Tyr306His, p.Thr373Cys and p.Val1386Leu acted as hypomorphic alleles in activating canonical Wnt pathway and inhibiting PCP signaling | [93] |
| 192 American SB infants | p.Ala3Val p.Tyr544Cys p.Pro1482Leu p.Arg1574Leu | SB | p.Tyr544Cys lost its ability to bind MESD and its membrane localization and acted as a hypomorhic allele in activating canonical Wnt signalling; p.Arg1574Leu acted as a hypermorphic allele in activating canonical Wnt signaling; p.Pro1482Leu lost its ability to inhibit PCP signaling | [96] | |
| PTK7 | 473 Italian and French Canadian patients (same as above) | p.Ile121Met p.Val291Ile p.Pro345Leu p.Gly348Ser p.Pro545Arg | MMC LipoMS MC MMC MMC | p.Ile121Met, p.Pro345Leu and p.Pro545Arg could act in a hypomorphic manner; p.Gly348Ser acted in a hypermorphic manner in overexpression assays in zebrafish;p.Pro545Arg affected stability of protein | [98,105] |
| 343 Chinese patients (70 AN, 19 CRS, 80 EC, 3 EX, 170 SB, 1 unknown) and 192 American SB infants | p.Asn128Ser p.Thr186Met p.Ala560Thr p.Arg630Ser p.Pro706Arg p.Tyr725Phe p.Gly765Arg p.Val775Met p.Arg790Leu | EX SB SB SB 2 AN, CRS, 2 SB SB 2 SB SB 2 SB, EC, AN | p.Arg630Ser affected protein stability and increased interaction with Dvl2; p.Thr186Met decreased PTK7 interactions with Dvl2 | [102] | |
| 510 Chinese patients (same as above) | p.Pro642Arg | SB | Not conducted | [101] | |
| SCRIB1 | 52 fetuses with CRS (same as above) | p.Arg1535Gln | CRS | p.Arg1535Gln affected membrane localization of Scrib1 | [90] |
| 192 American SB infants | p.Ala366Thr p.Thr552Met p.Pro1043Leu p.Pro1332Leu p.Leu1520Arg | SB | p.Pro1043Leu, p.Pro1332Leu and p.Leu1520Arg significantly affected protein membrane localization | [92] | |
| 473 Italian and French Canadian patients (same as above) | p.Gly263Ser p.Pro649His p.Gln808His p.Arg1150Gln p.Thr1422Met | MMC CA MMC VS MMC | P.Gly263Ser and p.Gln808His significantly affected the subcellular localization of SCRIB1 and failed in rescuing the localization defect of Par-3 and Vangl1 caused by knockdown of Scrib1; P.Gln808His and p.Arg1150Gln abolished the interaction of Scrib1 with Vangl2. | [68] | |
| 510 Chinese patients (same as above) | p.Lys618Arg p.Gly644Val p.Arg1044Gln p.Gly1108Glu | SB SB AN SB | Not conducted | [101] | |
AN, anencephaly; CA, caudal agenesis; CRS, craniorachischisis; DM, diastematomyelia; EC, encephalocele; Ex, exencephaly; HPS, holoprosencephaly; LipoMS, lipomyeloschisis; LMC, lipomyelocele; LMMC, lipomyelomeningocele; MC, meningocele; MCS, myelocystocele; MMC, myelomeningocele; NTD, neural tube defect; PCP, planar cell polarity; SB, spina bifida; TC, tethered cord; TFT, tight filum terminale; VS, vertebral schisis.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, M.; de Marco, P.; Capra, V.; Kibar, Z. Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects. Cells 2019, 8, 1198. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101198
AMA Style
Wang M, de Marco P, Capra V, Kibar Z. Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects. Cells. 2019; 8(10):1198. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101198
Chicago/Turabian StyleWang, Mingqin, Patrizia de Marco, Valeria Capra, and Zoha Kibar. 2019. "Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects" Cells 8, no. 10: 1198. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101198
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.