TGF-β and microRNA Interplay in Genitourinary Cancers


1
Department of Biochemistry and Molecular Biology, Centre of Postgraduate Medical Education; 01-813 Warsaw, Poland
2
II Department of Urology, Centre of Postgraduate Medical Education, 01-813 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Cells 2019, 8(12), 1619; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121619
Submission received: 20 November 2019
/
Revised: 9 December 2019
/
Accepted: 10 December 2019
/
Published: 12 December 2019
(This article belongs to the Special Issue microRNA as Therapeutic Target)
Abstract
:Genitourinary cancers (GCs) include a large group of different types of tumors localizing to the kidney, bladder, prostate, testis, and penis. Despite highly divergent molecular patterns, most GCs share commonly disturbed signaling pathways that involve the activity of TGF-β (transforming growth factor beta). TGF-β is a pleiotropic cytokine that regulates key cancer-related molecular and cellular processes, including proliferation, migration, invasion, apoptosis, and chemoresistance. The understanding of the mechanisms of TGF-β actions in cancer is hindered by the “TGF-β paradox” in which early stages of cancerogenic process are suppressed by TGF-β while advanced stages are stimulated by its activity. A growing body of evidence suggests that these paradoxical TGF-β actions could result from the interplay with microRNAs: Short, non-coding RNAs that regulate gene expression by binding to target transcripts and inducing mRNA degradation or inhibition of translation. Here, we discuss the current knowledge of TGF-β signaling in GCs. Importantly, TGF-β signaling and microRNA-mediated regulation of gene expression often act in complicated feedback circuits that involve other crucial regulators of cancer progression (e.g., androgen receptor). Furthermore, recently published in vitro and in vivo studies clearly indicate that the interplay between microRNAs and the TGF-β signaling pathway offers new potential treatment options for GC patients.
1. Introduction
Transforming growth factor-beta (TGF-β) emerges as one of the key regulators of tumor development and progression. It influences all crucial steps of cancer progression, including migration and invasion, with a prominent influence on the process of epithelial–mesenchymal transition (EMT). The mechanisms of TGF-β actions in cancer are complex. At the beginning of tumor development, TGF-β attenuates cancerous proliferation, limiting tumor growth. During cancer progression, these tumor suppressive effects reverse, and TGF-β starts to promote migration, invasion, and formation of distant metastasis [1]. This so called “TGF-β paradox”, universally occurring in cancers, has remained a mystery for many years. Recently, a growing body of evidence suggests that these paradoxical TGF-β actions could result from interplay with the activity of microRNAs, which emerge as important modulators and mediators of TGF-β effects in cancer cells. These small, non-coding RNAs are involved in the control of a wide array of critical biological processes, including cells development, proliferation, growth, differentiation, and apoptosis. Abnormalities in their expression or function contribute to the development of multiple disorders, including cancer [2]. microRNAs are transcribed as long, hairpin-shaped primary transcripts called pri-microRNAs. They are further processed and cleaved by Drosha ribonuclease and DGCR8 RNA binding protein, which form the microprocessor complex. The resulting pre-microRNA is exported outside the nucleus by exportin 5 for further processing in the cytoplasm where RNase dicer cleaves it into mature, short, double-stranded miRNA complex. Next, one of the miRNA strands is assembled with argonaute 2 (Ago2) protein within the RISC (RNA-induced silencing) complex, where miRNA functions as a guide enabling targeting mRNA to be degraded or translationally repressed by Ago2. The expression and functioning of miRNAs are modulated by multiple factors, including TGF-β, which regulates microprocessor activity by recruiting SMAD proteins, which enhance Drosha-mediated processing of pri-miRNA [3].
Genitourinary cancers (GCs) represent 25% of all solid tumors [4]. They derive from different types of cells located in the kidney, penis, testis, bladder, and prostate [5,6]. GCs are highly divergent, in terms of molecular pathology and prognosis, ranging from excellent outcomes for patients with testicular cancer to metastatic clear cell renal cell carcinoma, which is associated with poor outcomes. Recent studies revealed the importance of TGF-β signaling in GCs, including animal in vivo studies, bringing hope for new therapeutic approaches. On the other hand, multiple studies showed that microRNAs play an important role in the development and progression of many types of genitourinary tumors. Here, we discuss the interplay between TGF-β and microRNAs and demonstrate its significance as a new field for therapeutic interventions in genitourinary cancers.
2. Genitourinary Cancers (GC): Subtypes, Treatment, and Prognosis
Genitourinary cancers encompass a large group of tumors deriving from different types of cells in the kidney, bladder, prostate, testis, and penis. The particular GC types differ in molecular pathology and biological behavior, which leads to variant treatment options and prognosis. While in patients with testicular or bladder cancer there is a need for urgent aggressive multimodal treatment, in many cases of prostate or renal cancer, active surveillance can be a safe option. Although surgery plays a fundamental role in the radical treatment of all genitourinary cancers, it can be complemented by local or systemic chemotherapy, radiation therapy, local or systemic immunotherapy, hormonal therapy, or targeted therapies.
2.1. Renal Cancer
Renal tumors in adults arise mainly from the epithelium lining renal tubules and are classified as renal cell carcinomas (RCCs). From a histological perspective, there are three main types of RCCs: Clear cell RCC (ccRCC), papillary RCC (pRCC), and chromophobe RCC (chRCC). Novel RCC subtypes have recently been defined based on molecular features (e.g., succinate dehydrogenase (SDH)–deficient renal carcinomas) [5]. They differ in genetics and prognosis, with ccRCC being both the most common and the most aggressive one [7,8]. The key molecular alteration in ccRCC is von Hippel–Lindau (VHL) gene mutation, which leads to uncontrolled hypoxia-induced factor (HIF) expression, followed by the activation of several growth factor pathways, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and others [9].
With increasing use of imaging studies, nowadays, the majority of renal masses are diagnosed incidentally at a low stage [10]. Treatment of choice consists of surgical removal of the tumor, namely partial or radical nephrectomy [11]. The surgery can be either curative or cytoreductive in selected metastatic cases [12,13]. In metastatic disease, patients can be treated with systemic targeted therapy and/or immunotherapy. Targeted therapies act mainly via the VEGF pathway. Tyrosine kinase inhibitors (TKIs) are the most commonly used group, including sunitinib, pazopanib, cabozantinib, and others [11]. Regarding immunotherapy, monotherapy or the combination of nivolumab and ipilimumab are effective treatment options [14]. Nivolumab, an anti-PD1 antibody, restores antitumor activity of T cells while ipilimumab, an anti-CTLA4 antibody, potentiates this activity [15,16].
Prognosis of patients with RCC has significantly improved in the last years due to the progress in systematic therapies, including targeted anti-VEGF therapies and immunotherapies. In general, the 5-year overall survival in renal cancer patients is 49% [17,18]. Cancer-specific survival (CSS) depends on the tumor histology and stage of the disease. For ccRCC, 5-year CSS is estimated as 71% while it is much higher for chRCC and pRCC, reaching 88% and 91%, respectively [19]. Depending on the clinical stage, 5-year CSS in ccRCC can drop from 91% in clinical stage I to 32% in clinical stage IV [20].
2.2. Penile Cancer
Penile cancer is a rare tumor, accounting for about 0.2% of all cancer cases [21]. Usually, it is a squamous cell carcinoma arising from the glans penis or the inner prepuce of foreskin [22]. One-third of cases are associated with the human papilloma virus (HPV) infection, especially by so-called high-risk HPV genotypes 16, 18, 33, and 35 [23].
Treatment of patients with penile cancer covers two aspects: Primary lesion and lymph node management. Treatment of primary lesion requires surgery, with organ-sparing procedures now a standard of care whenever feasible and oncologically safe [24,25]. There are also alternatives to surgery, including topical treatments or laser ablation in low-stage and low-grade cases or radiation therapy in cases with small lesions [26,27,28,29]. Lymph node status and management have the strongest impact on patients’ survival [22]. In patients with clinically enlarged inguinal +/− pelvic lymph nodes, lymph node dissection (LND) is clearly indicated. In patients with clinically negative lymph nodes, surveillance can be considered in low-risk cases while invasive nodal staging with limited LND or sentinel node biopsy should be performed in the remaining cases [22]. In advanced and metastatic cases, systemic cisplatin-based chemotherapy is the treatment of choice [30].
For prognosis, stage of the disease, tumor size, and ethnicity were recently reported to be the most significant factors [31]. In general, 5-year overall survival in penile cancer patients is 61% [32]. However, the cancer-specific death rate does not exceed 19%, even in patients aged <40 years [33].
2.3. Testicular Cancer
Testicular cancer (TC) is a relatively rare tumor, representing approximately 1% of male neoplasms in the general population [34]. However, in a subset of young men, the incidence of testicular cancers is increasing, with 3 to 10 new cases diagnosed per 100,000 males/per year in industrialized countries [34]. The vast majority (90%–95%) of TCs are classified as testicular germ cell tumors (TGCTs), which are the most common malignancies diagnosed in males aged 15 to 44 years [35,36]. TGCTs develop from cells of germ cell neoplasia in situ (GCNIS), which originate from gonocytes that fail to undergo physiological spermatogenic differentiation [35]. They are commonly further divided into seminomas and non-seminoma tumors based on their histology and radiosensitivity [5]. An isochromosome of the short arm of chromosome 12 (12p) is a specific genetic marker of all GCTs [37]. Among numerous mutations described in testicular GCTs, the TP53 mutation is the most common [38].
The treatment of TC has been called one of the top five advances in 50 years of modern oncology [39]. Treatment usually starts with surgery, namely radical orchiectomy. In very selected cases, testis sparing surgery is an option to preserve hormonal and reproductive function of the gonad [40]. Excellent cure rates result mainly from the efficacy of systemic chemotherapy, usually based on cisplatin [41]. Testicular tumors are chemosensitive while seminomas are also radiosensitive. The vast majority of orchiectomized patients are candidates for adjuvant chemotherapy while final qualification is based on stage of the disease and estimated risk of relapse [42,43,44]. In clinical stage II seminomas, radiation therapy is an alternative to chemotherapy. Finally, retroperitoneal lymph node dissection can be an option for patients relapsing after chemotherapy or with residual retroperitoneal disease after chemotherapy or in the case of contraindications to chemotherapy [40].
Prognosis depends mainly on the stage of the disease, including the presence and location of metastases and the serum concentration of biomarkers after orchiectomy (alpha-fetoprotein (AFP), human chorionic gonadotropin (hCG), lactate dehydrogenase (LDH)). Depending on these factors, the 5-year overall survival in patients with metastatic disease ranges from 92% in the good prognosis group to 48% in the poor prognosis group of non-seminoma patients [44]. At the same time, the vast majority of testicular cancer cases are non-metastatic patients with a very good prognosis [45].
2.4. Bladder Cancer
Bladder cancer (BC) is the most common malignancy within the urinary tract [21]. The annual BC incidence reaches nearly 10 cases per 100,000 persons in developed regions, with 430,000 diagnosed cases and nearly 170,000 deaths annually worldwide [46]. The vast majority (up to 75%) of these tumors are urothelial carcinomas, arising from urothelium in the process of multistep heterogeneous mutations [6,47,48,49]. From a biological and clinical standpoint, BC is classified into non-muscle invasive (NMIBC), representing 70% to 80% of BC cases, and muscle invasive bladder cancer (MIBC) [50]. These two entities differ in terms of incidence, gene mutations, morphology, and aggressiveness [51,52,53,54]. Cases of NMIBC are further divided into three risk groups (low, intermediate, high) depending on the risk of recurrence and progression after resection [55,56]. While NMIBCs can be radically treated by endoscopic resection with or without adjuvant intravesical chemo- or immunotherapy, MIBCs require major surgery, namely radical cystectomy (removal of the urinary bladder, prostate, seminal vesicles, and pelvic lymph nodes in men; removal of the urinary bladder, uterus, adnexa, anterior wall of the vagina, and pelvic lymph nodes in women) with perioperative chemotherapy [50].
Up to 15% of bladder cancer patients are diagnosed upfront with metastatic disease [57,58]. In these cases, surgery is no longer a standard option and systemic, preferentially cisplatin-based, chemotherapy is the treatment of choice [59]. As neoantigen load and T cell infiltration in bladder cancers is high [60], new systemic treatment options with check-point inhibitors were shown to be effective [61]. Until now, the European Medical Agency has registered pembrolizumab, atezolizumab, and nivolumab for treatment of patients with advanced bladder cancer. Many further phase II and phase III trials are ongoing.
Prognosis in bladder cancer depends mainly on the stage of the disease. The survival rate in NMIBC is high while the 5-year risk of recurrence and progression after endoscopic resection reaches 31% to 78% and 1% to 45%, respectively [55]. Bladder sparing is possible and safe in the majority of these patients; however, they all require a close follow-up with repeated cystoscopies to detect disease relapse early [50]. On the contrary, prognosis in MIBC is poor. The 5-year recurrence rate after radical cystectomy in MIBC patients is 32% to 68% [62,63,64]. Systemic first-line chemotherapy in advanced and metastatic MIBCs offers a response rate of 46% to 49% and median overall survival of 14 to 15 months [65]. Unfortunately, over 50% of MIBC patients are cisplatin ineligible due to reduced renal function, performance status, heart failure, or other comorbidities [66]. Patients unfit for cisplatin or progressing after cisplatin therapy can be candidates for second-line treatment modalities, including carboplatin-based regimens, check-point inhibitors, monotherapy, or best supportive care.
2.5. Prostate Cancer
Prostate cancer (PCa) is the most common malignancy among men in America and Europe [21]. It is an androgen-dependent cancer, which develops typically in the peripheral zone of the prostate [67]. The introduction of PSA testing has dramatically increased diagnosis, with a limited positive impact on patients’ survival in the long run [68,69,70]. Prostate cancer is a disease of a different genetic mutation spectrum, different histological tumor grades, different progression potential, and different treatment options.
Standard curative treatment of prostate cancer patients involves surgery (radical prostatectomy) or radiation therapy (external beam radiation or/and brachytherapy) [71]. However, in many low- volume and low-grade cancers, potential progression is so slow that observation can be a safe alternative [72,73]. This is called active surveillance if radical treatment is considered or watchful waiting if only palliative therapy is an option [43,74,75]. In advanced cases, when radical treatment cannot be implemented, androgen deprivation therapy (ADT) is the treatment of choice. It involves bilateral orchiectomy or pharmacological castration with androgen receptor antagonists, GnRH agonists, or GnRH antagonists. In upfront metastatic patients, ADT should be combined with docetaxel chemotherapy or abiraterone [76,77,78,79,80]. At later disease stages, when prostate cancer is no longer castration sensitive (castration-resistant prostate cancer), there are several second-line treatment options, including chemotherapy (docetaxel and cabazitaxel), new antiandrogens (enzalutamide, apalutamide), steroidogenesis inhibitor (abiraterone), immunotherapy (sipuleucel-T), or radioactive compounds (radium 223) [81].
Prognosis in prostate cancer patients depends mainly on the stage of the disease and tumor grade. As these patients are usually elderly men with comorbidities, many of them die with prostate cancer but not from the cancer. Outcomes of local treatment in organ-confined disease by surgery and radiation therapy are described by a 5-year biochemical recurrence rate of 27% to 53% [81]. However, subsequent salvage therapy leads to undetectable PSA levels in the majority of patients. Moreover, many of the men with biochemical recurrence will never experience clinical progression [81]. In patients with advanced cancer, median overall survival depends on the response to ADT as measured by a PSA level decrease. In patients with a good response (PSA level after 7 months of ADT <0.2 ng/mL), it is 75 months while in patients with a poor response (PSA > 4.0 ng/mL), it is 13 months [82].
3. The Basics of TGF-β Signaling
The family of transforming factor β (TGF-β) proteins is a large group of 33 structurally related growth factors, including notably TGF-β1, TGF-β2, TGF-β3, activins/inhibins, bone morphogenetic proteins (BMPs), and growth differentiation factors (GDFs). TGF-β1 is the most studied isoform expressed in mammalian tissues. TGF-β1 is expressed as an inactive precursor protein that undergoes a series of posttranslational modifications (Figure 1).
The first step involves dimerization of two precursors and subsequent cleavage by furin endopeptidase, which produces two smaller proteins: The latency-associated peptide (LAP) and the mature TGF-β1. The two proteins form a non-covalently bound small latent complex in which TGF-β1 is enclosed and protected by the surrounding LAP. Finally, the small latent complex is covalently bound by latent TGF-β binding protein (LTBP) to form a large latent complex, which is secreted by the cell. Both LAP and the formation of latent complexes are required for proper folding, maturation, and secretion of TGF-β1. They also ensure TGF-β stability and prevent its inappropriate activation. TGF-β cytokine is activated by several mechanisms, including those triggered by plasmin, thrombospondin-1, integrins, matrix metalloproteinases, calpains, retinoic acids, fibroblast growth factor-2, and reactive oxygen species (ROS) [83,84].
TGF-β initiates intracellular signaling by binding to its cell surface receptor, TGFBR2, which forms a heterotetrameric complex with TGFBR1. This results in serine/threonine phosphorylation of the cytoplasmic GS domain of TGFBR1 and initiation of signaling cascades. In the canonical mechanism of TGF-β action, activation of TGFBR1 leads to phosphorylation of SMAD2/3 proteins that interact with SMAD4 and, following translocation to the nucleus, regulate transcription of TGF-β-target genes. Recruitment of SMAD2/3 to the TGF-β receptors is mediated by SARA protein. TGF-β signaling is negatively regulated by SMAD6 and SMAD7, which interfere with the activation of receptors, formation of the SMAD2/3-SMAD4 complex, and inhibit transcription of TGF-β-target genes by binding to the DNA sequence in their promoter regions [85].
In the non-canonical pathway, TGF-β activates ubiquitin ligase TRAF6 (tumor necrosis factor receptor-associated factor 6) that ubiquitinates TGFBR1, which in turn results in selective proteolytic cleavage by TACE and PS1 (presenilin), leading to liberation of the intracellular domain of TGFBR1. The relieved domain is translocated to the nucleus where it interacts with various molecules and activates multiple signaling pathways, including these involving Ras, RHOA (Ras homolog family member A), PI3K (phosphoinositide 3-kinase), PP2A (protein phosphatase 2A), MAPK (mitogen-activated protein kinase), TAKI1 (TGF-β-activated kinase), ERK1/2 (extracellular signal-regulated kinase ½), and JNK (c-Jun N-terminal kinase) [86,87]. The described mechanisms of TGF-β action are also regulated by multiple accessory receptor proteins, transcriptions factors, and transcriptional co-factors in a cell type- or context-specific manner. TGF-β-induced signaling contributes to the regulation of cell growth and differentiation, apoptosis, cell motility, production of the extracellular matrix, angiogenesis, and immunity.
TGF-β plays a dual role in carcinogenesis. In the early stages of tumor development, it inhibits cellular transformation and prevents cancer progression while in later stages, TGF-β plays the oncogenic role and promotes tumor progression by induction of epithelial–mesenchymal transition (EMT), stimulation of angiogenesis, and immunosuppression. This conversion in cancer-related TGF-β functions is known as the “TGF-β paradox” and has been described in detail in previous publications [1,86,87,88,89,90,91,92]. These dichotomous TGF-β actions occur universally in tumors, including breast [93], liver and gastrointestinal [94], colorectal [95], pancreatic [96], or lung cancers [97]. It appears that as tumors progress, cancer cells tend to acquire resistance to TGF-β growth inhibitory effects due to mutations and/or functional inactivation of TGF-β pathway elements. The most commonly mutated genes of the TGF-β signaling pathway include TGFBR1, TGFBR2, SMAD4, and SMAD2. The significance of the TGF-β signaling pathway in cancer was recently confirmed by a report of TCGA (The Cancer Genome Atlas) consortium. The analysis of tissue samples from 33 cancer types and > 9000 patients revealed that the genes encoding the TGF-β pathway elements were impaired in nearly 40% of the analyzed cancer cases [98]. Remarkably, in a subset of tumors that included genitourinary cancers (bladder urothelial carcinoma, clear cell renal cell carcinoma, papillary renal cell carcinoma, prostate adenocarcinoma, and testicular germ cell tumor), the activity of the TGF-β signaling pathway correlated with epithelial–mesenchymal transition. The same study also showed that miRNAs contribute to the transcriptional activity of the TGF-β pathway, indicating functional links between short-non-coding RNAs and transforming growth factor effects in cancer cells.
4. Alterations in TGF-β Signaling in Genitourinary Cancers
4.1. Renal Cancer
The proteins of the TGF-β family play an important role during kidney development and functioning. The gene encoding TGF-β is expressed in mammalian metanephros during tubular development [99]. Activin A and TGFB2 are involved in the formation of the ureteric bud [100,101] while TGFB2 knock-out results in kidney agenesis in mice [102].
TGF-β is a critical mediator of renal fibrosis by promoting intense production and accumulation of components of the extracellular matrix (ECM), resulting in renal damage [103]. Renal fibrosis contributes to development of chronic kidney diseases (CKDs), which in turn may progress to end-stage renal diseases (ESRDs). The role of TGF-β in renal fibrosis and other chronic kidney diseases was extensively reviewed [104,105,106,107]. Remarkably, despite the well-documented role of TGF-β1 in renal fibrosis, the role of TGF-β in renal cancer remains elusive due to the conflicting results of published studies.
RCC-derived cell lines and tumors consistently express and secrete increased amounts of TGF-β1 [108,109,110,111,112,113] (Table 1).
Increased TGF-β1 levels were also found in plasma and serum [114,115,116], as well as peripheral blood and tumor infiltrating lymphocytes (TILs) [117] of RCC patients. TGF-β1 expression directly correlates with tumor stage and grade, and is significantly increased in patients with metastatic RCC [116], indicating its importance in tumor progression [137]. Contradictory results were presented by Zheng et al., who demonstrated downregulation of TGF-β1 expression in metastatic renal cancer tissue compared to primary tumor samples, suggesting DNA hypermethylation as a possible cause of decreased expression. These results were recapitulated by a metastatic and primary renal cancer mice xenograft model. Pretreatment of cancer cells with 5-aza-2’-deoxycytidine (DNA methylation inhibitor) before inoculation in mice led to increased TGF-β1 expression, tumor size reduction, and an extended survival time of animals [138].
Sjolund et al. proved that elevated levels of TGFBR1 correlated with worse prognosis for ccRCC patients [139]. Similar results were obtained by Sitaram et al., who analyzed the expression of TGFBR1-FL (the full-length receptor) and TGFRB1-ICD (the intracellular domain of TGFRB1 receptor) in RCC [140]. Moreover, an analysis of 151 cases of urinary system cancers provided evidence that an intronic variant of TGFBR1, Int7G24A (rs334354), was associated with a higher risk of RCC development [141]. TGFBR2 expression was shown to decrease with cancer progression [142,143], which can partly explain the resistance of RCC cells to the TGF-β1 growth-inhibiting effect. However, in earlier research, conflicting findings showed that downregulation of TGFBR2 expression is associated with a better prognosis for ccRCC patients [144]. Similar results were obtained by Kominsky et al., who observed that reducing TGFBR2 expression and blocking the TGF-β pathway inhibited ccRCC metastasis to bone [145]. Furthermore, Ananth et al. demonstrated inactivation of TGF-β signaling in a 786-0 ccRCC-derived cell line due to the loss of the TGRBR2 receptor, whereas Sjolund et al. provided evidence that this pathway is functional in the same cell line [139,146]. Another study demonstrated that TGFBR3 has anticancer properties in ccRCC independent of TGF-β and its canonical mechanism of action, and that loss of this receptor may occur early during RCC carcinogenesis [143]. In accordance, downregulation of TGFBR3 expression in primary ccRCC was associated with poor survival of patients [139]. Recently, downregulation of TGFBR3 expression in advanced ccRCC tumor samples was confirmed by an independent study in which the loss of this receptor led to the stimulation of cell migration and formation of lung metastasis [147].
The expression and role of other components of the TGF-β signaling pathway in RCC were also investigated. Immunohistochemical analysis of 637 ccRCC tissue samples revealed a negative correlation between expression of SMAD3 and SMAD4, and the age of patients, nuclear grade, tumor size, as well as pTNM stage. Moreover, the expressions of these proteins were independent indicators of patients’ progression-free survival [148]. Increased expression of pSMAD2/3 (phosphorylated SMAD2 and SMAD3) and PAI-1 (plasminogen activator, TGF-β1 target gene) was observed in more differentiated ccRCC tumor samples and correlated with a larger tumor size and lower patient survival rate. Furthermore, stimulation of ccRCC cell lines, 786-0 and A498, with the exogenous TGF-β1 triggered activation of the TGFBR1 and TGF-β/SMAD/PAI-1 pathway and stimulated the invasive potential of ccRCC cells [140].
VHL inactivation is the key molecular aberration associated with ccRCC and several studies demonstrated regulation of TGF-β signaling by VHL status. VHL reduces TGF-β stability, resulting in suppression of its expression in the 786-0 ccRCC cell line [146]. In agreement with these findings, cells devoid of active VHL secrete more TGF-β1 than cells with functional VHL [149].
Pro-cancerous TGF-β effects were confirmed by antibody-mediated neutralization of TGF-β1, which led to tumor regression and inhibition of angiogenesis in a xenograft athymic mouse model [146]. Several studies aimed to investigate the mechanisms of TGF-β actions in RCC. TGF-β1 secretion was observed in a panel of ccRCC-derived cell lines (SKRC-7, SHRC-10, SKRC-17, and SKRC-52) while the functionality of the TGF-β1 pathway was confirmed through an observation of increased SMAD reporter activity in ccRCC cells treated with TGF-β1. It was also found that TGFBR1 inhibition using SB431542 compound stimulated the invasiveness of ccRCC cells. Microarray analysis revealed a signature of 157 genes regulated by TGF-β1 and correlating with poor prognosis for ccRCC patients. Many of these genes were shared by both the VHL/HIF and TGF-β1/SMAD signaling pathways. This overlap between the two pathways was further confirmed by VHL reconstitution in ccRCC cells, which attenuated both TGF-β1 secretion, as well as their responsiveness to TGF-β1-induced stimulation [150]. The links between VHL and TGF-β signaling were further confirmed be a study that analyzed angiogenic factors in formalin-fixed, paraffin-embedded ccRCC tissue samples. There was a significant correlation between expressions of VEGF, TGF-β1, and the clinical stage and nuclear grade of ccRCC tumors. Furthermore, TGF-β1 protein expression negatively correlated with microvessel density (MVD). Since high MVD values correlated with longer survival for patients with ccRCC, it may indicate that TGF-β1 overexpression leads to decreased MVD, which, in turn, might affects patients’ outcomes [151].
The role of TGF-β in the regulation of adhesion, EMT, migration, and invasion of ccRCC cells is well documented. Our previous studies showed that TGF-β1 stimulated the expression of genes involved in adhesion and extracellular matrix remodeling, including TGFBI, COL1A1, COL5A1, COL8A1, FN1, ITGA5, ITGAM, and TIMP1 [110]. TGF-β1 stimulation of RCC cell lines leads to decrease of E-cadherin and increase of vimentin and N-cadherin, synonymous with EMT activation, which is consistent with reports in other cancers [152,153,154]. TGF-β1 also induced the expression of αν integrins and mediators of the signaling pathway, including ILK (Integrin-linked kinase) and PINCH-1, known to contribute to EMT through binding and stimulation of latent TGF-β1 [155]. This in turn results in the activation of the Src/FAK complex, and EMT progression via inhibition of cadherin-dependent cell–cell interactions. Treatment of RCC cells with the synthetic integrin αVβ3 ligand RGD stimulated enhanced E-cadherin depletion, indicating synergistic cooperation between TGF-β1 and integrins. The same study revealed that these mutual TGF-β1/RGD actions are regulated by Snail1 transcription factor. The silencing of FAK and PINCH1 inhibited cooperation between TGF-β1 and RGD [156]. TGF-β1-induced migration and invasion of ccRCC cells is mediated by fascin1 [157]. Similar results were provided for bladder cancer cells [158], indicating that this could be a universal mechanism of TGF-β1-induced tumor progression.
TGF-β contributes to the formation of RCC metastasis to bone, occurring in about 40% of renal cell carcinoma patients [159]. TGFBR1 and TGFBR2 are expressed in human RCC bone metastases (RBM) while TGF-β1 is expressed and secreted by all cell lines derived from RBM tissues. Inhibition of TGF-β1 signaling attenuates tumor growth and osteolysis in mice with ccRCC xenografts. Unexpectedly, treatment of RBM cells with TGF-β1 does not influence or even reduces proliferation [145]. This observation is consistent with previous results obtained in primary RCC [109,111,112,118] and suggests that TGF-β1 stimulation of RBM growth might be indirect, e.g., via the paracrine interplay between tumor cells and the bone microenvironment [145]. In accordance with this suggestion, analysis of RBM tissue samples revealed TGF-β-1-induced overexpression of MMP13 (Matrix metallopeptidase 13), a protein involved in degradation of the extracellular matrix (ECM) [160].
4.2. Penile Cancer
The molecular basis of penile cancer is poorly understood, mainly due to limited studies of these rare tumors. However, recently published molecular characterization of several cell lines derived from penile squamous cell carcinoma revealed frequent disturbances of the TGF-β signaling pathway [161]. These alterations included multiple copy number losses or gains (e.g., of TGFB1, TGFB3, TGFBR2, BMPR1B, ACVR2B, SMAD4, SMAD2, SMAD1, SMAD7, PITX2) as well as mutations of TGFRB2 (R522X, S320X, S320X). The functional significance of these alterations, as well as their influence on penile cancer progression, await future analyses.
4.3. Testicular Cancer
TGF-β signaling is crucial for both the physiology and pathology of testis, contributing to its development and functioning [162,163]. Knock-out of genes encoding TGF-β1 and TGF-β2 in the mouse decreases the number of testis germ cells and seminiferous cords, respectively [164]. The development of testis and germline cells is regulated by activins and inhibins [165]. Analysis of mouse models revealed that expressions of nodal, activin, and TGF-β are regulated in a spatial and temporal manner during testis development, triggering mitotic arrest and expression of male fate markers [166]. In fetal testis, TGF-β attenuates proliferation [167], indicating that aberrances of the TGF-β pathway may lead to tumor development. Indeed, α-inhibin acts as a tumor suppressor and its knock-out results in the development of mixed or incompletely differentiated gonadal tumors, including intratubular, focally invasive gonadal stromal tumors in testis [168].
Regarding human studies, reports are limited mainly to simple analyses of the presence of components of the TGF-β signaling pathway in testicular tumors and cell lines, without comparisons with non-tumoral control tissues [169,170,171]. However, according to one study, expression of TGF-β1 and TGF-β2, as well as TGFBR1 and TGFBR2, was increased in testicular tumors compared with peritumoral non-neoplastic testis tissues [119]. TGF-β1, together with EGF and FGF4, synergistically contribute to differentiation of seminoma cells into the mixed non-seminoma-like cell type [172]. Since seminomas naturally progress into non-seminomas, this may suggest that TGF-β stimulates the progression of testicular tumors [173]. Furthermore TGF-β-induced signaling stimulates proliferation of TCam-2 seminoma cells [170].
The role of TGF-β1 in testis cancer is confirmed by analyses of its genetic variants. In a study involving 577 tumor cases and > 700 controls, the TGFB1 Ex5-73C > T variant was positively associated with TGCT risk while Ex1-282G and 509C > T variants were linked with increased risks of seminoma and non-seminoma, respectively (Purdue at al., 2007). In the TGF-β1 protein sequence, the Ex5-73C > T (rs1800472) variant results in a substitution of threonine with isoleucine (T263I) while Ex1-282G > C (rs1800471) changes the arginine to proline (P25R). The functional consequences of these alterations are unknown, although it was suggested they could influence TGF-β expression, with Ex5-73T causing a decrease, and Ex1-282G > C resulting in an increase of TGF-β protein levels. The -509C > T (rs1800469) variant does not change the TGF-β1 amino acid sequence; however, it was linked with elevated plasma concentrations of TGF-β1 [174]. The exact mechanism by which altered TGF-β functioning could affect the development of TGCT is currently unknown.
The alterations of the TGF-β pathway found in testicular cancers are also detected in other members of the TGF-β signaling pathway. A single nucleotide (thymine) insertion in SMAD4 was found in 2 out of 20 analyzed seminoma germ cell tumors [175]. This insertion resulted in a frameshift mutation that created a premature STOP codon, resulting in a loss of SMAD4 protein. The authors suggested that this mutation could be the cause of the unresponsiveness of seminoma cells to TGF-β signaling, resulting in a loss of its antiproliferative action.
A recent study showed that activin A stimulates expression of MMP2 and MMP9 in a seminoma-derived cell line [176]. MMP2 and MMP9 metalloproteinases are well-known stimulators of tumor invasion and progression, which suggests that activin/TGF-β signaling may contribute to TGCT development from GCNIS.
4.4. Bladder Cancer
The associations between bladder cancer’s clinical course and genetic variants of TGF-β1 and its receptors has been confirmed by several studies. TGFB1 c.29C > T substitution (rs1800470) correlates with an increased risk of bladder cancer [177]. This SNP (Single Nucleotide Polymorphism) is located in a region encoding the hydrophobic core of the TGF-β1 signal peptide and results in a substitution of proline with leucine in the 10th position of the amino acid sequence. The c.+29C results in increased TGF-β1 secretion in vitro and in vivo compared with c.+29T [178]. Chen et al. found Int7G24A (rs334354) intronic variant of the TGFBR1 gene frequently associates with transitional cell carcinoma (TCC) of the bladder as well as renal cell carcinoma [141]. Importantly, of the 65 analyzed cases of TCC and 86 of RCCs, both heterozygous and homozygous SNP carriers had a statistically significantly increased risk of developing tumors. The same study also reported one somatic mutation, resulting in a change of serine to phenylalanine at codon 57 of TGFBR1 [141]. Interestingly, the Int7G24A SNP is also associated with increased risks of osteosarcoma [179], colorectal, and breast cancer [180,181], suggestive of its general involvement in cancer predisposition.
The role of TGF-β signaling in BC development and progression has been extensively studied, bringing conflicting results. It is generally accepted that TGF-β1 expression is increased in bladder cancer compared with normal bladder epithelium and correlates with tumor progression, although a few studies reported the opposite (Table 1).
Highly inconsistent data were provided regarding the expressions of TGF-β receptors in bladder cancer. TGFBR1 expression was increased in invasive TCC compared with low-grade and superficial tumors, as well as tumors associated with Schistosoma infection [128]. However, it was also shown that a loss of TGFBR1 expression correlates with poor prognoses of bladder cancer patients [182] while loss of TGFBR1 and TGFBR2 correlated with increased bladder tumor grades [183], which agrees with the decreased TGFBR2 expression in invasive tumors compared with superficial transitional cell carcinomas [184]. The expression of TGFBR3 was generally reduced in bladder cancer tumors [185]; however, in muscle-invasive bladder cancer, TGFBR3 protein expression was increased while in non-muscle-invasive tumors, it was decreased when compared with corresponding paracarcinoma tissues.
The functional analyses of TGF-β1’s role in the development and progression of bladder cancer also brought inconsistent results. For instance, according to one report, TGF-β1 treatment of T24 bladder cancer cells inhibited proliferation and viability while inducing apoptosis, which was linked with activation of the p38 MAPK-JNK-Caspase9/8/3 pathway [186]. In contrast, another study reported that TGF-β1 stimulated proliferation, migration, and invasion of the T24 cell line, in a mechanism mediated by fascin1, a protein involved in the regulation of cell motility [158]. It is doubtful if the different TGF-β1 concentrations used in both studies could influence these opposite results ([158] used higher TGF-β1 concentrations (10 ng/mL) than those utilized by [186] (0.1–5 ng/mL)). Moreover, the latter study observed a clear dose-dependent inhibitory TGF-β1 effect [186].
TGF-β1 was reported as a powerful inhibitor of the growth of rat bladder cancer cells, with a prominent exception of several more tumorigenic and invasive cell lines that exerted TGF-β1 insensitivity [187,188]. This is in agreement with an early study in which TGF-β abolished mitogenic effects of aFGF (acidic fibroblast growth factor) in rat bladder cancer cells in vitro. Specifically, TGF-β1 inhibited aFGF-induced DNA synthesis [189]. Further analyses revealed that the loss of TGF-β1 responsiveness was linked with TGFBR1 deficiency. Restoration of TGFBR1 expression rescued TGF-β1’s ability to inhibit cell growth in vitro and attenuated the growth of bladder cancer tumors in a murine xenograft model [190,191]. In accordance with these findings, in a model of transgenic mice with COX-2 (cyclooxygenase-2)-induced transitional hyperplasia of the bladder progressing to invasive carcinoma, expressions of Tgfbi, Tgfb2, and Tgfb3 were significantly decreased compared with wild-type mice [192]. This is also in agreement with studies in bladder cancer patients. There, a loss of TGF-β1 receptors, TGFBR1 and TGFBR2, correlated with the invasive tumor stage, high grade, and lymphovascular invasion while enhanced TGF-β1 levels in bladder cancer tissues and plasma of patients were associated with tumor invasiveness [121,122].
On the other hand, multiple studies indicated that TGF-β1 and its receptors stimulate the progression of bladder cancer cells. Loss of the IQGAP1 tumor suppressor leads to increased expression of TGFBR2 and activated the TGF-β1 signaling pathway, thereby stimulating growth of human bladder cancer cells [193]. TGF-β1-stimulated migration and invasion of bladder cancer cells arise mediated by Src and FAK kinase [194] as well as transgelin (TAGLN), an actin-binding protein that stimulates colony formation, migration, and invasion, as well as epithelial–mesenchymal transition [195]. TGF-β1 can also stimulate bladder cancer progression by inducing mTORC2 signaling. Specifically, TGF-β1 activates SMAD2, which triggers mTORC2 activation, resulting in AKT phosphorylation and stimulation of the motility of bladder cancer cells [196]. TGF-β1 also stimulates bladder cancer progression by triggering Shh (sonic hedgehog signaling molecule)pathway activation. Blockade of Shh activity inhibits TGF-β1-induced migration, invasion, and clonogenic growth of bladder cancer cells. Pretreatment of bladder cancer cell lines with TGF-β1 accelerated growth of BC xenografts in mice [197].
One of the key processes linked to metastatic progression is epithelial–mesenchymal transition (EMT), in which cells lose their epithelial features and acquire a mesenchymal phenotype, detach from the basement membrane, and start to migrate and invade local and distant tissues. There is a general agreement that TGF-β induces EMT in T24 bladder carcinoma cells [198,199,200,201]. These effects are mediated by TGFBR1, as silencing of this receptor attenuates the migration and invasiveness of T24 cells, with concomitant downregulation of pro-invasive MMP9, as well as integrins α2, α3, and β1 [198]. TGF-β1-induced sumoylation of TGFBR1 is a prerequisite for induction of the epithelial–mesenchymal transition in bladder cancer cells [202]. This process is reversed by SENP2 (SUMO-specific protease-2), which removes SUMO from TGFBR1, thereby inhibiting TGF-β1-induced EMT, resulting in inhibition of the invasion of bladder cancer cells in vitro and tumor metastasis in vivo. The expression of SENP2 in bladder cancer patients is reduced, leading to attenuation of its suppressive effects on tumor progression. As a result, bladder cancer patients with reduced SENP2 expression have a greater chance for the development of more aggressive tumors and poor outcomes [202]. TGF-β1 actions in bladder cancer are also mediated by ARMc8 (Armadillo repeat-containing protein 8). Specifically, ARMc8 mediates TGF-β1-induced migration and invasion, as well as epithelial–mesenchymal transition [203]. Pro-cancerous TGF-β effects are counteracted by protein phosphatase PPM1A, which dephosphorylates SMAD2/3 TGF-β effectors. Loss of PPM1A promotes TGF-β1-induced EMT in vitro and correlates with bladder cancer progression and poor prognosis for patients [199]. TGF-β1 signaling is crucial in the context of the tumor microenvironment. EMT, migration, and invasion of bladder cancer cells are induced by TGF-β1 secreted by cancer-associated fibroblasts (CAFs), with ZEB2NAT, long non-coding RNA mediating TGF-β1-induced protumorous effects [201]. TGF-β1-induced EMT can be also mediated by other lncRNAs, such as Malat1 [204]. Silencing of Malat1 attenuates TGF-β1-induced migration and invasion of bladder cancer cells and inhibits progression of tumor xenografts in mice [204].
Interestingly, it was suggested that TGF-β1 could promote EMT in bladder cancer progression by altering the expression of genes involved in the synthesis of glycans that mediate cell-to-cell adhesion [205]. Specifically, treatment of HCV29 cells with TGF-β1 reduced the expression of α-mannosidase 2 and α-L-fucosidase, resulting in decreased levels of bi-, tri-, and tetra-antennary complex N-glycans; increased expression of hybrid-type N-glycans; and increased levels of fucosylated N-glycans [205].
The involvement of TGF-β1-signaling in the pathogenesis of bladder cancer is also confirmed by altered expression of its downstream effectors. Accordingly, the expression of TGFBI (TGF-β1-induced) is enhanced in muscle-invasive bladder cancers (MIBCs) compared to non-muscle invasive tumors (NMBCs) and correlates with poor survival of patients. TGFBI is secreted by bladder cancer cells to induce proliferation, migration, and invasion, as well as EMT in an autocrine manner. Moreover, TGFBI overexpression stimulates the growth of bladder cancer xenografts in mice [206]. Interestingly, enhanced TGF-β1 expression was suggested as a mechanism mediating cancerous effects of chronic long-term low-dose ionizing radiation exposure [207]. TGF-β1 expression was elevated in BC patients of the Chernobyl region compared with patients not exposed to radiation. The authors suggested that TGF-β1 could act as a sensor of excess ROS production resulting from radiation exposure [207].
TGF-β1 also acts as a mediator of other EMT-inducing proteins. For instance, EIF5A2 stimulates TGF-β1 expression via STAT3 to induce EMT and stimulate migration and invasion [208] while cancer-associated fibroblasts (CAFs) secrete Kindlin-2, which induces EMT in a TGF-β-dependent manner [209]. The TGF-β1/SMAD2 pathway is also utilized by Golgi membrane protein 73 (GP73) to induce EMT and promote invasion and metastasis of bladder cancer [210]. TGF-β1 mediates signaling initiated by collagens. Huang et al. showed that expression of COL6A3 is increased in bladder tumors, stimulates proliferation and angiopoiesis as well as epithelial–mesenchymal transition, with possible involvement of TGF-β. COL6A3 silencing resulted in reduced expression of TGF-β as well as phosphorylation of SMAD2 and SMAD3 [211]. Trim59, the expression of which is upregulated in bladder tumors, promotes proliferation, EMT, migration, and invasion of bladder cancer cells. Silencing of Trim59 attenuates migration and invasion of bladder cancer cells while the presence of TGF-β1 relieves the suppressive effect of Trim59 knock-out [212].
There is strong evidence that TGF-β signaling contributes to the tumor microenvironment and affects anti-tumor immunity. As revealed by in vivo studies in rabbits, the growth and progression of bladder cancer is promoted by mesenchymal stem/stromal cells (MSCs) that stimulate secretion of growth factors and ECM-remodeling proteins, including TGF-β1 [213]. The tumor-growth stimulatory effects of MSC are mediated by TGF-β receptor and SMAD2 protein [214]. This particular study, however, has several limitations, including the lack of a direct evaluation of TGF-β receptor and SMAD2 expression following silencing [214]. It is thus difficult to conclude to what extent the silencing of both genes contributed to the attenuation of MSC tumor growth stimulatory effects. Intensified tumor-derived TGF-β secretion facilitates evasion of immune surveillance. Specifically, in patients with superficial transitional cell carcinoma, increased plasma TGF-β1 attenuates cytotoxicity of NK (Natural Killer) cells [215]. High expressions of TGF-β1 and TGFBR2 attenuate T cell penetration into the center of metastatic urothelial carcinoma tumors [216], bringing the rationale for combination therapies involving immune checkpoint inhibitors and TGF-β1 blockers (further discussed in Chapter 6). Furthermore, it was found that TGF-β mediates S1P1-induced tumor-associated expansion of Treg cells in bladder cancer patients. S1P1 is a receptor of sphingosine-1-phosphate, a lipid involved in the regulation of the tumor immune microenvironment. Specifically, S1P1 triggers phosphorylation of SMAD2/3, which activates the TGF-β signaling pathway and production of TGF-β by bladder cancer cells. This in turn results in enhanced production of Tregs from naive T cells [217]. Interestingly, most (up to 86%) of TGF-β1 secreted by bladder cancer cells is encapsulated in exosomes that target healthy fibroblasts and stimulate their differentiation into cancer-associated fibroblasts (CAFs) by triggering SMAD2 phosphorylation [218]. Interestingly, the expression of TGF-β1 and TGFBR1 in peripheral blood mononuclear cells (PBMNCs) of bladder cancer patients is increased compared with healthy individuals, which coincides with the role of TGF-β1 signaling in bladder cancer immunity. According to the same study, however, the levels of TGFBR1 in urine sediments of bladder cancer patients were decreased, with a concomitant increase of TGF-β1, which indicates independent mechanisms of TGF-β1 signaling in cancer and immune cells [219].
The importance of TGF-β receptors in bladder cancer is underscored by studies that demonstrated that Tgfbr2 knock-out or Tgfbr1 inhibition attenuates growth and progression of chemically-induced bladder tumors in mice [220] while TGFBR3 knock-down in a human T24 bladder cancer cell line results in reduced viability, colony formation, migration, and invasion [185]. Deletion of Tgfbr2 decreased the population of cancer stem cells, attenuated proliferation, and induced apoptosis of bladder cancer cells in vivo. Furthermore, conditional knock-out of Tgfbr2 decreased EMT as indicated by lowered expression of mesenchymal markers, including vimentin, Slug, Snai1, Twist, and Zeb1, with concomitant upregulation of epithelial E-cadherin [220]. Notably, TGFBR2 mutations are frequently found in bladder cancer patients. Glu269 to Lys mutation (G→A) facilitates TGF-β1-induced invasion of bladder cancer cells [221].
The expressions of other members of the TGF-β protein family associate with the tumorigenicity of cell lines derived from bladder cancer xenografts in mice [222]. Accordingly, the expression of BMP2 was increased in a tumorigenic and invasive cell line compared with nontumorigenic cells while inhibin-βB expression was enhanced in invasive bladder cancer cell lines compared with nontumorigenic and tumorigenic cells. The promoter of BAMBI (BMP and activin membrane-bound inhibitor) is hypermethylated in a subset of high-grade bladder cancers, leading to its low expression [222,223]. BAMBI acts as a TGFR1/BMPR1-related pseudoreceptor and interferes with TGF-β/BMP signaling by hindering the formation of functional receptor complexes. In accordance with this function, forced BAMBI expression attenuated the migration of cancer cells induced by TGF-β1 or BMP2 [223].
BMP9 is overexpressed in bladder cancer and stimulates proliferation and migration of cancer cells by increasing the expression of lncRNA UCA1, which in turn leads to activation of AKT. BMP9 also stimulates the growth of bladder cancer tumors in vivo [224]. Another member of bone morphogenetic proteins (BMPs), the growth differentiation factor-9 (GDF-9), was suggested as a possible tumor suppressor in bladder cancer. Specifically, ectopic expression of GDF-9 in bladder cancer cell lines attenuated cell growth, and reduced migration and adhesion. The study also suggested that GDF-9 expression, while clearly present in normal bladder tissue, was absent from bladder cancer tissue samples [225]. A similar tumor suppressive function was also found for GDF-15, which inhibited the proliferation and invasion of bladder cancer cells in vitro, and attenuated the growth of bladder cancer xenografts in vivo. Remarkably, GDF-15 knock-out resulted in reverse effects. These effects were mediated by GDF-15-induced blocking of EMT and polarization of F-actin. The GDF-15 evaluation in bladder cancer cell lines suggested that its expression could be reduced in bladder tumors, possibly due to DNA hypermethylation as well p53 inactivation in bladder cancer cells [226].
4.5. Prostate Cancer
The role of TGF-β in prostate tumors is a broad topic that has been extensively earlier reviewed [227,228,229,230], thus here we only provide a short outline of the mechanism of TGF-β actions in prostatic cancer cells.
TGF-β signaling regulates proliferation, growth, differentiation, and apoptosis of prostatic stromal and epithelial cells [231,232]. As in the other genitourinary cancers, altered expression of TGF-β1 family proteins has been observed in prostate tumors. Increased TGF-β1 levels were found in tumor tissue samples [130,131,132,133,134,135], serum [136], and urine [133] of PCa patients. TGF-β1 overexpression correlates with the prostate cancer stage and grade, patients’ survival rate as well as the degree of angiogenesis induction and the presence of bone metastasis [134,233]. Downregulation or loss of TGF-β receptors was observed in about 30% of cases of prostate cancer compared with normal prostate tissues while expressions of TGFBR1 and TGFBR2 were lower in metastatic compared with primary tumors [234]. Upregulation of TGFBR2 enables pro-apoptotic activity of TGF-β1 and inhibits the growth of prostate cancer cells through a caspase-1-mediated mechanism, whereas downregulation of this receptor leads to stimulation of malignant transformation [235,236]. The resistance of prostate cancer cells to TGF-β-mediated growth inhibition is thus possibly a result of decreased expression of TGFBR2, which acts as a tumor suppressor gene [237,238]. Downregulation of TGFBR2 in PCa cells might result from hypoxic activation of DNA methyltransferases, which leads to the hypermethylation of the promoter region of TGFBR2 [239]. Contrarily, earlier research reported a lack of promoter methylation and mutations in the TGFBR2 gene [240]. TGFBR3 downregulation is the most common modification of the TGF-β signaling pathway in prostate cancer and contributes to stimulation of cancer cells’ motility and invasiveness in vitro as well as enhanced tumorigenicity in vivo [241]. In normal prostate epithelial cells, decreased TGFBR3 expression and/or activity results in morphological changes suggestive of inhibition of cell–cell contacts and stimulation of the cancer stem cell phenotype [242].
TGF-β signaling in prostate cancer interplays with the activity of the androgen receptor (AR). AR belongs to the family of nuclear receptors and is activated by testosterone and dihydrotestosterone (DHT). Following activation, cytoplasmic AR is translocated to the nucleus to regulate the expression of target genes, either directly (by binding to AREs, androgen receptor response elements in genes’ promoters) or indirectly (by interacting with transcription co-regulators). The expression of TGF-β-regulated genes in prostate adenocarcinoma cells is affected by the interaction of AR with SMAD3, which interferes with the binding of the latter to SBEs (SMAD binding elements) [243]. Furthermore, the expression of TGFBR2 is suppressed by DHT, which attenuates the binding of Sp1 to its promoter. The resulting decreased TGFBR2 expression leads to upregulation of TGF-β-target genes, Bcl-xL and CyclinDs, caspase-3 activation, and triggering of apoptosis [244]. AR mutations or loss, which lead to androgen resistance in differentiated prostate cancers, contribute to TGF-β overexpression, stimulation of growth, viability, and aggressiveness of prostate cancer cells [231,245]. TGF-β and AR synergistically stimulate apoptosis in prostate cancer cells overexpressing TGFBR2. This activation is associated with AR interaction with SMAD4 proteins [246]. Treatment of PC-3 prostate cancer cells with DHT leads to inhibition of E-cadherin and β-catenin, with concomitant overexpression of N-cadherin, indicating EMT activation. These effects are possibly mediated by Snail, which is induced by DHT [247]. Moreover, TGF-β upregulates AR signaling via activation of Twist1, which results in the induction of EMT and stimulation of invasiveness of prostate cancer cells [248,249,250].
Similar to other types of tumors, TGF-β signaling induces EMT in prostate cancer. In particular, TGF-β secreted by stromal cells triggers EMT of benign prostatic hyperplasia cells by activation of SMAD signaling [251]. Tumorigenic prostate epithelial cells progressing to malignancy avoid growth-inhibiting TGF-β activity and acquire constitutive activation of the AKT pathway. In turn, AKT modulates the response of cancer cells to TGF-β by blocking nuclear translocation of SMAD3 and p21, an important mediator of TGF-β signaling, responsible for cell cycle inhibition. TGF-β-induced EMT is mediated by the PI3K/AKT pathway. Specifically, PI3K/AKT inhibition attenuates TGF-β-induced expression of vimentin, downregulation of keratin, and increased cell motility [252]. TGF-β-induced EMT is also mediated by NF-κβ. Overexpression of TGF-β leads to upregulation of vimentin and NF-κβ in prostate tumors with high and intermediate Gleason grades. Inhibition of either TGF-β or NF-κβ suppressed the invasion of cancer cells and the EMT process. These inhibitory NF-κβ effects could not be reversed by the incubation of cells with TGF-β, suggesting that these factors may synergistically induce invasion and EMT in prostate cancer cells [253]. TGF-β-mediated EMT can also be inhibited by Elf5 (through suppression of SMAD3 phosphorylation [254] or FoxA1 [255], and induced by SOX5 [105], CML, CRM1 [256], TRPM7 [257], or SENP1. Interestingly, the latter effect involves SENP1-induced deSUMOylation of SMAD4, leading to its inhibition and induction of EMT [258].
TGF-β is involved in the regulation of the prostate tumor microenvironment, formed by myofibroblasts, carcinoma-associated fibroblasts (CAFs), endothelial cells, lymphocytes, and cancer epithelial cells that promote tumor growth and progression [259]. TGF-β acts as a chemoattractant, which triggers the migration of mesenchymal stem cells (MSC) into the vicinity of prostate cancer cells insensitive to androgen. Moreover, TGF-β plays an important role in the induction of MSC trans-differentiation into CAF-like cells, which are able to induce EMT. It was therefore suggested that blocking of TGF-β signaling in the prostate tumor microenvironment could inhibit cancer progression [260]. TGF-β was also shown to recruit immunosuppressive Treg cells in the prostate tumor environment [261]. Furthermore, TGF-β induces expression of vascular endothelial growth factor (VEGF), a crucial proangiogenic agent, in a hypoxia-dependent manner [262,263].
TGF-β is a crucial stimulator of prostate cancer metastases to bone by promoting the growth and survival of metastasizing cancer cells [264]. Suppression of TGF-β signaling in prostate cancer cells results in inhibition of its metastases to bone [265,266,267]. Knock-out of Tgfbr1 in mice inhibits bone metastases of prostate cancer cells by disrupting their interactions with the bone microenvironment. TGF-β upregulates the expression of multiple genes involved in the progression of bone metastases, including PTHRP, IL11, and PMEPA1. Remarkably, PMEAP1 knock-out induces TGF-β signaling and the formation of bone metastases, indicating negative feedback regulation between the two genes [267]. The stimulatory TGF-β effects on prostate cancer progression are counteracted by PICK1. Specifically, PICK1 upregulation in prostate cancer cells inhibited nuclear translocation of pSMAD2/3 and suppressed the expression of TGF-β target genes both in the presence and absence of TGF-β. Moreover, overexpression of PICK1 reduced the motility and invasiveness of prostate cancer cells. In accordance with these findings, the expression of PICK1 decreases in prostate cancer cells metastasizing to bone and negatively correlates with PSA levels, Gleason grade, and the presence of bone metastasis in patients. In vivo experiments conducted on male nude mice inoculated with PC-3 cells overexpressing PICK1 led to a reduction of metastatic foci, osteolytic areas of metastatic tumors along with prolonged overall survival of the animals and delayed bone metastasis formation [268].
5. MicroRNAs and TGF-β Signaling in GC
TGF-β signaling is regulated by miRNAs. Mechanistically, there are three types of interactions between TGF-β signaling and microRNAs: i) TGF-β regulates expressions of miRNAs, ii) miRNAs regulate the expression of genes involved in TGF-β signaling (Figure 2), and iii) miRNAs interfere with the TGF-β-induced process, such as EMT [269,270,271,272,273]. In the following chapters, we discuss the importance of these complex networks of interactions that contribute to the development and progression of genitourinary cancers, as well as responses to therapy.
5.1. Renal Cancer
The potential significance of miRNAs as diagnostic and prognostic biomarkers in renal cell carcinoma has been confirmed by multiple studies [274,275,276]. These small molecules participate in the regulation of RCC cell growth, cell cycle, apoptosis, angiogenesis, tissue invasion, and metastasis [277]. Noteworthy, miRNAs may be useful markers, allowing for the selection of patients to treatment. For instance, increased serum levels of miR-183 correlate with patients’ resistance to NK cytotoxicity [278] while miR-942 confers resistance of RCC patients to sunitinib [279]. It was also shown that miR-381 enhances the sensitivity of RCC cells to 5-fluorouracil [280].
Most studies addressing the interplay between TGF-β and microRNAs in renal cancer focus on miRNA-mediated regulation of the expression of genes involved in the TGF-β signaling pathway. Several reports showed that miRNAs stimulate ccRCC progression by targeting SMAD4. Accordingly, miR-19a, the expression of which is enhanced in ccRCC tumors, activates proliferation of ccRCC cells while suppressing the expression of SMAD4 and PTEN. Furthermore, the expression of miR-19a significantly correlated with the tumor stage and poor prognosis of ccRCC patients [281]. miR-452-5p stimulates migration and invasion of RCC cells by decreasing SMAD4 expression. Interestingly, these effects are counteracted by treatment with sunitinib, which attenuates migration and invasion of RCC cells by suppressing miR-452-5p expression. Increased miR-452-5p expression is associated with a poor prognosis for RCC patients [282]. SMAD4 and SMAD5 are also directly targeted by miR-224, which induces degradation of their transcripts [283]. Furthermore, miRNAs can also regulate SMAD4 expression indirectly. ccRCC tumors overexpress miR-629, which targets TRIMP33 (tripartite motif-containing 33), an inhibitor of the TGF-β/SMAD signaling pathway. TRIMP33 regulates SMAD4 expression and cellular localization or competes with SMAD4 for binding with SMAD2/3. Inhibition of miR-629 results in suppression of TGF-β-dependent induction of SMAD2/3 and SMAD4 through upregulation of TRIM33 expression. In contrast, transfection of RCC cells with miR-629 mimic potentiated the impact of TGF-β on EMT, motility, and invasion [284]. ACVR2B, another member of the TGF-β signaling pathway, is regulated by miR-192, miR-215, miR-194, miR-141, and miR-200c in nephroblastomas. The expression of these miRNAs is decreased in these pediatric tumors compared with mature kidneys [285]. TAK1 (TGF-β-activated kinase 1) is directly targeted by miR-486-5p in RCC. Remarkably, induced expression of TAK1 leads to stimulation of tumor growth, which can be inhibited by overexpression of miR-486-5p [286].
miRNAs can also interfere with TGF-β-induced signaling in renal cancer cells. Treatment of RCC cell lines with TGF-β1 induces the expression of RBL2, a member of the Rb family involved in TGF-β1-dependent inhibition of proliferation and cell cycle progression. RBL2 is also directly targeted by miR-93, which downregulates its expression. This in turn prevents TGF-β-induced activation of RBL2 expression. Consequently, miR-93 abolishes tumor-suppressive TGF-β1 effects, providing an explanation for TGF-β1 resistance in RCC. Intriguingly, TGF-β1 reduces miR-93 expression, which leads to increased RBL2 expression, indicating reciprocal feedback regulation in the TGF-β1-RBL2-miR-93 axis [287]. Furthermore, miR-429 disrupts TGF-β-dependent inhibition of E-cadherin expression, suggestive of an attenuation of TGF-β-induced EMT. miR-429 expression is decreased in ccRCC tumors and may function as a potential tumor suppressor [288].
Remarkably, TGF-β1 controls the expression of miRNAs involved in the regulation of cellular adhesion in ccRCC. Treatment of RCC cells with TGF-β1 inhibits expressions of miR-30a-5p and miR-328 while increasing miR-25-3p levels, contributing to an altered expression of adhesion proteins, including COL5A1 and ITGA5, and changes in the adhesive properties of ccRCC cells. Inhibition of miR-25-3p potentiates the TGF-β1-stimulatory effect on the expression of adhesion proteins, indicating its interference with the cellular effects of transforming growth factor [289].
5.2. Penile Cancer
There are limited studies that combine simultaneous TGF-β1 and microRNA analysis in penile cancers, mainly due to the rare diagnoses of these tumors. The first NGS analysis of small RNAs expressed in penile cancers was published in 2015. Interestingly, the TGF-β signaling pathway was among the top enriched pathways predicted to be targeted by miRNAs differently expressed in penile cancer tissues compared with matched adjacent non-cancerous tissues [290]. Furthermore, TGFB3 and TGFBR2 were found within the top pathways involving miRNA-targeted genes disrupted in penile cancer [291]. TGFBR2 was also predicted as one of the 47 candidate driver genes targeted by multiple miRNAs, the expressions of which were altered in penile cancer samples compared with normal penile tissue [292]. None of these studies, however, included functional analysis to validate the miRNA-mediated regulation of TGF-β pathway genes, nor the TGF-β-mediated regulation of miRNAs. Thus, the functional links between TGF-β and miRNAs and their role in penile cancer pathogenesis await future analyses.
5.3. Testicular Cancer
Multiple studies revealed altered expressions of miRNA in TC, both tumor tissue expressed and circulating in plasma/serum [293]. In particular, circulating miRNAs offer great promise as potential prognostic markers and are currently explored in clinical trials focusing on TGCT [293]. Surprisingly, the studies addressing the interplay between TGF-β signaling and miRNAs in testicular cancer are scarce. The global analysis of 782 miRNAs in germ-cell tumors revealed that TGF-β signaling was one of the two predicted pathways most highly targeted by miRNAs that were differentially expressed in yolk sack tumors (YSTs) compared with germinomatous tumors (GERs). The expressions of 34 genes of the TGF-β/BMP signaling pathway (including ligands, receptors, SMAD proteins, key target genes) were altered in YST compared with GER. Based on this study, however, it is difficult to draw conclusions regarding testicular tumors because the YST group included tumors located not only in testis but also liver, peri-rectum, and ovary [171]. The functional associations between TGF-β signaling and miRNAs in testis are supported by a study showing TGF-β-induced changes in miRNAs expressions in a mouse GC-spg cell line derived from spermatogonia [294]. It can thus be expected, that disturbed TGF-β pathway functioning in testicular cancer should be associated with altered expressions of target miRNAs. This hypothesis, however, awaits future experimental evaluation.
5.4. Bladder Cancer
The functional associations between TGF-β signaling and microRNAs in bladder cancer were initially supported by a study showing significant correlations between expressions of miRNAs detected in urine sediments of bladder cancer patients and transcripts of genes involved in the TGF-β signaling pathway [295]. Later reports revealed that microRNAs could be both mediators and modulators of TGF-β effects in bladder cancer cells. TGF-β1 stimulates the expression of ZEB1-AS1, which acts as a sponge of tumor-suppressive miR-200b, leading to a decrease of its expression. As a result, the expression of the miR-200b target, fascin1, is induced in bladder cancer cells, leading to activated migration and invasion. Induced ZEB1-AS1 expression inhibits apoptosis, promotes the cell cycle, and activates proliferation, as well as stimulates the growth of bladder cancer xenografts in mice [296]. According to another study, miR-200b targets and inhibits the expression of MMP16. TGF-β1-mediated repression miR-200b leads to activation of MMP16 expression and stimulation of the migration of bladder cancer cells [297]. TGF-β1-induced expression of oncogenic miR-221 triggers EMT and stimulates the migration and invasion of bladder cancer cells [298]. On the other hand, microRNAs can interfere with TGF-β signaling. miR-520f, which is capable of reversing EMT in bladder cancer cells, downregulates expression of TGFBR2 [299].
The miRNA-TGF-β interference also contributes to the chemoresistance of bladder cancer cells (further discussed in Chapter 6).
5.5. Prostate Cancer
There are multiple studies showing altered expression of microRNAs in prostate cancer tissues and urine, suggesting their potential as diagnostic and prognostic biomarkers [300,301,302]. miRNAs are also involved in the regulation of the AR signaling pathway. For instance, miR-125b overexpression is associated with androgen-independent growth of PCa cells in castrate mice [303].
The interplay between TGF-β signaling and miRNA activity in prostate cancer has been documented by multiple studies. miR-15a/16 target and downregulate expressions of SMAD3 and ACVR2A, resulting in attenuated expression of TGF-β dependent genes, including MMP2, E-cadherin, Snail, and Twist, and leading to inhibition of EMT and invasion of prostate cancer cells [304]. Prostate tumors overexpress the TR4 transcription regulator, which attenuates the expression of miR-373-3p, leading to enhanced invasion of prostate cancer cells. Remarkably, miR-373-3p is capable of inhibiting the expression of TGFBR2 and its downstream pSMAD3. In consequence, TR4 knock-out facilitates miR-373-3p-mediated downregulation of TGFBR2 and pSMAD3. Inoculation of mice with prostate cancer cells overexpressing both miR-373-3p and TR4 results in the development of metastases, suggesting that silencing of miR-373-3p and/or TR4 might be used in prostate cancer treatment [305]. The expression of TGFBR2 is also regulated by miR-93. During hypoxia, miR-93 expression is induced, resulting in downregulation of TGFBR2. The expression of miR-93 is increased in prostate tumors compared with non-tumorous control samples and correlates with cancer progression. Transfection of prostate cancer cell lines with miR-93 mimics stimulates their proliferation, migration, and invasion [239]. Complex regulation of the TGF-β signaling pathway is exerted by miR-34b, which modulates expressions of TGF-β, TGFBR1, and pSMAD4, p53. Transfection of PC3 prostate cancer cells with miR-34b mimics results in the inhibition of cell growth, migration, and invasion [306]. The expression of SMAD4 is also regulated by multiple other microRNAs, including miR-1260b, miR-301a, miR-888, and miR-183 [307,308,309,310,311]. Interestingly, expression of miR-1260b is downregulated by genistein, an isoflavone naturally occurring in plants (e.g., lupin, fava beans, soybeans, and coffee), exerting chemopreventive and anticancer activities [312,313,314]. Genistein can also directly suppress SMAD4 expression by inducing changes in DNA methylation and histone modifications [307]. miR-301a represses the expression of SMAD4, promoting the proliferation of prostate cancer cells in vitro and tumor growth in vivo. Interestingly, the expression of miR-301a is regulated by high glucose levels [308], which fits the known association between hyperglycemia and the development of prostate cancer, as well as the prostate cancer grade, Gleason score, and increased risk of prostate cancer recurrence [315,316]. MicroRNAs also regulate the expression of SMAD2. Overexpression of miR-486-5p in prostate cancer results in downregulation of SMAD2 while promoting proliferation, migration, and colony formation. In vivo, inhibition of miR-486-5p attenuates tumor development [317]. Decreased expression of miR-133b in prostate tumors and bone metastases leads to upregulation of TGFBR1 and TGFBR2, enabling TGF-β signaling, which contributes to the migration and invasion of prostate cancer cells. Importantly, the induction of miR-133b expression inhibits the formation of prostate cancer bone metastasis in the mouse model [318]. Similarly, decreased expression of miR-505-3p contributes to prostate cancer progression by targeting SMAD2 and SMAD3, and stimulating migration and invasion. Furthermore, miR-505-3p expression in prostate cancer bone metastases negatively correlates with serum PSA, Gleason grade, and bone metastatic-free survival of patients [319].
TGF-β signaling can also be indirectly regulated by miRNAs. For instance, miR-539 interferes with TGF-β-induced EMT activation by targeting DLX1, a transcription factor that stimulates the expression of SMAD4. The expression of miR-539 in prostate tumors is decreased, contributing to elevated expression of DLX1, which in turn facilitates TGF-β-induced changes in expression of e-cadherin, vimentin, Snail1, and Slug, synonymous with epithelial–mesenchymal transition. Re-introduction of miR-539 into prostate cancer cells inhibits expression of DLX1, leading to attenuation of TGF-β signaling, inhibition of proliferation, migration, and invasion, as well as growth of prostate cancer xenografts in mice [320]. TGF-β signaling in prostate cancer is regulated by a complex axis involving AR, miR-2909, and STAT1. Specifically, miR-2909 attenuates the expression of SOC3, a negative regulator of STAT1, resulting in elevated expression of the latter and leading to overexpression of SMAD7. This in turn results in decreased phosphorylation and activation of SMAD3, contributing to inhibition of TGF-β signaling. On the other hand, miR-2909 directly targets and reduces the expression of TGFBR2, which disables pSMAD2/3 activation, resulting in enhanced expressions of c-Myc and CCND1, reduced p21CIP, and escape of TGF-β suppressive effects on proliferation and viability. Furthermore, miR-2909 and androgen receptor are involved in a positive feedback regulation in which AR stimulates the expression of miR-2909 while miR-2909 enhances the expression of AR, providing further reinforcement of TGF-β signaling inhibition [321]. Strikingly, similar positive feedback was found between AR and miR-21, which targets and downregulates TGFBR2, preventing TGF-β antitumor activity [322]. miR-331-3p stimulates TGF-β1-induced EMT by decreasing expressions of NRP2 and NACC1, multifunctional proteins involved in cancerous transformation and progression. Specifically, a miR-331-3p-induced decrease of NRP2 and NACC1 results in upregulations of TGF-β1 and SMAD4, triggering EMT of prostate cancer cells [323]. Furthermore, loss of tumor suppressive miR-19a-3p leads to activation of SMAD2 and SMAD4, contributing to the formation of bone metastasis in a mouse model [324]. Loss of miR-132/212 in prostate cancer results in upregulation of targeted SOX4, thereby contributing to activation of TGF-β signaling and induction of EMT. Re-expression of miR-132/212 in prostate cancer cells suppresses their invasion and migration, and attenuates colony formation by inhibiting TGF-β-induced EMT [325]. Interestingly, the crosstalk between TGF-β and microRNAs is also utilized by the tumor microenvironment for stimulation of cancer progression. Accordingly, it was shown that pre-adipocytes induce miR-301a expression in prostate cancer cells, leading to suppression of its target, AR, and subsequent activation of TGF-β/SMAD/MMP-9 signaling and promotion of cancer invasiveness in vivo [326].
The spatio-temporal changes in the expression of microRNAs may provide a mechanical explanation for the “TGF-β paradox” in prostate cancer. It was found that expressions of miR-582-3p and miR-582-5p are increased in prostate tumors compared with adjacent normal tumors while being downregulated in bone metastatic tissue compared with metastases in other organs. Remarkably, overexpression of these microRNAs repressed the migration and invasion of prostate cancer cells in vitro and the formation of bone metastases in vivo. These effects were mediated by microRNA-induced repression of key mediators of the TGF-β signaling pathway, including TGFBR1, TGFBR2, SMAD2, and SMAD4, which led to the reprogramming of multiple genes involved in the formation of bone metastases [327]. Loss of miR-15 and miR-16 in prostate cancer cells potentiates TGF-β signaling by upregulating USP9X (a gene encoding an enzyme deubiquitinating SMAD4), as well as activin RIIA, an activin receptor, contributing to the survival of cancer cells in bone marrow and the formation of bone metastasis. Furthermore, these effects synergize with the activity of miR-21, the enhanced expression of which results in suppression of SMAD7. As a result, depletion of miR-15/miR-16 with concomitant upregulation of miR-21 in prostate cancer cells stimulates the formation of bone lesions in mice [328]. Remarkably, TGF-β upregulates miR-21 expression by stimulating the processing of its primary transcript by ribonuclease Drosha [329]. This suggests that the reciprocal feedback regulation between miRNAs and TGF-β in prostate cancer may be involved in the regulation of metastasis.
Interestingly, TGF-β-mediated regulation of microRNAs emerges as an important mechanism contributing to the progression of prostate cancer. In this regard, activin A is a powerful regulator of miRNAs, triggering changes in the expression of nine microRNAs (miR-222-3p, miR-15b-5p, miR-93-5p, miR-18a-5p, miR-30a/30d-5p, let-7c, and miR-196b-5p). Activin A is a member of the TGF-β signaling pathway, recognized as an important negative regulator of the growth and migration of prostate cancer cells [330]. TGF-β regulates the expression of miR-96 via activation of the SMAD 2/3/4 complex, which interacts with SBEs (SMAD2/3-binding elements) within the promoter region of pri-miR-96 precursor and stimulates its expression. miR-96 regulates the expression of AKT1S1, which inhibits mTOR and promotes the proliferation and formation of bone metastasis in mice [331]. TGF-β attenuates the expression of miR-1 and miR-200b, leading to increased expression of Slug, EMT stimulation, and progression of prostate cancer in a mice model. Remarkably, this regulation involves a negative feedback circuit in which microRNAs inhibit the expression of Slug while the latter attenuates the expression of microRNAs in a TGF-β-dependent manner. TGF-β-induced reduction of miR-1 and miR-200b disrupts this miRNA-Slug balance and triggers a cascade of changes in the expression of genes involved in EMT and cancer progression [332]. Interestingly, it was suggested that the miR-200 family and miR-205 could oppose the reversal of EMT in a benign prostatic hyperplasia epithelial cell line [333].
6. TGF-β1 and microRNAs and Treatment of GC
The TGF-β signaling pathway is apparently an attractive option for therapeutic approaches in cancer. One of the early studies reporting TGF-β as a target for a therapeutic approach in urinary bladder cancer was published in 1998. In that study, antisense oligonucleotides attenuated TGF-β secretion by bladder cancer cells, reduced colony growth in soft agar, and inhibited the growth of tumors inoculated in mice. Unfortunately, that study did not include a statistical analysis of the presented data, so the conclusions remain elusive [334]. TGF-β is also a target of hispolon (6 -(3,4-dihydroxyphenyl)-4-hydroxyhexa-3,5-dien-2-one), HPL), a compound isolated from Phellinus linteus, exerting anticancer properties against multiple tumor types, including cancers of the cervix, colon, and kidney [335,336], as well as melanoma [337]. Hong et al. showed that HPL attenuates TGF-β-induced EMT of bladder cancer cells, resulting in reduced migration and invasion [338].
The significance of TGF-β signaling as a clinical target emerged through recent studies on the combined therapies involving targeting TGF-β and immune checkpoints. An example of such an approach is M7824, a bi-functional fusion protein consisting of avemulab, a monoclonal antibody directed against PD-L1 (programmed death-ligand 1) and the extracellular domain of TGFBR2 [339]. Avelumab acts as an immune checkpoint inhibitor, enabling the cytotoxic activity of T-cells towards tumor cells, and was approved by the FDA (Food and Drug Administration) for treatment of urothelial carcinoma in 2017 [340]. The extracellular TGFBR2 domain counteracts the functioning of all three TGF-β isoforms by acting as a ‘trap’, efficiently eliminating the TGF-β pool available for endogenous TGF-β receptors and thus preventing the immunosuppressive activity of high TGF-β levels produced by tumor cells. The study demonstrated that the TGFBR2 component of M7824 increased the sensitivity of urothelial transitional cell carcinoma cells towards TRAIL-mediated lysis. Compared to the sole PD-L1 blockade, M7824 also stimulated antigen-specific CD8+-mediated tumor cell lysis. Mechanistically, treatment of bladder cancer cells with M7824 altered the expression of genes involved in the angiogenesis process, remodeling of the extracellular matrix, EMT, as well as extracellular markers involved in immunogenic modulation [339].
TGF-β signaling determines patients’ responses to immunotherapies. TGF-β +C28.>T polymorphism (in other studies described as c.-1347C > T, -509C > T, and rs1800469) was linked with patients’ outcome to BCG (Bacillus Calmette–Guérin) immunotherapy. Specifically, homozygotic TT patients had a lower risk of recurrence following BCG treatment [341]. Interestingly, this genotype is associated with increased TGF-β expression when compared with CC homozygotes, and individuals with T substitution have twice higher TGF-β1 plasma concentrations when compared with CC carriers [174,178]. TGF-β modulates the functioning of myeloid-derived suppressor cells (MDSCs) of bladder cancer patients. MDSCs regulate the antitumor immune responses and suppress T-cell function. Yuan et al. found that compared with healthy controls, bladder cancer patients have an increased population of CD14+HLA-DR−/low cells, which secreted TGF-β to suppress T-cell proliferation and production of IFN-γ (interferon-gamma) [342].
The expression of TGF-β1 and TGFBR2 is increased in metastatic urothelial cancer patients who are not responding to atezolizumab, a blocker of PD-L1 [216]. In a mouse EMT6 mammary carcinoma model, therapeutic concomitant blockade of both PD-L1 and TGF-β increased tumor infiltration by T cells (in particular, CD8+ Teff cells) and significantly inhibited tumor growth. Remarkably, these effects were not observed when either PD-L1 or TGF-β inhibitors were introduced. The immunomodulatory TGF-β actions are initiated in the tumor microenvironment, not directly in tumor cells. TGF-β acts through peritumoral stromal fibroblasts to reprogram anti-tumor immunity, as indicated by altered downstream TGF-β signaling (including pSMAD2/3) in fibroblasts [216]. These findings open new possibilities for increasing the efficiency of therapies involving immune checkpoint inhibitors.
TGF-β/miRNA crosstalk is involved in the responses of cancer cells to therapies. For instance, treatment of bladder cancer cells with celecoxib, a selective inhibitor of COX-2, attenuates proliferation, migration, invasion, as well as epithelial–mesenchymal transition. These effects are mediated by increased expression of miR-145, which directly targets and downregulates expressions of TGFBR2 and SMAD3 [343]. On the other hand, miR-145 is involved in TGF-β1-induced gemcitabine resistance of bladder cancer cells. Prolonged treatment of bladder cancer cells with gemcitabine results in induced expression of TGF-β1, which triggers SMAD-mediated repression of lncRNA-LET expression. This in turn relieves lncRNA-mediated repression of NF90, a protein involved in the regulation of transcript stability. Upregulated NF90 suppresses the biogenesis of miR-145, a suppressor of cancer stemness. As a consequence, prolonged treatment of bladder cancer cells leads to enrichment of cancer stem cells via TGF-β1 activation and the resulting dysregulation of the lncRNA-LET/NF90/miR-145 axis. The potential therapeutic significance of this mechanism has been confirmed by treatment of chemoresistant tumors with a TGF-β1 inhibitor, which re-sensitized bladder cancer cells to gemcitabine [344].
MicroRNA-mediated regulation of SMAD2 is involved in the mechanism triggered by arsenic trioxide (As2O3), a potential inhibitor of prostate cancer angiogenesis, which increases miR-155 expression by inducing DNA demethylation. In turn, re-activated miR-155 inhibits TGF-β signaling by directly targeting and decreasing expression of SMAD2, which results in suppression of VEGF secretion and inhibition of angiogenesis [345].
MicroRNAs that directly target genes of the TGF-β signaling pathway are also considered as therapeutic options in the treatment of GCs. As mentioned, re-introduction of both miR-582-3p and miR-582-5p in prostate cancer cells attenuated migration and invasion in vitro, and reduced the formation of bone metastases in vivo, by targeting SMAD2, SMAD4, TGFBRI, and TGFBRII, and leading to an inhibition of TGF-β signaling. In prostate cancer patients, decreased expressions of miR-582-3p and miR-582-5p correlated with poor bone metastasis-free survival, indicating that these miRNAs could be considered as a therapeutic option for prostate cancer patients [327].
7. Conclusions
Both the TGF-β signaling pathway and microRNAs clearly contribute to the development and progression of genitourinary cancers. In particular, the interplay between microRNAs and TGF-β signaling may provide a mechanistic explanation for the TGF-β paradox universally occurring in cancer cells. In the first model, microRNAs targeting key mediators of the TGF-β signaling pathway, including TGF-β receptors and SMADs, suppress pro-cancerous TGF-β actions in primary tumors. During cancer progression, the expression of these miRNAs becomes downregulated, releasing the expression of downstream TGF-β effectors and promoting metastatic growth. According to the second model, TGF-β actions may be mediated by miRNAs that either promote or inhibit cancerous progression by direct targeting of genes regulating cancerous proliferation, adhesion, migration, and invasion. In primary tumors, higher expression of tumor-suppressive miRNA overcomes the effects of oncogenic miRNAs, attenuating tumor growth. In metastatic tumors, increased expression of oncogenic miRNAs overcomes the effects of suppressive miRNAs, leading to cancer progression. Thus, in both models, the “switching” of miRNAs’ expression between primary and secondary tumor lesions would be the key mechanism contributing to the TGF-β paradox in cancer.
Clinically, the most relevant are findings on the role of TGF-β1 signaling in modulating the tumor microenvironment and immunity. Changes in the expression of TGF-β1 and microRNAs have potential as informative diagnostic and prognostic biomarkers. However, further studies are needed to validate the obtained data, in particular regarding the expressions of TGF-β1 and microRNAs in serum and urine. The interplay between the TGF-β signaling pathway and microRNAs emerges as an important field for therapeutic interventions. Therapeutic microRNAs are already being tested in clinical trials [346]. It can thus be expected that miRNAs targeting the TGF-β signaling pathway will be implemented in future clinical tests of new therapies for GC. Given the results of the in vivo studies, microRNAs that modulate TGF-β-mediated progression of prostate cancer offer great promise for the treatment of metastatic tumors.
Funding
This research was funded by The Polpharma Scientific Foundation’s grant (to J.B.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wakefield, L.M.; Hill, C.S. Beyond TGF beta: Roles of other TGF beta superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Oner, M.G.; Meuwissen, R.L.; Genc, S. The role of microRNAs in human diseases. Methods Mol. Biol. 2014, 1107, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, R.M. Genitourinary oncology: Current status and future challenges. Front. Oncol. 2011, 1, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A.; Moch, H.; Cubilla, A.L.; Ulbright, T.M.; Reuter, V.E. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part B: Prostate and Bladder Tumours. Eur. Urol. 2016, 70, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Capitanio, U.; Cloutier, V.; Zini, L.; Isbarn, H.; Jeldres, C.; Shariat, S.F.; Perrotte, P.; Antebi, E.; Patard, J.J.; Montorsi, F.; et al. A critical assessment of the prognostic value of clear cell, papillary and chromophobe histological subtypes in renal cell carcinoma: A population-based study. BJU Int. 2009, 103, 1496–1500. [Google Scholar] [CrossRef]
- Keegan, K.A.; Schupp, C.W.; Chamie, K.; Hellenthal, N.J.; Evans, C.P.; Koppie, T.M. Histopathology of surgically treated renal cell carcinoma: Survival differences by subtype and stage. J. Urol. 2012, 188, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J. Molecular genetics of clear-cell renal cell carcinoma. J. Clin. Oncol. 2014, 32, 1968–1976. [Google Scholar] [CrossRef] [Green Version]
- Jayson, M.; Sanders, H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998, 51, 203–205. [Google Scholar] [CrossRef]
- Ljungberg, B.; Albiges, L.; Abu-Ghanem, Y.; Bensalah, K.; Dabestani, S.; Fernandez-Pello, S.; Giles, R.H.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; et al. European Association of Urology Guidelines on Renal Cell Carcinoma: The 2019 Update. Eur. Urol. 2019, 75, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, R.C.; Mickisch, G.; Sylvester, R.; Tangen, C.; Van Poppel, H.; Crawford, E.D. Cytoreductive nephrectomy in patients with metastatic renal cancer: A combined analysis. J. Urol. 2004, 171, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejean, A.; Ravaud, A.; Thezenas, S.; Colas, S.; Beauval, J.B.; Bensalah, K.; Geoffrois, L.; Thiery-Vuillemin, A.; Cormier, L.; Lang, H.; et al. Sunitinib Alone or after Nephrectomy in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Wallis, C.J.D.; Klaassen, Z.; Bhindi, B.; Ye, X.Y.; Chandrasekar, T.; Farrell, A.M.; Goldberg, H.; Boorjian, S.A.; Leibovich, B.; Kulkarni, G.S.; et al. First-line Systemic Therapy for Metastatic Renal Cell Carcinoma: A Systematic Review and Network Meta-analysis. Eur. Urol. 2018, 74, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Necchi, A.; Rosen, G.; Hariharan, S.; Apolo, A.B. Anti-Programmed Cell Death 1/Ligand 1 (PD-1/PD-L1) Antibodies for the Treatment of Urothelial Carcinoma: State of the Art and Future Development. Clin. Genitourin Cancer 2018, 16, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Nunno, V.D.; Mollica, V.; Montironi, R.; Cheng, L.; Cimadamore, A.; Blanca, A.; Lopez-Beltran, A. Immunotherapy in renal cell carcinoma from poverty to the spoiled of choice. Immunotherapy 2019. [Google Scholar] [CrossRef]
- Wahlgren, T.; Harmenberg, U.; Sandstrom, P.; Lundstam, S.; Kowalski, J.; Jakobsson, M.; Sandin, R.; Ljungberg, B. Treatment and overall survival in renal cell carcinoma: A Swedish population-based study (2000-2008). Br. J. Cancer 2013, 108, 1541–1549. [Google Scholar] [CrossRef]
- Li, P.; Wong, Y.N.; Armstrong, K.; Haas, N.; Subedi, P.; Davis-Cerone, M.; Doshi, J.A. Survival among patients with advanced renal cell carcinoma in the pretargeted versus targeted therapy eras. Cancer Med. 2016, 5, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Leibovich, B.C.; Lohse, C.M.; Crispen, P.L.; Boorjian, S.A.; Thompson, R.H.; Blute, M.L.; Cheville, J.C. Histological subtype is an independent predictor of outcome for patients with renal cell carcinoma. J. Urol. 2010, 183, 1309–1315. [Google Scholar] [CrossRef]
- Tsui, K.H.; Shvarts, O.; Smith, R.B.; Figlin, R.A.; deKernion, J.B.; Belldegrun, A. Prognostic indicators for renal cell carcinoma: A multivariate analysis of 643 patients using the revised 1997 TNM staging criteria. J. Urol. 2000, 163, 1090–1095, quiz 1295. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakenberg, O.W.; Comperat, E.M.; Minhas, S.; Necchi, A.; Protzel, C.; Watkin, N. EAU guidelines on penile cancer: 2014 update. Eur. Urol. 2015, 67, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Stratton, K.L.; Culkin, D.J. A Contemporary Review of HPV and Penile Cancer. Oncology 2016, 30, 245–249. [Google Scholar] [PubMed]
- Minhas, S.; Kayes, O.; Hegarty, P.; Kumar, P.; Freeman, A.; Ralph, D. What surgical resection margins are required to achieve oncological control in men with primary penile cancer? BJU Int. 2005, 96, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, A.; Chipollini, J.; Yan, S.; Ottenhof, S.R.; Tang, D.H.; Draeger, D.; Protzel, C.; Zhu, Y.; Ye, D.W.; Hakenberg, O.W.; et al. Penile Sparing Surgery for Penile Cancer: A Multicenter International Retrospective Cohort. J. Urol. 2018, 199, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.H.; Yan, S.; Ottenhof, S.R.; Draeger, D.; Baumgarten, A.S.; Chipollini, J.; Protzel, C.; Zhu, Y.; Ye, D.W.; Hakenberg, O.W.; et al. Laser ablation as monotherapy for penile squamous cell carcinoma: A multi-center cohort analysis. Urol. Oncol. 2018, 36, 147–152. [Google Scholar] [CrossRef]
- Kamel, M.H.; Bissada, N.; Warford, R.; Farias, J.; Davis, R. Organ Sparing Surgery for Penile Cancer: A Systematic Review. J. Urol. 2017, 198, 770–779. [Google Scholar] [CrossRef]
- Hasan, S.; Francis, A.; Hagenauer, A.; Hirsh, A.; Kaminsky, D.; Traughber, B.; Abouassaly, R.; Ellis, R. The role of brachytherapy in organ preservation for penile cancer: A meta-analysis and review of the literature. Brachytherapy 2015, 14, 517–524. [Google Scholar] [CrossRef]
- Crook, J.; Ma, C.; Grimard, L. Radiation therapy in the management of the primary penile tumor: An update. World J. Urol. 2009, 27, 189–196. [Google Scholar] [CrossRef]
- Pond, G.R.; Di Lorenzo, G.; Necchi, A.; Eigl, B.J.; Kolinsky, M.P.; Chacko, R.T.; Dorff, T.B.; Harshman, L.C.; Milowsky, M.I.; Lee, R.J.; et al. Prognostic risk stratification derived from individual patient level data for men with advanced penile squamous cell carcinoma receiving first-line systemic therapy. Urol. Oncol. 2014, 32, 501–508. [Google Scholar] [CrossRef]
- Yang, J.; Pan, Z.; He, Y.; Zhao, F.; Feng, X.; Liu, Q.; Lyu, J. Competing-risks model for predicting the prognosis of penile cancer based on the SEER database. Cancer Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, M.N.; Deal, A.M.; Ferguson, J.E., III; Wang, Y.; Smith, A.B.; Nielsen, M.E.; Pruthi, R.S.; Woods, M.E. Contemporary survival trends in penile cancer: Results from the National Cancer Database. Urol. Oncol. 2017, 35, e671–674. [Google Scholar] [CrossRef] [PubMed]
- Paiva, G.R.; de Oliveira Araujo, I.B.; Athanazio, D.A.; de Freitas, L.A. Penile cancer: Impact of age at diagnosis on morphology and prognosis. Int. Urol. Nephrol. 2015, 47, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Albers, P.; Albrecht, W.; Algaba, F.; Bokemeyer, C.; Cohn-Cedermark, G.; Fizazi, K.; Horwich, A.; Laguna, M.P.; Nicolai, N.; Oldenburg, J. EAU Guidelines on Testicular Cancer; European Association of Urology: Arnhem, The Netherlands, 2017. [Google Scholar]
- Batool, A.; Karimi, N.; Wu, X.N.; Chen, S.R.; Liu, Y.X. Testicular germ cell tumor: A comprehensive review. Cell Mol. Life Sci. 2019, 76, 1713–1727. [Google Scholar] [CrossRef]
- La Vecchia, C.; Bosetti, C.; Lucchini, F.; Bertuccio, P.; Negri, E.; Boyle, P.; Levi, F. Cancer mortality in Europe, 2000-2004, and an overview of trends since 1975. Ann. Oncol. 2010, 21, 1323–1360. [Google Scholar] [CrossRef]
- Bosl, G.J.; Motzer, R.J. Testicular germ-cell cancer. N. Engl. J. Med. 1997, 337, 242–253. [Google Scholar] [CrossRef]
- Kuczyk, M.A.; Serth, J.; Bokemeyer, C.; Jonassen, J.; Machtens, S.; Werner, M.; Jonas, U. Alterations of the p53 tumor suppressor gene in carcinoma in situ of the testis. Cancer 1996, 78, 1958–1966. [Google Scholar] [CrossRef]
- Goldberg, K. ASCO 50th Anniversary Poll Names the Top 5 Advances from the Past 50 Years. Results Released Ahead of the “Rally for Medical Research” on Capitol Hill, September 18th – New Cancer Research Funding Urgently Needed. Available online: https://www.asco.org/about-asco/press-center/news-releases/asco-50th-anniversary-poll-names-top-5-advances-past-50-years (accessed on 30 August 2019).
- Albers, P.; Albrecht, W.; Algaba, F.; Bokemeyer, C.; Cohn-Cedermark, G.; Fizazi, K.; Horwich, A.; Laguna, M.P.; Nicolai, N.; Oldenburg, J.; et al. Guidelines on Testicular Cancer: 2015 Update. Eur. Urol. 2015, 68, 1054–1068. [Google Scholar] [CrossRef]
- Hoffmann, R.; Plug, I.; McKee, M.; Khoshaba, B.; Westerling, R.; Looman, C.; Rey, G.; Jougla, E.; Lang, K.; Parna, K.; et al. Innovations in health care and mortality trends from five cancers in seven European countries between 1970 and 2005. Int. J. Public Health 2014, 59, 341–350. [Google Scholar] [CrossRef]
- Warde, P.; Specht, L.; Horwich, A.; Oliver, T.; Panzarella, T.; Gospodarowicz, M.; von der Maase, H. Prognostic factors for relapse in stage I seminoma managed by surveillance: A pooled analysis. J. Clin. Oncol. 2002, 20, 4448–4452. [Google Scholar] [CrossRef] [PubMed]
- Albers, P.; Siener, R.; Kliesch, S.; Weissbach, L.; Krege, S.; Sparwasser, C.; Schulze, H.; Heidenreich, A.; de Riese, W.; Loy, V.; et al. Risk factors for relapse in clinical stage I nonseminomatous testicular germ cell tumors: Results of the German Testicular Cancer Study Group Trial. J. Clin. Oncol. 2003, 21, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Mead, G.M.; Stenning, S.P. The International Germ Cell Consensus Classification: A new prognostic factor-based staging classification for metastatic germ cell tumours. Clin. Oncol. 1997, 9, 207–209. [Google Scholar] [CrossRef]
- Klepp, O.; Flodgren, P.; Maartman-Moe, H.; Lindholm, C.E.; Unsgaard, B.; Teigum, H.; Fossa, S.D.; Paus, E. Early clinical stages (CS1, CS1Mk+ and CS2A) of non-seminomatous testis cancer. Value of pre- and post-orchiectomy serum tumor marker information in prediction of retroperitoneal lymph node metastases. Swedish-Norwegian Testicular Cancer Project (SWENOTECA). Ann. Oncol. 1990, 1, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Nordentoft, I.; Lamy, P.; Birkenkamp-Demtroder, K.; Shumansky, K.; Vang, S.; Hornshoj, H.; Juul, M.; Villesen, P.; Hedegaard, J.; Roth, A.; et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014, 7, 1649–1663. [Google Scholar] [CrossRef] [Green Version]
- van Tilborg, A.A.; de Vries, A.; de Bont, M.; Groenfeld, L.E.; van der Kwast, T.H.; Zwarthoff, E.C. Molecular evolution of multiple recurrent cancers of the bladder. Hum. Mol. Genet. 2000, 9, 2973–2980. [Google Scholar] [CrossRef] [Green Version]
- Babjuk, M.; Burger, M.; Comperat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Roupret, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma In Situ)-2019 Update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef]
- Poletajew, S.; Biernacki, R.; Buraczynski, P.; Chojnacki, J.; Czarniecki, S.; Gajewska, D.; Pohaba, T.; Sondka, J.; Skrzypczyk, M.; Suchojad, T.; et al. Stage of bladder cancer in Central Europe-Polish perspective. Neoplasma 2016, 63, 642–647. [Google Scholar] [CrossRef] [Green Version]
- Martin-Doyle, W.; Kwiatkowski, D.J. Molecular biology of bladder cancer. Hematol. Oncol. Clin. North. Am. 2015, 29, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczyk, M.A.; Nyk, L.; Szostek, P.; Szemplinski, S.; Borowka, A.; Dobruch, J. The role of endoscopic bladder tumour assessment in the management of patients subjected to transurethral bladder tumour resection. Eur. J. Cancer Care 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, R.J.; van der Meijden, A.P.; Oosterlinck, W.; Witjes, J.A.; Bouffioux, C.; Denis, L.; Newling, D.W.; Kurth, K. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: A combined analysis of 2596 patients from seven EORTC trials. Eur. Urol. 2006, 49, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gomez, J.; Madero, R.; Solsona, E.; Unda, M.; Martinez-Pineiro, L.; Gonzalez, M.; Portillo, J.; Ojea, A.; Pertusa, C.; Rodriguez-Molina, J.; et al. Predicting nonmuscle invasive bladder cancer recurrence and progression in patients treated with bacillus Calmette-Guerin: The CUETO scoring model. J. Urol. 2009, 182, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Carroll, P.R.; Small, E.J. Update on chemotherapy for advanced bladder cancer. J. Urol. 2005, 174, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Poletajew, S.; Biernacki, R.; Buraczynski, P.; Chojnacki, J.; Czarniecki, S.; Gajewska, D.; Pohaba, T.; Sondka, J.; Skrzypczyk, M.; Suchojad, T.; et al. Patterns of care in patients with muscle-invasive bladder cancer - a retrospective cohort study. Contemp. Oncol. 2016, 20, 341–343. [Google Scholar] [CrossRef]
- Alfred Witjes, J.; Lebret, T.; Comperat, E.M.; Cowan, N.C.; De Santis, M.; Bruins, H.M.; Hernandez, V.; Espinos, E.L.; Dunn, J.; Rouanne, M.; et al. Updated 2016 EAU Guidelines on Muscle-invasive and Metastatic Bladder Cancer. Eur. Urol. 2017, 71, 462–475. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Rouanne, M.; Roumiguie, M.; Houede, N.; Masson-Lecomte, A.; Colin, P.; Pignot, G.; Larre, S.; Xylinas, E.; Roupret, M.; Neuzillet, Y. Development of immunotherapy in bladder cancer: Present and future on targeting PD(L)1 and CTLA-4 pathways. World J. Urol. 2018, 36, 1727–1740. [Google Scholar] [CrossRef]
- Stein, J.P.; Lieskovsky, G.; Cote, R.; Groshen, S.; Feng, A.C.; Boyd, S.; Skinner, E.; Bochner, B.; Thangathurai, D.; Mikhail, M.; et al. Radical cystectomy in the treatment of invasive bladder cancer: Long-term results in 1,054 patients. J. Clin. Oncol. 2001, 19, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Karakiewicz, P.I.; Palapattu, G.S.; Lotan, Y.; Rogers, C.G.; Amiel, G.E.; Vazina, A.; Gupta, A.; Bastian, P.J.; Sagalowsky, A.I.; et al. Outcomes of radical cystectomy for transitional cell carcinoma of the bladder: A contemporary series from the Bladder Cancer Research Consortium. J. Urol. 2006, 176, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, M.; Dybowski, B.; Kuczkiewicz-Siemion, O.; Osiecki, R.; Smigielska, K.; Gonczar, S.; Poletajew, S.; Radziszewski, P. Factors affecting one-year survival after radical cystectomy: A prospective study. Cent. Eur.opean J. Urol. 2017, 70, 238–244. [Google Scholar] [CrossRef] [Green Version]
- von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef]
- Dash, A.; Galsky, M.D.; Vickers, A.J.; Serio, A.M.; Koppie, T.M.; Dalbagni, G.; Bochner, B.H. Impact of renal impairment on eligibility for adjuvant cisplatin-based chemotherapy in patients with urothelial carcinoma of the bladder. Cancer 2006, 107, 506–513. [Google Scholar] [CrossRef]
- McNeal, J.E. The zonal anatomy of the prostate. Prostate 1981, 2, 35–49. [Google Scholar] [CrossRef]
- Ilic, D.; Neuberger, M.M.; Djulbegovic, M.; Dahm, P. Screening for prostate cancer. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Hayes, J.H.; Barry, M.J. Screening for prostate cancer with the prostate-specific antigen test: A review of current evidence. JAMA 2014, 311, 1143–1149. [Google Scholar] [CrossRef]
- Schroder, F.H.; Hugosson, J.; Roobol, M.J.; Tammela, T.L.; Zappa, M.; Nelen, V.; Kwiatkowski, M.; Lujan, M.; Maattanen, L.; Lilja, H.; et al. Screening and prostate cancer mortality: Results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet 2014, 384, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.M.; Joniau, S.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef]
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilt, T.J.; Brawer, M.K.; Jones, K.M.; Barry, M.J.; Aronson, W.J.; Fox, S.; Gingrich, J.R.; Wei, J.T.; Gilhooly, P.; Grob, B.M.; et al. Radical prostatectomy versus observation for localized prostate cancer. N. Engl. J. Med. 2012, 367, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulewicz, R.S.; Weiner, A.B.; Schaeffer, E.M. Active Surveillance for Prostate Cancer. JAMA 2017, 318, 2152. [Google Scholar] [CrossRef] [PubMed]
- Loeb, S.; Zhou, Q.; Siebert, U.; Rochau, U.; Jahn, B.; Muhlberger, N.; Carter, H.B.; Lepor, H.; Braithwaite, R.S. Active Surveillance Versus Watchful Waiting for Localized Prostate Cancer: A Model to Inform Decisions. Eur. Urol. 2017, 72, 899–907. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Gravis, G.; Fizazi, K.; Joly, F.; Oudard, S.; Priou, F.; Esterni, B.; Latorzeff, I.; Delva, R.; Krakowski, I.; Laguerre, B.; et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 149–158. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef]
- Hussain, M.; Tangen, C.M.; Higano, C.; Schelhammer, P.F.; Faulkner, J.; Crawford, E.D.; Wilding, G.; Akdas, A.; Small, E.J.; Donnelly, B.; et al. Absolute prostate-specific antigen value after androgen deprivation is a strong independent predictor of survival in new metastatic prostate cancer: Data from Southwest Oncology Group trial 9346 (INT-0162). J. Clin. Oncol. 2006, 24, 3984–3990. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sakai, T. Biological significance of local TGF-beta activation in liver diseases. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-beta-an excellent servant but a bad master. J. Transl. Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimi, R.A.; Leof, E.B. TGF-beta signaling: A tale of two responses. J. Cell Biochem. 2007, 102, 593–608. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. TGF beta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; ten Dijke, P. The dynamic roles of TGF-beta in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef]
- Cantelli, G.; Crosas-Molist, E.; Georgouli, M.; Sanz-Moreno, V. TGFB-induced transcription in cancer. Semin. Cancer Biol. 2017, 42, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Ikushima, H.; Miyazono, K. TGF beta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef]
- Drabsch, Y.; ten Dijke, P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metast Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGF beta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.Z.; Neil, J.R.; Schiemann, W.P. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2011, 23, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarzynska, J.M. Two faces of TGF-beta1 in breast cancer. Mediat. Inflamm 2014, 2014, 141747. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.H.; Likhter, M.; Jogunoori, W.; Belkin, M.; Ohshiro, K.; Mishra, L. TGF-beta signaling in liver and gastrointestinal cancers. Cancer Lett. 2016, 379, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Pasche, B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16, R14–R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truty, M.J.; Urrutia, R. Basics of TGF-beta and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef]
- Jeon, H.S.; Jen, J. TGF-beta signaling and the role of inhibitory Smads in non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Korkut, A.; Zaidi, S.; Kanchi, R.S.; Rao, S.; Gough, N.R.; Schultz, A.; Li, X.; Lorenzi, P.L.; Berger, A.C.; Robertson, G.; et al. A Pan-Cancer Analysis Reveals High-Frequency Genetic Alterations in Mediators of Signaling by the TGF-beta Superfamily. Cell Syst. 2018, 7, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Thompson, N.L.; Flanders, K.C.; Smith, J.M.; Ellingsworth, L.R.; Roberts, A.B.; Sporn, M.B. Expression of Transforming Growth Factor-Beta-1 in Specific Cells and Tissues of Adult and Neonatal Mice. J. Cell Biol. 1989, 108, 661–669. [Google Scholar] [CrossRef]
- Shah, M.M.; Sampogna, R.V.; Sakurai, H.; Bush, K.T.; Nigam, S.K. Branching morphogenesis and kidney disease. Development 2004, 131, 1449–1462. [Google Scholar] [CrossRef] [Green Version]
- Plisov, S.Y.; Yoshino, K.; Dove, L.F.; Higinbotham, K.G.; Rubin, J.S.; Perantoni, A.O. TGF beta 2, LIF and FGF2 cooperate to induce nephrogenesis. Development 2001, 128, 1045–1057. [Google Scholar]
- Sanford, L.P.; Ormsby, I.; GittenbergerdeGroot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGF beta 2 knockout mice have multiple developmental defects that are nonoverlapping with other TGF beta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Chung, A.C.K.; Lan, H.Y. Molecular Mechanisms of TGF-β Signaling in Renal Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nature Reviews Nephrology 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-beta/Smad signaling in tissue fibrosis. Chem-Biol. Int.eract. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaka, Y. Targeting TGF-beta Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-beta: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49. [Google Scholar] [CrossRef]
- Derynck, R.; Goeddel, D.V.; Ullrich, A.; Gutterman, J.U.; Williams, R.D.; Bringman, T.S.; Berger, W.H. Synthesis of Messenger-Rnas for Transforming Growth Factor-Alpha and Factor-Beta and the Epidermal Growth-Factor Receptor by Human-Tumors. Cancer Res. 1987, 47, 707–712. [Google Scholar]
- Gomella, L.G.; Sargent, E.R.; Wade, T.P.; Anglard, P.; Linehan, W.M.; Kasid, A. Expression of Transforming Growth Factor-Alpha in Normal Human Adult Kidney and Enhanced Expression of Transforming Growth Factor-Alpha and Factor-Beta-1 in Renal-Cell Carcinoma. Cancer Res. 1989, 49, 6972–6975. [Google Scholar]
- Boguslawska, J.; Kedzierska, H.; Poplawski, P.; Rybicka, B.; Tanski, Z.; Piekielko-Witkowska, A. Expression of Genes Involved in Cellular Adhesion and Extracellular Matrix Remodeling Correlates with Poor Survival of Patients with Renal Cancer. J. Urol. 2016, 195, 1892–1902. [Google Scholar] [CrossRef]
- Ramp, U.; Jaquet, K.; Reinecke, P.; Nitsch, T.; Gabbert, H.E.; Gerharz, C.D. Acquisition of TGF-beta(1) resistance: An important progression factor in human renal cell carcinoma. Lab. Investig. 1997, 76, 739–749. [Google Scholar] [CrossRef]
- Ramp, U.; Jaquet, K.; Reinecke, P.; Schardt, C.; Friebe, U.; Nitsch, T.; Marx, N.; Gabbert, H.E.; Gerharz, C.D. Functional intactness of stimulatory and inhibitory autocrine loops in human renal carcinoma cell lines of the clear cell type. J. Urol. 1997, 157, 2345–2350. [Google Scholar] [CrossRef]
- Knoefel, B.; Nuske, K.; Steiner, T.; Junker, K.; Kosmehl, H.; Rebstock, K.; Reinhold, D.; Junker, U. Renal cell carcinomas produce IL-6, IL-10, IL-11, and TGF-beta 1 in primary cultures and modulate T lymphocyte blast transformation. J. Interf. Cytok Res. 1997, 17, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, H.; Steiner, T.; Junker, U.; Knofel, B.; Schlichter, A.; Schubert, J. Serum transforming growth factor-beta 1 in patients with renal cell carcinoma. J. Urol. 1997, 157, 1602–1603. [Google Scholar] [CrossRef]
- Wunderlich, H.; Steiner, T.; Kosmehl, H.; Junker, U.; Reinhold, D.; Reichelt, O.; Zermann, D.H.; Schubert, J. Increased transforming growth factor beta 1 plasma level in patients with renal cell carcinoma: A tumor-specific marker? Urol. Int 1998, 60, 205–207. [Google Scholar] [CrossRef]
- Hegele, A.; Varga, Z.; von Knobloch, R.; Heidenreich, A.; Kropf, J.; Hofmann, R. TGF-beta 1 in patients with renal cell carcinoma. Urol. Res. 2002, 30, 126–129. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, Y.; Kwon, T.; Yoon, J.H.; Kim, K.H.; You, D.; Hong, J.H.; Ahn, H.; Jeong, I.G. Regulatory T cells and TGF-beta 1 in clinically localized renal cell carcinoma: Comparison with age-matched healthy controls. Urol. Oncol.-Semin Orig. 2015, 33. [Google Scholar] [CrossRef]
- Gomella, L.G.; Sargent, E.R.; Linehan, W.M.; Kasid, A. Transforming Growth Factor-Beta Inhibits the Growth of Renal-Cell Carcinoma Invitro. J. Urol. 1989, 141, 1240–1244. [Google Scholar] [CrossRef]
- Cardillo, M.R.; Petrangeli, E.; Ravenna, L.; Salvatori, L.; Di Silverio, F. Transforming growth factor-beta expression in human testicular neoplasms. Anal. Quant. Cytol. Histol. 1998, 20, 461–469. [Google Scholar]
- Eder, I.E.; Stenzl, A.; Hobisch, A.; Cronauer, M.V.; Bartsch, G.; Klocker, H. Expression of transforming growth factors beta-1, beta 2 and beta 3 in human bladder carcinomas. Brit. J. Cancer 1997, 75, 1753–1760. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Shariat, S.F.; Kim, I.Y.; Menesses-Diaz, A.; Tokunaga, H.; Wheeler, T.M.; Lerner, S.P. Predictive value of expression of transforming growth factor-beta(1) and its receptors in transitional cell carcinoma of the urinary bladder. Cancer 2001, 92, 1475–1483. [Google Scholar] [CrossRef]
- Shariat, S.F.; Kim, J.H.; Andrews, B.; Kattan, M.W.; Wheeler, T.M.; Kim, I.Y.; Lerner, S.P.; Slawin, K.M. Preoperative plasma levels of Transforming growth factor beta(1) strongly predict clinical outcome in patients with bladder carcinoma. Cancer 2001, 92, 2985–2992. [Google Scholar] [CrossRef]
- Eissa, S.; Salem, A.M.; Zohny, S.F.; Hegazy, M.G. The diagnostic efficacy of urinary TGF-beta1 and VEGF in bladder cancer: Comparison with voided urine cytology. Cancer Biomark 2007, 3, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Stojnev, S.; Krstic, M.; Kokoris, J.C.; Conic, I.; Petkovic, I.; Ilic, S.; Milosevic-Stevanovic, J.; Velickovic, L.J. y Prognostic Impact of Canonical TGF-beta Signaling in Urothelial Bladder Cancer. Med. -Lith. 2019, 55. [Google Scholar] [CrossRef] [Green Version]
- Eder, I.E.; Stenzl, A.; Hobisch, A.; Cronauer, M.V.; Bartsch, G.; Klocker, H. Transforming growth factors-beta 1 and beta 2 in serum and urine from patients with bladder carcinoma. J. Urol. 1996, 156, 953–957. [Google Scholar] [CrossRef]
- Choi, Y.D.; Cho, N.H.; Ahn, H.S.; Cho, K.S.; Cho, S.Y.; Yang, W.J. Matrix metalloproteinase expression in the recurrence of superficial low grade bladder transitional cell carcinoma. J. Urol. 2007, 177, 1174–1178. [Google Scholar] [CrossRef]
- Miyamoto, H.; Kubota, Y.; Shuin, T.; Torigoe, S.; Dobashi, Y.; Hosaka, M. Expression of Transforming Growth-Factor-Beta-1 in Human Bladder-Cancer. Cancer 1995, 75, 2565–2570. [Google Scholar] [CrossRef]
- Shaker, O.; Hammam, O.; Wishahi, M.; Roshdi, M. TGF-B1 pathway as biological marker of bladder carcinoma schistosomal and non-schistosomal. Urol. Oncol. 2013, 31, 372–378. [Google Scholar] [CrossRef]
- Baharlou, R.; Ahmadi Vasmehjani, A.; Dehghani, A.; Ghobadifar, M.A.; Khoubyari, M. Reduced interleukin-17 and transforming growth factor Beta levels in peripheral blood as indicators for following the course of bladder cancer. Immune Netw. 2014, 14, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Merz, V.W.; Arnold, A.M.; Studer, U.E. Differential Expression of Transforming Growth-Factor-Beta-1 and Beta-3 as Well as C-Fos Messenger-Rna in Normal Human Prostate, Benign Prostatic Hyperplasia and Prostatic-Cancer. World J. Urol. 1994, 12, 96–98. [Google Scholar] [CrossRef]
- Eastham, J.A.; Truong, L.D.; Rogers, E.; Kattan, M.; Flanders, K.C.; Scardino, P.T.; Thompson, T.C. Transforming Growth-Factor-Beta-1-Comparative Immunohistochemical Localization in Human Primary and Metastatic Prostate-Cancer. Lab. Investig. 1995, 73, 628–635. [Google Scholar]
- Tu, H.C.; Jacobs, S.C.; Borkowski, A.; Kyprianou, N. Incidence of apoptosis and cell proliferation in prostate cancer: Relationship with TGF-beta(1) and bcl-2 expression. Int. J. Cancer 1996, 69, 357–363. [Google Scholar] [CrossRef]
- Perry, K.T.; Anthony, C.T.; Steiner, M.S. Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in normal and malignant human prostate. Prostate 1997, 33, 133–140. [Google Scholar] [CrossRef]
- dos Reis, S.T.; Pontes, J.; Antunes, A.A.; de Sousa-Canavez, J.M.; Abe, D.K.; da Cruz, J.A.S.; Dall'Oglio, M.F.; Crippa, A.; Passerotti, C.C.; Ribeiro, L.A.; et al. Tgf-beta 1 expression as a biomarker of poor prognosis in prostate cancer. Clinics 2011, 66, 1143–1147. [Google Scholar] [CrossRef]
- Liu, G.L.; Yang, H.J.; Liu, T.; Lin, Y.Z. Expression and significance of E-cadherin, N-cadherin, transforming growth factor-beta1 and Twist in prostate cancer. Asian. Pac. J. Trop Med. 2014, 7, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Kakehi, Y.; Oka, H.; Mitsumori, K.; Itoh, N.; Ogawa, O.; Yoshida, O. Elevation of serum transforming growth factor-β1 Level in patients with metastatic prostate cancer. Urol. Oncol. 1996, 2, 131–135. [Google Scholar] [CrossRef]
- Mitropoulos, D.; Kiroudi, A.; Christelli, E.; Serafetinidis, E.; Zervas, A.; Anastasiou, I.; Dimopoulos, C. Expression of transforming growth factor beta in renal cell carcinoma and matched non-involved renal tissue. Urol. Res. 2004, 32, 317–322. [Google Scholar] [CrossRef]
- Zheng, J.B.; Mei, Y.H.; Xiang, P.; Zhai, G.S.; Zhao, N.; Xu, C.B.; Liu, M.; Pan, Z.S.; Tang, K.; Jia, D.S. DNA methylation affects metastasis of renal cancer and is associated with TGF-beta/RUNX3 inhibition. Cancer Cell Int. 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Sjolund, J.; Bostrom, A.K.; Lindgren, D.; Manna, S.; Moustakas, A.; Ljungberg, B.; Johansson, M.; Fredlund, E.; Axelson, H. The Notch and TGF-beta Signaling Pathways Contribute to the Aggressiveness of Clear Cell Renal Cell Carcinoma. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Sitaram, R.T.; Mallikarjuna, P.; Landstrom, M.; Ljungberg, B. Transforming growth factor-beta promotes aggressiveness and invasion of clear cell renal cell carcinoma. Oncotarget 2016, 7, 35917–35931. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Jackson, C.; Costello, B.; Singer, N.; Colligan, B.; Douglass, L.; Pemberton, J.; Deddens, J.; Graff, J.R.; Carter, J.H. An intronic variant of the TGFBR1 gene is associated with carcinomas of the kidney and bladder. Int. J. Cancer 2004, 112, 420–425. [Google Scholar] [CrossRef]
- Miyajima, A.; Asano, T.; Seta, K.; Asano, T.; Kakoi, N.; Hayakawa, M. Loss of expression of transforming growth factor-beta receptor as a prognostic factor in patients with renal cell carcinoma. Urology 2003, 61, 1072–1077. [Google Scholar] [CrossRef]
- Copland, J.A.; Luxon, B.A.; Ajani, L.; Maity, T.; Campagnaro, E.; Guo, H.P.; LeGrand, S.N.; Tamboli, P.; Wood, C.G. Genomic profiling identifies alterations in TGF beta signaling through loss of TGF beta receptor expression in human renal cell carcinogenesis and progression. Oncogene 2003, 22, 8053–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.S.; Lohse, C.M.; Wu, K.; Kreinest, P.; Copland, J.A.; Hilton, T.; Wehle, M.; Cheville, J.C.; Blute, M. Lower expression levels of the transforming growth factor beta receptor type II protein are associated with a less aggressive tumor phenotype and improved survival among patients with clear cell renal cell carcinoma. Hum. Pathol 2007, 38, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, S.L.; Doucet, M.; Brady, K.; Weber, K.L. TGF-beta promotes the establishment of renal cell carcinoma bone metastasis. J. Bone Miner. Res. 2007, 22, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Ananth, S.; Knebelmann, B.; Gruning, W.; Dhanabal, M.; Walz, G.; Stillman, I.E.; Sukhatme, V.P. Transforming growth factor beta 1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res. 1999, 59, 2210–2216. [Google Scholar] [PubMed]
- Nishida, J.; Miyazono, K.; Ehata, S. Decreased TGFBR3/betaglycan expression enhances the metastatic abilities of renal cell carcinoma cells through TGF-beta-dependent and-independent mechanisms. Oncogene 2018, 37, 2197–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Lee, C.; Suh, J.H.; Chae, J.Y.; Moon, K.C. Nuclear expression of Smad proteins and its prognostic significance in clear cell renal cell carcinoma. Hum. Pathol 2013, 44, 2047–2054. [Google Scholar] [CrossRef]
- Shang, D.H.; Liu, Y.T.; Yang, P.Q.; Chen, Y.Q.; Tian, Y. TGFBI-promoted Adhesion, Migration and Invasion of Human Renal Cell Carcinoma Depends on Inactivation of von Hippel-Lindau Tumor Suppressor. Urology 2012, 79. [Google Scholar] [CrossRef]
- Bostrom, A.K.; Lindgren, D.; Johansson, M.E.; Axelson, H. Effects of TGF-beta signaling in clear cell renal cell carcinoma cells. Biochem. Bioph. Res. Co. 2013, 435, 126–133. [Google Scholar] [CrossRef]
- Yagasaki, H.; Kawata, N.; Takimoto, Y.; Nemoto, N. Histopathological analysis of angiogenic factors in renal cell carcinoma. Int. J. Urol. 2003, 10, 220–227. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross Talk among TGF-beta Signaling Pathways, Integrins, and the Extracellular Matrix. Csh. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Feldkoren, B.; Hutchinson, R.; Rapaport, Y.; Mahajan, A.; Margulis, V. Integrin signaling potentiates transforming growth factor-beta 1 (TGF-beta 1) dependent down-regulation of E-Cadherin expression - Important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp. Cell Res. 2017, 355, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cen, S.; Kang, X.L.; Wang, W.F.; Wang, Y.; Chen, X. TGF-beta 1-induced Fascin1 promotes cell invasion and metastasis of human 786-0 renal carcinoma cells. Acta Histochem. 2016, 118, 144–151. [Google Scholar] [CrossRef]
- Zhang, N.W.; Bi, X.J.; Zeng, Y.; Zhu, Y.Y.; Zhang, Z.; Liu, Y.; Wang, J.F.; Li, X.J.; Bi, J.B.; Kong, C.Z. TGF-beta 1 promotes the migration and invasion of bladder carcinoma cells by increasing fascin1 expression. Oncol. Rep. 2016, 36, 977–983. [Google Scholar] [CrossRef]
- Zekri, J.; Ahmed, N.; Coleman, R.E.; Hancock, B.W. The skeletal metastatic complications of renal cell carcinoma. Int. J. Oncol. 2001, 19, 379–382. [Google Scholar] [CrossRef]
- Kominsky, S.L.; Doucet, M.; Thorpe, M.; Weber, K.L. MMP-13 is over-expressed in renal cell carcinoma bone metastasis and is induced by TGF-beta 1. Clin. Exp. Metastasis 2008, 25, 865–870. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Deng, C.Z.; Li, Z.S.; Chen, J.P.; Yao, K.; Huang, K.B.; Liu, T.Y.; Liu, Z.W.; Qin, Z.K.; Zhou, F.J.; et al. Molecular characterization and integrative genomic analysis of a panel of newly established penile cancer cell lines. Cell Death. Dis. 2018, 9, 684. [Google Scholar] [CrossRef] [Green Version]
- Gnessi, L.; Fabbri, A.; Spera, G. Gonadal peptides as mediators of development and functional control of the testis: An integrated system with hormones and local environment. Endocr. Rev. 1997, 18, 541–609. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Galuska, S.P.; Kudipudi, P.K.; Riaz, M.A.; Loveland, K.L.; Konrad, L. Signaling by TGF-betas in tubule cultures of adult rat testis. Am. J. Transl. Res. 2017, 9, 1173–1182. [Google Scholar] [PubMed]
- Memon, M.A.; Anway, M.D.; Covert, T.R.; Uzumcu, M.; Skinner, M.K. Transforming growth factor beta (TGFbeta1, TGFbeta2 and TGFbeta3) null-mutant phenotypes in embryonic gonadal development. Mol. Cell Endocrinol. 2008, 294, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, B.; Itman, C.; Mendis, S.H.; Loveland, K.L. Activins and inhibins in mammalian testis development: New models, new insights. Mol. Cell Endocrinol. 2012, 359, 66–77. [Google Scholar] [CrossRef]
- Spiller, C.; Burnet, G.; Bowles, J. Regulation of fetal male germ cell development by members of the TGFbeta superfamily. Stem. Cell Res. 2017, 24, 174–180. [Google Scholar] [CrossRef]
- Devouassoux-Shisheboran, M.; Mauduit, C.; Tabone, E.; Droz, J.P.; Benahmed, M. Growth regulatory factors and signalling proteins in testicular germ cell tumours. APMIS 2003, 111, 212–224. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Su, J.G.; Hsueh, A.J.; Bradley, A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 1992, 360, 313–319. [Google Scholar] [CrossRef]
- Cobellis, L.; Cataldi, P.; Reis, F.M.; De Palo, G.; Raspagliesi, F.; Pilotti, S.; Arcuri, F.; Petraglia, F. Gonadal malignant germ cell tumors express immunoreactive inhibin/activin subunits. Eur. J. Endocrinol. 2001, 145, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Young, J.C.; Jaiprakash, A.; Mithraprabhu, S.; Itman, C.; Kitazawa, R.; Looijenga, L.H.; Loveland, K.L. TCam-2 seminoma cell line exhibits characteristic foetal germ cell responses to TGF-beta ligands and retinoic acid. Int. J. Androl. 2011, 34, e204–e217. [Google Scholar] [CrossRef]
- Fustino, N.; Rakheja, D.; Ateek, C.S.; Neumann, J.C.; Amatruda, J.F. Bone morphogenetic protein signalling activity distinguishes histological subsets of paediatric germ cell tumours. Int. J. Androl. 2011, 34, E218–E233. [Google Scholar] [CrossRef] [Green Version]
- Nettersheim, D.; Gillis, A.J.; Looijenga, L.H.; Schorle, H. TGF-beta1, EGF and FGF4 synergistically induce differentiation of the seminoma cell line TCam-2 into a cell type resembling mixed non-seminoma. Int. J. Androl. 2011, 34, e189–e203. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Schorle, H. The plasticity of germ cell cancers and its dependence on the cellular microenvironment. J. Cell. Mol. Med. 2017, 21, 1463–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, D.J.; Heathcote, K.; Chiano, M.; Snieder, H.; Kemp, P.R.; Metcalfe, J.C.; Carter, N.D.; Spector, T.D. Genetic control of the circulating concentration of transforming growth factor type beta1. Hum. Mol. Genet. 1999, 8, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouras, M.; Tabone, E.; Bertholon, J.; Sommer, P.; Bouvier, R.; Droz, J.P.; Benahmed, M. A novel SMAD4 gene mutation in seminoma germ cell tumors. Cancer Res. 2000, 60, 922–928. [Google Scholar] [PubMed]
- Szarek, M.; Bergmann, M.; Konrad, L.; Schuppe, H.C.; Kliesch, S.; Hedger, M.P.; Loveland, K.L. Activin A target genes are differentially expressed between normal and neoplastic adult human testes: Clues to gonocyte fate choice. Andrology 2019, 7, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, K.A.; Pooja, S.; Sankhwar, S.N.; Sankhwar, P.L.; Goel, A.; Rajender, S. c. 29C > T polymorphism in the transforming growth factor-beta 1 (TGFB1) gene correlates with increased risk of urinary bladder cancer. Cytokine 2015, 75, 344–348. [Google Scholar] [CrossRef]
- Martelossi Cebinelli, G.C.; Paiva Trugilo, K.; Badaro Garcia, S.; Brajao de Oliveira, K. TGF-beta1 functional polymorphisms: A review. Eur. Cytokine Netw. 2016, 27, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.S.; Pan, Y.; Li, W.H.; Zhang, Y.; Li, J.; Ma, B.A. Int7G24A variant of transforming growth factor-beta receptor 1 is associated with osteosarcoma susceptibility in a Chinese population. Med. Oncol. 2011, 28, 622–625. [Google Scholar] [CrossRef]
- Castillejo, A.; Mata-Balaguer, T.; Guarinos, C.; Castillejo, M.I.; Martinez-Canto, A.; Barbera, V.M.; Montenegro, P.; Ochoa, E.; Lazaro, R.; Guillen-Ponce, C.; et al. The Int7G24A variant of transforming growth factor-beta receptor type I is a risk factor for colorectal cancer in the male Spanish population: A case-control study. BMC Cancer 2009, 9, 406. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tong, Y.; Wei, X.; Zhao, Q.; Pan, X.; Yu, G.; Lu, Q. Association between Int7G24A rs334354 polymorphism and cancer risk: A meta-analysis of case-control studies. Sci. Rep. 2015, 5, 11350. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, H.; Lee, D.H.; Kim, I.Y.; Wheeler, T.M.; Lerner, S.P. Decreased expression of transforming growth factor beta receptor type I is associated with poor prognosis in bladder transitional cell carcinoma patients. Clin. Cancer Res. 1999, 5, 2520–2525. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Yang, S.C.; Hong, S.J.; Chung, B.H.; Chung, H.J.; Tokunaga, H.; Kim, I.Y.; Song, Y.S.; Lerner, S.P.; Morton, R.A. The loss of expression of transforming growth factor-beta receptors correlates with the histopathologic tumor grade in bladder transitional cell carcinoma patients. Yonsei Med. J. 1999, 40, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarvey, T.W.; Tait, E.; Tomaszewski, J.E.; Malkowicz, S.B. Expression of transforming growth factor-beta receptors and related cell-cycle components in transitional-cell carcinoma of the bladder. Mol. Urol. 1999, 3, 371–379. [Google Scholar] [PubMed]
- Liu, X.L.; Xue, B.X.; Lei, Z.; Yang, D.R.; Zhang, Q.C.; Shan, Y.X.; Zhang, H.T. TGFBR3 Co-Downregulated With GATA3 Is Associated With Methylation of the GATA3 Gene in Bladder Urothelial Carcinoma. Anat. Rec. 2013, 296, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Al-Azayzih, A.; Gao, F.; Goc, A.; Somanath, P.R. TGF beta 1 induces apoptosis in invasive prostate cancer and bladder cancer cells via Akt-independent, p38 MAPK and JNK/SAPK-mediated activation of caspases. Biochem. Bioph. Res. Co. 2012, 427, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, H.; Azuma, M.; Kameyama, S.; Li, N.; Oyasu, R. Effect of Epidermal Growth-Factor Transforming Growth Factor-Alpha and Transforming Growth Factor-Beta-1 on Growth-Invitro of Rat Urinary-Bladder Carcinoma-Cells. Cell Growth Differ. 1992, 3, 819–825. [Google Scholar]
- Kawamata, H.; Kameyama, S.; Li, N.; Kawai, K.; Oyasu, R. Effect of Epidermal Growth-Factor and Transforming Growth-Factor-Beta-1 on Growth and Invasive Potentials of Newly Established Rat Bladder-Carcinoma Cell-Lines. Int. J. Cancer 1993, 55, 968–973. [Google Scholar] [CrossRef]
- Boyer, B.; Thiery, J.P. Cyclic-Amp Distinguishes between 2 Functions of Acidic Fgf in a Rat Bladder-Carcinoma Cell-Line. J. Cell Biol. 1993, 120, 767–776. [Google Scholar] [CrossRef]
- Okamoto, M.; Oyasu, R. Overexpression of transforming growth factor beta type I receptor abolishes malignant phenotype of a rat bladder carcinoma cell line. Cell Growth Differ. 1997, 8, 921–926. [Google Scholar]
- Hattori, K.; Okamoto, M.; Oyasu, R. Transforming growth factor beta type I receptor acts as a potent tumor suppressor in rat bladder carcinoma. Carcinogenesis 1997, 18, 1867–1870. [Google Scholar] [CrossRef]
- Wang, X.Y.; Colby, J.K.L.; Rengel, R.C.; Fischer, S.M.; Clinton, S.K.; Klein, R.D. Overexpression of Cyclooxygenase-2 (COX-2) in the Mouse Urinary Bladder Induces the Expression of Immune- and Cell Proliferation-Related Genes. Mol. Carcinog. 2009, 48, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensel, J.; Duex, J.E.; Owens, C.; Dancik, G.M.; Edwards, M.G.; Frierson, H.F.; Theodorescu, D. Patient Mutation Directed shRNA Screen Uncovers Novel Bladder Tumor Growth Suppressors. Mol. Cancer Res. 2015, 13, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.B.; Chen, F.; Sima, N. Focal adhesion kinases crucially regulate TGFw beta-induced migration and invasion of bladder cancer cells via Src kinase and E-cadherin. Oncotargets Ther. 2017, 10, 1783–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.C.; He, S.M.; Zhan, Y.H.; He, A.B.; Fang, D.; Gong, Y.Q.; Li, X.S.; Zhou, L.Q. TGF-beta-induced transgelin promotes bladder cancer metastasis by regulating epithelial-mesenchymal transition and invadopodia formation. Ebiomedicine 2019, 47, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Hau, A.M.; Al-Ahmadie, H.A.; Harwalkar, J.; Shoskes, A.C.; Elson, P.; Beach, J.R.; Hussey, G.S.; Schiemann, W.P.; Egelhoff, T.T.; et al. Transforming Growth Factor-beta Is an Upstream Regulator of Mammalian Target of Rapamycin Complex 2-Dependent Bladder Cancer Cell Migration and Invasion. Am. J. Pathol. 2016, 186, 1351–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, S.S.; Mokhtari, R.B.; Noman, A.S.; Uddin, M.; Rahman, M.Z.; Azadi, M.A.; Zlotta, A.; van der Kwast, T.; Yeger, H.; Farhat, W.A. Sonic hedgehog (Shh) signaling promotes tumorigenicity and stemness via activation of epithelial-to-mesenchymal transition (EMT) in bladder cancer. Mol. Carcinog. 2016, 55, 537–551. [Google Scholar] [CrossRef]
- Li, Y.B.; Yang, K.; Mao, Q.Q.; Zheng, X.Y.; Kong, D.B.; Xie, L.P. Inhibition of TGF-beta receptor I by siRNA suppresses the motility and invasiveness of T24 bladder cancer cells via modulation of integrins and matrix metalloproteinase. Int. Urol. Nephrol. 2010, 42, 315–323. [Google Scholar] [CrossRef]
- Geng, J.; Fan, J.; Ouyang, Q.; Zhang, X.P.; Zhang, X.L.; Yu, J.; Xu, Z.D.; Li, Q.Y.; Yao, X.D.; Liu, X.P.; et al. Loss of PPM1A expression enhances invasion and the epithelial-to-mesenchymal transition in bladder cancer by activating the TGF-beta/Smad signaling pathway. Oncotarget 2014, 5, 5700–5711. [Google Scholar] [CrossRef] [Green Version]
- Brito, R.B.O.; Malta, C.S.; Souza, D.M.; Matheus, L.H.G.; Matos, Y.S.T.; Silva, C.S.; Ferreira, J.M.; Nunes, V.S.; Franca, C.M.; Delle, H. 1-Methyl-D-Tryptophan Potentiates TGF-beta-Induced Epithelial-Mesenchymal Transition in T24 Human Bladder Cancer Cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Zhuang, J.L.; Lu, Q.; Shen, B.; Huang, X.J.; Shen, L.; Zheng, X.; Huang, R.M.; Yan, J.; Guo, H.Q. TGF beta 1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.Y.; Zhang, D.G.; Zhang, E.C.; Xu, D.L.; Liu, Z.H.; Qiu, J.X.; Fan, Y.; Shen, B. SENP2 suppresses epithelial-mesenchymal transition of bladder cancer cells through deSUMOylation of TGF-RI. Mol. Carcinog. 2017, 56, 2332–2341. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Men, Q.L.; Li, Y.W.; Li, H.C.; Chong, T.; Li, Z.L. Silencing of Armadillo Repeat-Containing Protein 8 (ARMc8) Inhibits TGF-beta-Induced EMT in Bladder Carcinoma UMUC3 Cells. Oncol. Res. 2017, 25, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Shen, B.; Tan, M.Y.; Mu, X.Y.; Qin, Y.; Zhang, F.; Liu, Y. TGF-beta-Induced Upregulation of malat1 Promotes Bladder Cancer Metastasis by Associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Li, X.; Tan, Z.Q.; Lu, W.; Yang, G.L.; Guan, F. Alteration of N-glycans and Expression of Their Related Glycogenes in the Epithelial-Mesenchymal Transition of HCV29 Bladder Epithelial Cells. Molecules 2014, 19, 20073–20090. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Huang, R.Y.; Li, H.J.; Wang, B.; Chen, Y.F.; Chen, S.W.; Ou, K.F.; Wang, X.T. Secreted TGF-beta-induced protein promotes aggressive progression in bladder cancer cells. Cancer Manag. Res. 2019, 11, 6995–7006. [Google Scholar] [CrossRef] [Green Version]
- Romanenko, A.; Morimura, K.; Kinoshita, A.; Wanibuchi, H.; Vozianov, A.; Fukushima, S. Aberrant expression of E-cadherin and beta-catenin in association with transforming growth factor-beta 1 in urinary bladder lesions in humans after the Chernobyl accident. Cancer Sci. 2006, 97, 45–50. [Google Scholar] [CrossRef]
- Wei, J.H.; Cao, J.Z.; Zhang, D.; Liao, B.; Zhong, W.M.; Lu, J.; Zhao, H.W.; Zhang, J.X.; Tong, Z.T.; Fan, S.; et al. EIF5A2 predicts outcome in localised invasive bladder cancer and promotes bladder cancer cell aggressiveness in vitro and in vivo. Brit. J. Cancer 2014, 110, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.T.; Yu, C.C.; Cai, L.; Lu, Y.Y.; Jiang, L.; Liu, C.; Li, Y.W.; Feng, F.; Gao, Z.L.; Zhu, Z.; et al. Effects of increased Kindlin-2 expression in bladder cancer stromal fibroblasts. Oncotarget 2017, 8, 50692–50703. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.J.; Liu, G.L.; Liu, B.; Liu, T. GP73 promotes invasion and metastasis of bladder cancer by regulating the epithelial-mesenchymal transition through the TGF-beta 1/Smad2 signalling pathway. J. Cell. Mol. Med. 2018, 22, 1650–1665. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, G.; Wang, K.; Mu, Z.Y.; Xie, Q.P.; Qu, H.C.; Lv, H.; Hu, B. Collagen Type VI Alpha 3 Chain Promotes Epithelial-Mesenchymal Transition in Bladder Cancer Cells via Transforming Growth Factor beta (TGF-beta)/Smad Pathway. Med. Sci. Monitor 2018, 24, 5346–5354. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, K.; Miao, C.K.; Xu, A.M.; Zhang, J.Z.; Zhu, J.D.; Su, S.F.; Wang, Z.J. Silencing Trim59 inhibits invasion/migration and epithelial-to-mesenchymal transition via TGF-beta/Smad2/3 signaling pathway in bladder cancer cells. Oncotargets Ther. 2017, 10, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ma, L.; Zhang, N.Z.; Zhu, Y.F.; Zhang, K.Q.; Xu, Z.S.; Wang, Q. Mesenchymal Stem Cells Promote Tumor Progression via Inducing Stroma Remodeling on Rabbit VX2 Bladder Tumor Model. Int. J. Biol. Sci. 2018, 14, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Y.; Chen, J.; Zhu, Y.F.; Xu, Z.S. Mesenchymal Stem Cells Accelerate the Remodeling of Bladder VX2 Tumor Interstitial Microenvironment by TGF beta 1-Smad Pathway. J. Cancer 2019, 10, 4532–4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.T.; Yu, M.T.; Li, C.; Ho, Y.C.; Shen, C.H.; Liu, D.W.; Chang, D.C.; Wu, S.F. Dysfunction of natural killer cells in patients with transitional cell carcinoma. Cancer Lett. 2010, 291, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.L.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF beta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544. [Google Scholar] [CrossRef]
- Liu, Y.N.; Zhang, H.; Zhang, L.; Cai, T.T.; Huang, D.J.; He, J.; Ni, H.H.; Zhou, F.J.; Zhang, X.S.; Li, J. Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: Leading to poor survival in bladder cancer. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGF beta Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Helmy, A.; Hammam, O.A.; El Lithy, T.R.; El Deen Wishahi, M.M. The role of TGF-beta-1 protein and TGF-beta-R-1 receptor in immune escape mechanism in bladder cancer. MedGenMed 2007, 9, 34. [Google Scholar]
- Liang, Y.; Zhu, F.Y.; Zhang, H.J.; Chen, D.M.; Zhang, X.H.; Gao, Q.; Li, Y. Conditional ablation of TGF-beta signaling inhibits tumor progression and invasion in an induced mouse bladder cancer model. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Bian, J.; Li, B.; Zeng, X.Y.; Hu, H.Y.; Hong, Y.; Ouyang, H.; Zhang, X.X.; Wang, Z.H.; Zhu, H.F.; Lei, P.; et al. Mutation of TGF-beta receptor II facilitates human bladder cancer progression through altered TGF-beta 1 signaling pathway. Int. J. Oncol. 2013, 43, 1549–1559. [Google Scholar] [CrossRef]
- Hung, T.T.; Wang, H.; Kingsley, E.A.; Risbridger, G.P.; Russell, P.J. Molecular profiling of bladder cancer: Involvement of the TGF-beta pathway in bladder cancer progression. Cancer Lett. 2008, 265, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Khin, S.S.; Kitazawa, R.; Win, N.; Aye, T.T.; Mori, K.; Kondo, T.; Kitazawa, S. BAMBI gene is epigenetically silenced in subset of high-grade bladder cancer. Int. J. Cancer 2009, 125, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Gou, L.Y.; Liu, M.Y.; Xia, J.; Wan, Q.; Jiang, Y.Y.; Sun, S.L.; Tang, M.; Zhou, L.; He, T.C.; Zhang, Y. BMP9 Promotes the Proliferation and Migration of Bladder Cancer Cells through Up-Regulating lncRNA UCA1. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Ye, L.; Li, H.; Ruge, F.; Yang, Y.; Jiang, W.G. Growth differentiation factor-9 expression is inversely correlated with an aggressive behaviour in human bladder cancer cells. Int. J. Mol. Med. 2012, 29, 428–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsui, K.H.; Hsu, S.Y.; Chung, L.C.; Lin, Y.H.; Feng, T.H.; Lee, T.Y.; Chang, P.L.; Juang, H.H. Growth differentiation factor-15: A p53-and demethylation-upregulating gene represses cell proliferation, invasion, and tumorigenesis in bladder carcinoma cells. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Ahel, J.; Hudorovic, N.; Vicic-Hudorovic, V.; Nikles, H. Tgf-Beta in the Natural History of Prostate Cancer. Acta Clin. Croat. 2019, 58, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol. 2018, 1095, 101–110. [Google Scholar] [CrossRef]
- Loomans, H.A.; Andl, C.D. Intertwining of Activin A and TGF Signaling: Dual Roles in Cancer Progression and Cancer Cell Invasion. Cancers 2015, 7, 70–91. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Jia, Z.Y.; Rahmatpanah, F.; Zhang, Q.; Zi, X.L.; McClelland, M.; Mercola, D. Role of the Adjacent Stroma Cells in Prostate Cancer Development and Progression: Synergy between TGF-beta and IGF Signaling. Biomed. Res. Int. 2014, 10.1155/2014/502093. [Google Scholar] [CrossRef] [Green Version]
- Barrack, E.R. TGF beta in prostate cancer: A growth inhibitor that can enhance tumorigenicity. Prostate 1997, 31, 61–70. [Google Scholar] [CrossRef]
- Wikstrom, P.; Damber, J.E.; Bergh, A. Role of transforming growth factor-beta 1 in prostate cancer. Microsc. Res. Techniq. 2001, 52, 411–419. [Google Scholar] [CrossRef]
- Wikstrom, P.; Stattin, P.; Franck-Lissbrant, I.; Damber, J.E.; Bergh, A. Transforming growth factor beta 1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 1998, 37, 19–29. [Google Scholar] [CrossRef]
- Guo, Y.P.; Jacobs, S.C.; Kyprianou, N. Down-regulation of protein and mRNA expression for transforming growth factor-beta (TGF-beta 1) type I and type II receptors in human prostate cancer. Int. J. Cancer 1997, 71, 573–579. [Google Scholar] [CrossRef]
- Guo, Y.P.; Kyprianou, N. Restoration of transforming growth factor beta signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer Res. 1999, 59, 1366–1371. [Google Scholar] [PubMed]
- Pu, H.; Collazo, J.; Jones, E.; Gayheart, D.; Sakamoto, S.; Vogt, A.; Mitchell, B.; Kyprianou, N. Dysfunctional transforming growth factor-beta receptor II accelerates prostate tumorigenesis in the TRAMP mouse model. Cancer Res. 2009, 69, 7366–7374. [Google Scholar] [CrossRef] [Green Version]
- Brattain, M.G.; Markowitz, S.D.; Willson, J.K. The type II transforming growth factor-beta receptor as a tumor-suppressor gene. Curr. Opin. Oncol. 1996, 8, 49–53. [Google Scholar] [CrossRef]
- Chowdhury, S.; Ammanamanchi, S.; Howell, G.M. Epigenetic Targeting of Transforming Growth Factor β Receptor II and Implications for Cancer Therapy. Mol. Cell Pharm. 2009, 1, 57–70. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, G.Q.; Ma, X.Y.; Xiao, J.; Yu, G.; Yang, C.G.; Xu, N.; Zhang, B.; Zhou, J.; Ye, Z.Q.; et al. Attenuation of TGFBR2 expression and tumour progression in prostate cancer involve diverse hypoxia-regulated pathways. J. Exp. Clin. Canc Res. 2018, 37. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Takahashi, S.; McDonell, N.; Watanabe, N.; Niwa, T.; Hosoya, K.; Tsujino, Y.; Shirai, T.; Ushijima, T. Methylation silencing of transforming growth factor-beta receptor type II in rat prostate cancers. Cancer Res. 2008, 68, 2112–2121. [Google Scholar] [CrossRef] [Green Version]
- Turley, R.S.; Finger, E.C.; Hempel, N.; How, T.; Fields, T.A.; Blobe, G.C. The type III transforming growth factor-beta receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007, 67, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, N.; Hurt, E.M.; Kawasaki, B.T.; Farrar, W.L. TGFBR3 loss and consequences in prostate cancer. Prostate 2007, 67, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Cornelius, S.C.; Pultz, N.J.; Jorgensen, J.S.; Bonham, M.J.; Kim, S.J.; Danielpour, D. The androgen receptor represses transforming growth factor-beta signaling through interaction with Smad3. J. Biol. Chem. 2002, 277, 1240–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Wang, H.; Krebs, T.L.; Kim, S.J.; Danielpour, D. Androgenic control of transforming growth factor-beta signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-beta receptor II. Cancer Res. 2008, 68, 8173–8182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, M.S.; Barrack, E.R. Transforming Growth Factor-Beta-1 Overproduction in Prostate-Cancer - Effects on Growth-Invivo and Invitro. Mol. Endocrinol. 1992, 6, 15–25. [Google Scholar] [CrossRef]
- Zhu, M.L.; Partin, J.V.; Bruckheimer, E.M.; Strup, S.E.; Kyprianou, N. TGF-beta signaling and androgen receptor status determine apoptotic cross-talk in human prostate cancer. Prostate 2008, 68, 287–295. [Google Scholar] [CrossRef]
- Zhu, M.L.; Kyprianou, N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. Faseb. J. 2010, 24, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Kashiwagi, E.; Masubuchi, D.; Eto, M.; Uchiumi, T.; Naito, S. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 2010, 29, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Itsumi, M.; Takeuchi, A.; Imada, K.; Yokomizo, A.; Kuruma, H.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Oda, Y.; et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr-Relat Cancer 2015, 22, 889–900. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Zardan, A.; Takeuchi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Zoubeidi, A.; Gleave, M.E. Clusterin Mediates TGF-beta-Induced Epithelial-Mesenchymal Transition and Metastasis via Twist1 in Prostate Cancer Cells. Cancer Res. 2012, 72, 5261–5272. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Yu, W.; Lv, T.J.; Chang, C.S.; Li, X.; Jin, J. Evidence of TGF-beta 1 mediated epithelial-mesenchymal transition in immortalized benign prostatic hyperplasia cells. Mol. Membr Biol. 2014, 31, 103–110. [Google Scholar] [CrossRef]
- Ao, M.F.; Williams, K.; Bhowmick, N.A.; Hayward, S.W. Transforming growth factor-beta promotes invasion in tumorigenic but not in nontumorigenic human prostatic epithelial cells. Cancer Res. 2006, 66, 8007–8016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Helfand, B.T.; Jang, T.L.; Zhu, L.H.J.; Chen, L.; Yang, X.M.J.; Kozlowski, J.; Smith, N.; Kundu, S.D.; Yang, G.Y.; et al. Nuclear Factor-KB-Mediated Transforming Growth Factor-beta-Induced Expression of Vimentin Is an Independent Predictor of Biochemical Recurrence after Radical Prostatectomy. Clin. Cancer Res. 2009, 15, 3557–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, B.; Zhao, J.S.; Li, Y.L.; Li, H.; Hu, Z.J.; Pan, P.; Zhang, Y.R.; Du, E.; Liu, R.L.; Xu, Y. Elf5 Inhibits TGF-beta-Driven Epithelial-Mesenchymal Transition in Prostate Cancer by Repressing SMAD3 Activation. Prostate 2015, 75, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Park, S.H.; Zhao, J.C.; Fong, K.W.; Li, S.Z.; Lee, Y.; Yang, Y.A.; Sridhar, S.; Lu, X.D.; Abdulkadir, S.A.; et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J. Clin. Investig. 2019, 129, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Buczek, M.E.; Miles, A.K.; Green, W.; Johnson, C.; Boocock, D.J.; Pockley, A.G.; Rees, R.C.; Hulman, G.; van Schalkwyk, G.; Parkinson, R.; et al. Cytoplasmic PML promotes TGF-beta-associated epithelial-mesenchymal transition and invasion in prostate cancer. Oncogene 2016, 35, 3465–3475. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.Y.; Schaar, A.; Sukumaran, P.; Dhasarathy, A.; Singh, B.B. TGF beta-induced epithelial-to-mesenchymal transition in prostate cancer cells is mediated via TRPM7 expression. Mol. Carcinogen 2018, 57, 752–761. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Wang, H.; Wang, H.; Xiao, F.J.; Seth, P.; Xu, W.D.; Jia, Q.H.; Wu, C.; Yang, Y.F.; Wang, L.S. SUMO-Specific Cysteine Protease 1 Promotes Epithelial Mesenchymal Transition of Prostate Cancer Cells via Regulating SMAD4 deSUMOylation. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr-Relat Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef] [Green Version]
- Barcellos-de-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-1. Stem Cells 2016, 34, 2536–2547. [Google Scholar] [CrossRef]
- Wu, C.T.; Chang, Y.H.; Lin, W.Y.; Chen, W.C.; Chen, M.F. TGF Beta1 Expression Correlates with Survival and Tumor Aggressiveness of Prostate Cancer. Ann. Surg. Oncol. 2015, 22, S1587–S1593. [Google Scholar] [CrossRef]
- Darrington, E.; Zhong, M.; Vo, B.H.; Khan, S.A. Vascular endothelial growth factor A, secreted in response to transforming growth factor-beta 1 under hypoxic conditions, induces autocrine effects on migration of prostate cancer cells. Asian J. Androl. 2012, 14, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, K.S.; Kang, M.J.; Lee, J.H.; Ryu, B.K.; Lee, M.G.; Her, N.G.; Ha, T.K.; Han, J.; Kim, Y.K.; Chi, S.G. Opposite functions of HIF-alpha isoforms in VEGF induction by TGF-beta 1 under non-hypoxic conditions. Oncogene 2011, 30, 1213–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.B.; Gupta, J.; Zhang, Z.W.; Gerseny, H.; Berg, A.; Seth, P. Systemic Delivery of Oncolytic Adenoviruses Targeting Transforming Growth Factor Beta Inhibits Established Bone Metastasis in a Prostate Cancer Mouse Model. Mol. Ther. 2012, 20, S118–S119. [Google Scholar]
- Wan, X.H.; Li, Z.G.; Yingling, J.M.; Yang, J.; Starbuck, M.W.; Ravoori, M.K.; Kundra, V.; Vazquez, E.; Navone, N.M. Effect of transforming growth factor beta (TGF-beta) receptor I kinase inhibitor on prostate cancer bone growth. Bone 2012, 50, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Fournier, P.G.J.; Juarez, P.; Jiang, G.L.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.L.; Mohammad, K.S.; et al. The TGF-beta Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.H.; Ren, D.; Yang, Q.; Cui, Y.M.; Guo, W.; Lai, Y.R.; Du, H.; Lin, C.Y.; Li, J.; Song, L.B.; et al. The TGF-beta signalling negative regulator PICK1 represses prostate cancer metastasis to bone. Brit. J. Cancer 2017, 117, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I. MicroRNA Control of TGF-beta Signaling. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Butz, H.; Racz, K.; Hunyady, L.; Patocs, A. Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharm. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef]
- Bowen, T.; Jenkins, R.H.; Fraser, D.J. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J. Pathol. 2013, 229, 274–285. [Google Scholar] [CrossRef]
- Guo, L.L.; Zhang, Y.S.; Zhang, L.F.; Huang, F.B.; Li, J.F.; Wang, S.L. MicroRNAs, TGF-beta signaling, and the inflammatory microenvironment in cancer. Tumor. Biol. 2016, 37, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janakiraman, H.; House, R.P.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H.; Palanisamy, V. The Long (lncRNA) and Short (miRNA) of It: TGF beta-Mediated Control of RNA-Binding Proteins and Noncoding RNAs. Mol. Cancer Res. 2018, 16, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, Y.M.; White, N.M.A.; Grigull, J.; Krizova, A.; Samy, C.; Mejia-Guerrero, S.; Evans, A.; Yousef, G.M. Accurate Molecular Classification of Kidney Cancer Subtypes Using MicroRNA Signature. Eur.Opean Urol. 2011, 59, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Fridman, E.; Dotan, Z.; Barshack, I.; Ben David, M.; Dov, A.; Tabak, S.; Zion, O.; Benjamin, S.; Benjamin, H.; Kuker, H.; et al. Accurate Molecular Classification of Renal Tumors Using MicroRNA Expression. J. Mol. Diagn. 2010, 12, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, R.M.; Costa-Pinheiro, P.; Luis, A.; Antunes, L.; Lobo, F.; Oliveira, J.; Henrique, R.; Jeronimo, C. MicroRNA profile: A promising ancillary tool for accurate renal cell tumour diagnosis. Brit. J. Cancer 2013, 109, 2646–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mytsyk, Y.; Dosenko, V.; Skrzypczyk, M.A.; Borys, Y.; Diychuk, Y.; Kucher, A.; Kowalskyy, V.; Pasichnyk, S.; Mytsyk, O.; Manyuk, L. Potential clinical applications of microRNAs as biomarkers for renal cell carcinoma. Cent. Eur.opean J. Urol. 2018, 71, 295–303. [Google Scholar] [CrossRef]
- Zhang, Q.M.; Di, W.Y.; Dong, Y.Q.; Lu, G.J.; Yu, J.; Li, J.S.; Li, P.F. High serum miR-183 level is associated with poor responsiveness of renal cancer to natural killer cells. Tumor. Biol. 2015, 36, 9245–9249. [Google Scholar] [CrossRef]
- Prior, C.; Perez-Gracia, J.L.; Garcia-Donas, J.; Rodriguez-Antona, C.; Guruceaga, E.; Esteban, E.; Suarez, C.; Castellano, D.; del Alba, A.G.; Lozano, M.D.; et al. Identification of Tissue microRNAs Predictive of Sunitinib Activity in Patients with Metastatic Renal Cell Carcinoma. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Chen, B.H.; Duan, L.J.; Yin, G.M.; Tan, J.; Jiang, X.Z. miR-381, a novel intrinsic WEE1 inhibitor, sensitizes renal cancer cells to 5-FU by up-regulation of Cdc2 activities in 786-O. J. Chemother. 2013, 25, 229–238. [Google Scholar] [CrossRef]
- Ma, Q.; Peng, Z.Q.; Wang, L.; Li, Y.M.; Wang, K.Z.; Zheng, J.F.; Liang, Z.Y.; Liu, T.H. miR-19a correlates with poor prognosis of clear cell renal cell carcinoma patients via promoting cell proliferation and suppressing PTEN/SMAD4 expression. Int. J. Oncol. 2016, 49, 2589–2599. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Li, S.Y.; Zhang, J.; Chen, Y.H.; Ma, J.J.; Kong, W.; Gong, D.K.; Zheng, J.H.; Xue, W.; Xu, Y.F. Sunitinib-suppressed miR-452-5p facilitates renal cancer cell invasion and metastasis through modulating SMAD4/SMAD7 signals. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichner, Z.; Mejia-Guerrero, S.; Ignacak, M.; Krizova, A.; Bao, T.T.; Girgis, A.H.F.; Youssef, Y.M.; Yousef, G.M. Pleiotropic Action of Renal Cell Carcinoma-Dysregulated miRNAs on Hypoxia-Related Signaling Pathways. Am. J. Pathol. 2012, 180, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Jingushi, K.; Ueda, Y.; Kitae, K.; Hase, H.; Egawa, H.; Ohshio, I.; Kawakami, R.; Kashiwagi, Y.; Tsukada, Y.; Kobayashi, T.; et al. miR-629 Targets TRIM33 to Promote TGF beta/Smad Signaling and Metastatic Phenotypes in ccRCC. Mol. Cancer Res. 2015, 13, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senanayake, U.; Das, S.; Vesely, P.; Alzoughbi, W.; Frohlich, L.F.; Chowdhury, P.; Leuschner, I.; Hoefler, G.; Guertl, B. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis 2012, 33, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Liu, J.Z.; Wang, Y.J.; Zhu, X.L.; Fan, Z.C.; Li, C.B.; Yin, H.; Liu, Y. Role of miR-486-5p in regulating renal cell carcinoma cell proliferation and apoptosis via TGF-beta-activated kinase 1. J. Cell Biochem. 2019, 120, 2954–2963. [Google Scholar] [CrossRef]
- Shi, J.; Zhuang, Y.; Liu, X.K.; Zhang, Y.X.; Zhang, Y. TGF-beta induced RBL2 expression in renal cancer cells by down-regulating miR-93. Clin. Transl. Oncol. 2014, 16, 986–992. [Google Scholar] [CrossRef]
- Machackova, T.; Mlcochova, H.; Stanik, M.; Dolezel, J.; Fedorko, M.; Pacik, D.; Poprach, A.; Svoboda, M.; Slaby, O. MiR-429 is linked to metastasis and poor prognosis in renal cell carcinoma by affecting epithelial-mesenchymal transition. Tumor. Biol. 2016, 37, 14653–14658. [Google Scholar] [CrossRef]
- Boguslawska, J.; Rodzik, K.; Poplawski, P.; Kedzierska, H.; Rybicka, B.; Sokol, E.; Tanski, Z.; Piekielko-Witkowska, A. TGF-beta 1 targets a microRNA network that regulates cellular adhesion and migration in renal cancer. Cancer Lett. 2018, 412, 155–169. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, P.F.; Shen, X.D.; Zhang, Y.W.; Xu, B.; Zhou, J.; Fan, S.; Hao, Z.Y.; Shi, H.Q.; Zhang, X.S.; et al. MicroRNA Expression Profile in Penile Cancer Revealed by Next-Generation Small RNA Sequencing. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Kuasne, H.; Barros-Filho, M.C.; Busso-Lopes, A.; Marchi, F.A.; Pinheiro, M.; Munoz, J.J.M.; Scapulatempo-Neto, C.; Faria, E.F.; Guimaraes, G.C.; Lopes, A.; et al. Integrative miRNA and mRNA analysis in penile carcinomas reveals markers and pathways with potential clinical impact. Oncotarget 2017, 8, 15294–15306. [Google Scholar] [CrossRef]
- Marchi, F.A.; Martins, D.C.; Barros, M.C.; Kuasne, H.; Lopes, A.F.B.; Brentani, H.; Trindade, J.C.S.; Guimaraes, G.C.; Faria, E.F.; Scapulatempo-Neto, C.; et al. Multidimensional integrative analysis uncovers driver candidates and biomarkers in penile carcinoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Nappi, L.; Nichols, C. MicroRNAs as Biomarkers for Germ Cell Tumors. Urol. Clin. N. Am. 2019, 46, 449. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.X.; Li, D.; Liu, X.W.; Liu, Z.Y.; Wu, D.B.; Liu, X.M. Screening for miRNAs and their potential targets in response to TGF-beta 1 based on miRNA microarray and comparative proteomics analyses in a mouse GC-1 spg germ cell line. Int. J. Mol. Med. 2015, 35, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chan, E.S.Y.; Kwan, B.C.H.; Li, P.K.T.; Yip, S.K.H.; Szeto, C.C.; Ng, C.F. Expression of microRNAs in the Urine of Patients With Bladder Cancer. Clin. Genitourin Cancer 2012, 10, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.X.; Zhang, N.W.; Yang, J.Y.; Zhu, Y.Y.; Zhang, Z.; Wang, J.F.; Xu, X.L.; Li, Z.L.; Liu, X.K.; Li, Z.H.; et al. Long non-coding RNA ZEB1-AS1 regulates miR-200b/FSCN1 signaling and enhances migration and invasion induced by TGF-beta 1 in bladder cancer cells. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.F.; Zeng, F.; Qi, L.; Zui, X.B.; Wang, J.; Liu, L.F.; Li, Y. Transforming growth factor-beta 1 induces epithelial-mesenchymal transition and increased expression of matrix metalloproteinase-16 via miR-200b downregulation in bladder cancer cells. Mol. Med. Rep. 2014, 10, 1549–1554. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cao, J.; Zhao, X.K. miR-221 facilitates the TGFbeta1-induced epithelial-mesenchymal transition in human bladder cancer cells by targeting STMN1. BMC Urol. 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- van Kampen, J.G.M.; van Hooij, O.; Jansen, C.F.; Smit, F.P.; van Noort, P.I.; Schultz, I.; Schaapveld, R.Q.J.; Schalken, J.A.; Verhaegh, G.W. miRNA-520f Reverses Epithelial-to-Mesenchymal Transition by Targeting ADAM9 and TGFBR2. Cancer Res. 2017, 77, 2008–2017. [Google Scholar] [CrossRef] [Green Version]
- Porkka, K.P.; Pfeiffer, M.J.; Waltering, K.K.; Vessella, R.L.; Tammela, T.L.J.; Visakorpi, T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007, 67, 6130–6135. [Google Scholar] [CrossRef] [Green Version]
- Haj-Ahmad, T.A.; Abdalla, M.A.K.; Haj-Ahmad, Y. Potential Urinary miRNA Biomarker Candidates for the Accurate Detection of Prostate Cancer among Benign Prostatic Hyperplasia Patients. J. Cancer 2014, 5, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Brase, J.C.; Johannes, M.; Schlomm, T.; Falth, M.; Haese, A.; Steuber, T.; Beissbarth, T.; Kuner, R.; Sultmann, H. Circulating miRNAs are correlated with tumor progression in prostate cancer. Int. J. Cancer 2011, 128, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.B.; Xue, L.R.; Ma, A.H.; Tepper, C.G.; Kung, H.J.; White, R.W.D. miR-125b Promotes Growth of Prostate Cancer Xenograft Tumor Through Targeting Pro-Apoptotic Genes. Prostate 2011, 71, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Chen, F.J.; Wang, K.F.; Song, Y.; Fei, X.; Wu, B. miR-15a/miR-16 cluster inhibits invasion of prostate cancer cells by suppressing TGF-beta signaling pathway. Biomed. Pharm. 2018, 104, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.F.; Zhu, J.; Sun, Y.; Fan, K.; Yang, D.R.; Li, G.H.; Yang, G.S.; Chang, C.S. TR4 nuclear receptor increases prostate cancer invasion via decreasing the miR-373-3p expression to alter TGF beta R2/p-Smad3 signals. Oncotarget 2015, 6, 15397–15409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.L.; Sun, B.F.; Huang, L.R.; Yuan, H.B.; Zhang, S.; Chen, J.; Yu, Z.J.; Luo, H. Potent Inhibition of miR-34b on Migration and Invasion in Metastatic Prostate Cancer Cells by Regulating the TGF-beta Pathway. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Tanaka, Y.; Tabatabai, Z.L.; Dahiya, R. Genistein downregulates onco-miR-1260b and upregulates sFRP1 and Smad4 via demethylation and histone modification in prostate cancer cells. Brit. J. Cancer 2014, 110, 1645–1654. [Google Scholar] [CrossRef]
- Li, X.J.; Li, J.; Cai, Y.; Peng, S.B.; Wang, J.; Xiao, Z.M.; Wang, Y.; Tao, Y.R.; Li, J.; Leng, Q.; et al. Hyperglycaemia-induced miR-301a promotes cell proliferation by repressing p21 and Smad4 in prostate cancer. Cancer Lett. 2018, 418, 211–220. [Google Scholar] [CrossRef]
- Lewis, H.; Lance, R.; Troyer, D.; Beydoun, H.; Hadley, M.; Orians, J.; Benzine, T.; Madric, K.; Semmes, O.J.; Drake, R.; et al. miR-888 is an expressed prostatic secretions-derived microRNA that promotes prostate cell growth and migration. Cell Cycle 2014, 13, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Ueno, K.; Hirata, H.; Shahryari, V.; Deng, G.; Tanaka, Y.; Tabatabai, Z.L.; Hinoda, Y.; Dahiya, R. microRNA-183 is an oncogene targeting Dkk-3 and SMAD4 in prostate cancer. Brit. J. Cancer 2013, 108, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Glavich, G.J.; Pahuski, M.; Short, A.; Semmes, O.J.; Yang, L.F.; Galkin, V.; Drake, R.; Esquela-Kerscher, A. Characterization and Evidence of the miR-888 Cluster as a Novel Cancer Network in Prostate. Mol. Cancer Res. 2018, 16, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Ding, Y.Z.; Catalona, W.J.; Yang, X.M.J.; Anderson, W.F.; Jovanovic, B.; Wellman, K.; Killmer, J.; Huang, X.K.; Scheidt, K.A.; et al. MEK4 Function, Genistein Treatment, and Invasion of Human Prostate Cancer Cells. JNCI-J. Natl Cancer Inst. 2009, 101, 1141–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozten-Kandas, N.; Bosland, M.C. Chemoprevention of prostate cancer: Natural compounds, antiandrogens, and antioxidants - In vivo evidence. J. Carcinog 2011, 10, 27. [Google Scholar] [CrossRef]
- Ahmad, A.; Biersack, B.; Li, Y.W.; Bao, B.; Kong, D.J.; Ali, S.; Banerjee, S.; Sarkar, F.H. Perspectives on the Role of Isoflavones in Prostate Cancer. Aaps. J. 2013, 15, 991–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitin, T.; Chen, M.; Moran, B.J.; Dosoretz, D.E.; Katin, M.J.; Braccioforte, M.H.; Salenius, S.; D'Amico, A.V. Diabetes mellitus, race, and the odds of high-grade prostate cancer in men diagnosed with prostate cancer in the United States. J. Clin. Oncol. 2011, 29. [Google Scholar] [CrossRef]
- Moreira, D.M.; Anderson, T.; Gerber, L.; Thomas, J.A.; Banez, L.L.; McKeever, M.G.; Hoyo, C.; Grant, D.; Jayachandran, J.; Freedland, S.J. The association of diabetes mellitus and high-grade prostate cancer in a multiethnic biopsy series. Cancer Cause Control. 2011, 22, 977–983. [Google Scholar] [CrossRef]
- Yang, Y.; Ji, C.W.; Guo, S.H.; Su, X.; Zhao, X.Z.; Zhang, S.W.; Liu, G.X.; Qiu, X.F.; Zhang, Q.; Guo, H.Q.; et al. The miR-486-5p plays a causative role in prostate cancer through negative regulation of multiple tumor suppressor pathways. Oncotarget 2017, 8, 72835–72846. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wa, Q.D.; Pan, J.C.; Peng, X.S.; Ren, D.; Li, Q.J.; Dai, Y.H.; Yang, Q.; Huang, Y.; Zhang, X.; et al. Transcriptional downregulation of miR-133b by REST promotes prostate cancer metastasis to bone via activating TGF-beta signaling. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Tang, Y.B.; Wu, B.W.; Huang, S.; Peng, X.S.; Li, X.; Huang, X.F.; Zhou, W.; Xie, P.G.; He, P.H. Downregulation of miR-505-3p predicts poor bone metastasis-free survival in prostate cancer. Oncol. Rep. 2019, 41, 57–66. [Google Scholar] [CrossRef]
- Sun, B.G.; Fan, Y.Y.; Yang, A.J.; Liang, L.N.; Cao, J.H. MicroRNA-539 functions as a tumour suppressor in prostate cancer via the TGF-beta/Smad4 signalling pathway by down-regulating DLX1. J. Cell. Mol. Med. 2019, 23, 5934–5948. [Google Scholar] [CrossRef] [Green Version]
- Ayub, S.G.; Kaul, D.; Ayub, T. An androgen-regulated miR-2909 modulates TGF beta signalling through AR/miR-2909 axis in prostate cancer. Gene 2017, 631, 1–9. [Google Scholar] [CrossRef]
- Mishra, S.; Deng, J.J.; Gowda, P.S.; Rao, M.K.; Lin, C.L.; Chen, C.L.; Huang, T.; Sun, L.Z. Androgen receptor and microRNA-21 axis downregulates transforming growth factor beta receptor II (TGFBR2) expression in prostate cancer. Oncogene 2014, 33, 4097–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Shimada, K.; Tatsumi, Y.; Tanaka, N.; Fujimoto, K.; Konishi, N. Syndecan-1 up-regulates microRNA-331-3p and mediates epithelial-to-mesenchymal transition in prostate cancer. Mol. Carcinog. 2016, 55, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Wa, Q.D.; Li, L.; Lin, H.C.; Peng, X.S.; Ren, D.; Huang, Y.; He, P.H.; Huang, S. Downregulation of miR-19a-3p promotes invasion, migration and bone metastasis via activating TGF-beta signaling in prostate cancer. Oncol. Rep. 2018, 39, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.W.; Tao, T.; Qi, M.; Wang, L.; Hu, J.; Li, X.J.; Xing, N.D.; Du, R.; Han, B. MicroRNA-132/212 Upregulation Inhibits TGF-beta-Mediated Epithelial-Mesenchymal Transition of Prostate Cancer Cells by Targeting SOX4. Prostate 2016, 76, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.J.; Li, L.; Zhu, G.D.; Dang, Q.; Ma, Z.K.; He, D.L.; Chang, L.K.; Song, W.B.; Chang, H.C.; Krolewski, J.J.; et al. Infiltrated pre-adipocytes increase prostate cancer metastasis via modulation of the miR-301a/androgen receptor (AR)/TGF-beta 1/Smad/MMP9 signals. Oncotarget 2015, 6, 12326–12339. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zou, C.Y.; Tang, Y.B.; Wa, Q.D.; Peng, X.S.; Chen, X.; Yang, C.X.; Ren, D.; Huang, Y.; Liao, Z.W.; et al. miR-582-3p and miR-582-5p Suppress Prostate Cancer Metastasis to Bone by Repressing TGF-beta Signaling. Mol. Ther.-Nucl. Acids 2019, 16, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonci, D.; Coppola, V.; Patrizii, M.; Addario, A.; Cannistraci, A.; Francescangeli, F.; Pecci, R.; Muto, G.; Collura, D.; Bedini, R.; et al. A microRNA code for prostate cancer metastasis. Oncogene 2016, 35, 1180–1192. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56. [Google Scholar] [CrossRef] [Green Version]
- Ottley, E.C.; Nicholson, H.D.; Gold, E.J. Activin A regulates microRNAs and gene expression in LNCaP cells. Prostate 2016, 76, 951–963. [Google Scholar] [CrossRef]
- Siu, M.K.; Tsai, Y.C.; Chang, Y.S.; Yin, J.J.; Suau, F.; Chen, W.Y.; Liu, Y.N. Transforming growth factor-beta promotes prostate bone metastasis through induction of microRNA-96 and activation of the mTOR pathway. Oncogene 2015, 34, 4767–4776. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.N.; Yin, J.J.; Abou-Kheir, W.; Hynes, P.G.; Casey, O.M.; Fang, L.; Yi, M.; Stephens, R.M.; Seng, V.; Sheppard-Tillman, H.; et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene 2013, 32, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slabakova, E.; Pernicova, Z.; Slavickova, E.; Starsichova, A.; Kozubik, A.; Soucek, K. TGF-beta 1-Induced EMT of Non-Transformed Prostate Hyperplasia Cells Is Characterized by Early Induction of SNAI2/Slug. Prostate 2011, 71, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Tzai, T.S.; Lin, C.I.; Shiau, A.L.; Wu, C.L. Antisense oligonucleotide specific for transforming growth factor-beta 1 inhibit both in vitro and in vivo growth of MBT-2 murine bladder cancer. Anticancer Res. 1998, 18, 1585–1589. [Google Scholar] [PubMed]
- Hsin, M.C.; Hsieh, Y.H.; Wang, P.H.; Ko, J.L.; Hsin, I.L.; Yang, S.F. Hispolon suppresses metastasis via autophagic degradation of cathepsin S in cervical cancer cells. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, Y.C.; Park, B. Hispolon from Phellinus linteus induces apoptosis and sensitizes human cancer cells to the tumor necrosis factor-related apoptosis-inducing ligand through upregulation of death receptors. Oncol. Rep. 2016, 35, 1020–1026. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.S.; Lee, S.M.; Lin, C.C.; Liu, C.Y. Hispolon Decreases Melanin Production and Induces Apoptosis in Melanoma Cells through the Downregulation of Tyrosinase and Microphthalmia-Associated Transcription Factor (MITF) Expressions and the Activation of Caspase-3,-8 and-9. Int. J. Mol. Sci. 2014, 15, 1201–1215. [Google Scholar] [CrossRef]
- Hong, D.R.; Park, M.J.; Jang, E.H.; Jung, B.; Kim, N.J.; Kim, J.H. Hispolon as an inhibitor of TGF-beta-induced epithelial-mesenchymal transition in human epithelial cancer cells by co-regulation of TGF-beta-Snail/Twist axis. Oncol. Lett. 2017, 14, 4866–4872. [Google Scholar] [CrossRef] [Green Version]
- Grenga, I.; Donahue, R.N.; Gargulak, M.L.; Lepone, L.M.; Roselli, M.; Bilusic, M.; Schlom, J. Anti-PD-L1/TGFI3R2 (M7824) fusion protein induces immunogenic modulation of human urothelial carcinoma cell lines, rendering them more susceptible to immune-mediated recognition and lysis. Urol. Oncol.-Semin Orig. 2018, 36. [Google Scholar] [CrossRef]
- FDA. FDA Grants Accelerated Approval to Avelumab for Urothelial Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-avelumab-urothelial-carcinoma (accessed on 18 September 2019).
- Ahirwar, D.K.; Agrahari, A.; Mandhani, A.; Mittal, R.D. Cytokine gene polymorphisms are associated with risk of urinary bladder cancer and recurrence after BCG immunotherapy. Biomarkers 2009, 14, 213–218. [Google Scholar] [CrossRef]
- Yuan, X.K.; Zhao, X.K.; Xia, Y.C.; Zhu, X.; Xiao, P. Increased Circulating Immunosuppressive CD14(+)HLA-DR-/low Cells Correlate with Clinical Cancer Stage and Pathological Grade in Patients with Bladder Carcinoma. J. Int. Med. Res. 2011, 39, 1381–1391. [Google Scholar] [CrossRef]
- Liu, X.Q.; Wu, Y.L.; Zhou, Z.T.; Huang, M.C.; Deng, W.; Wang, Y.B.; Zhou, X.C.; Chen, L.Y.; Li, Y.; Zeng, T.; et al. Celecoxib inhibits the epithelial-to-mesenchymal transition in bladder cancer via the miRNA-145/TGFBR2/Smad3 axis. Int. J. Mol. Med. 2019, 44, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.L.; Shen, L.; Yang, L.; Huang, X.J.; Lu, Q.; Cui, Y.Y.; Zheng, X.; Zhao, X.Z.; Zhang, D.Z.; Huang, R.M.; et al. TGF beta 1 Promotes Gemcitabine Resistance through Regulating the LncRNA-LET/NF90/miR-145 Signaling Axis in Bladder Cancer. Theranostics 2017, 7, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, Y.; Jiang, F.; Wang, X.X.; Zhang, J.P.; Shen, J.; Yang, X.J. Inhibition of transforming growth factor beta/SMAD signal by MiR-155 is involved in arsenic trioxide-induced anti-angiogenesis in prostate cancer. Cancer Sci. 2014, 105, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossein, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
TGF-β maturation. The inactive TGF-β precursor dimerizes and the resulting dimer is cleaved by furin endopeptidase resulting in mature TGF-β and the latency-associated peptide (LAP) that bind non-covalently to produce small latent complex. The latter is next bound by latent TGF-β binding protein (LTBP), resulting in a large latent complex.
Figure 1.
TGF-β maturation. The inactive TGF-β precursor dimerizes and the resulting dimer is cleaved by furin endopeptidase resulting in mature TGF-β and the latency-associated peptide (LAP) that bind non-covalently to produce small latent complex. The latter is next bound by latent TGF-β binding protein (LTBP), resulting in a large latent complex.

Figure 2.
MicroRNAs regulating key genes of the TGF-β signaling pathway. MicroRNAs acting in bladder, prostate, and renal cancer are shown in colors (yellow, purple, and red, respectively). The details of miRNA-mediated regulation of the TGF-β signaling pathway are provided in the text
Figure 2.
MicroRNAs regulating key genes of the TGF-β signaling pathway. MicroRNAs acting in bladder, prostate, and renal cancer are shown in colors (yellow, purple, and red, respectively). The details of miRNA-mediated regulation of the TGF-β signaling pathway are provided in the text


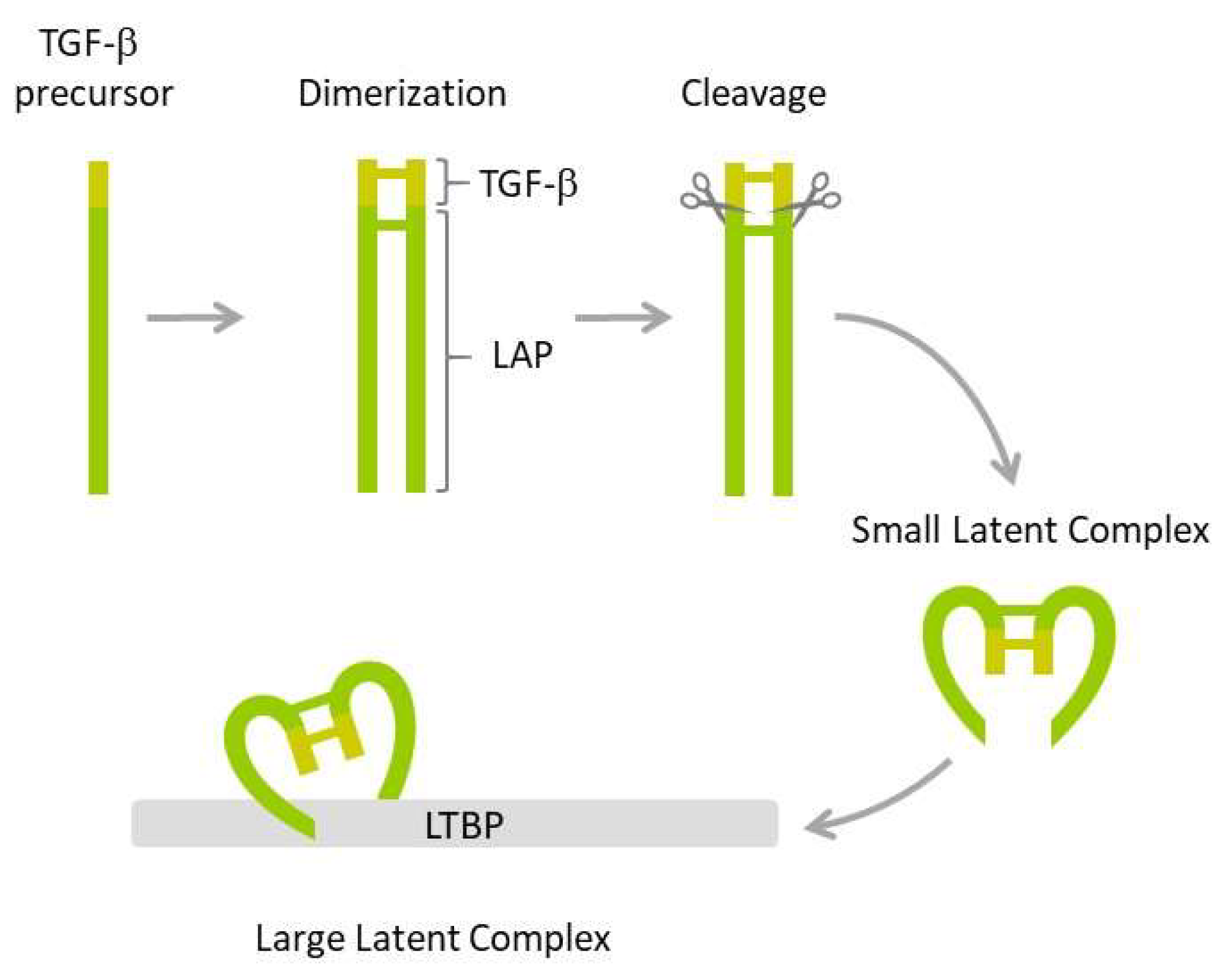{kind=link}
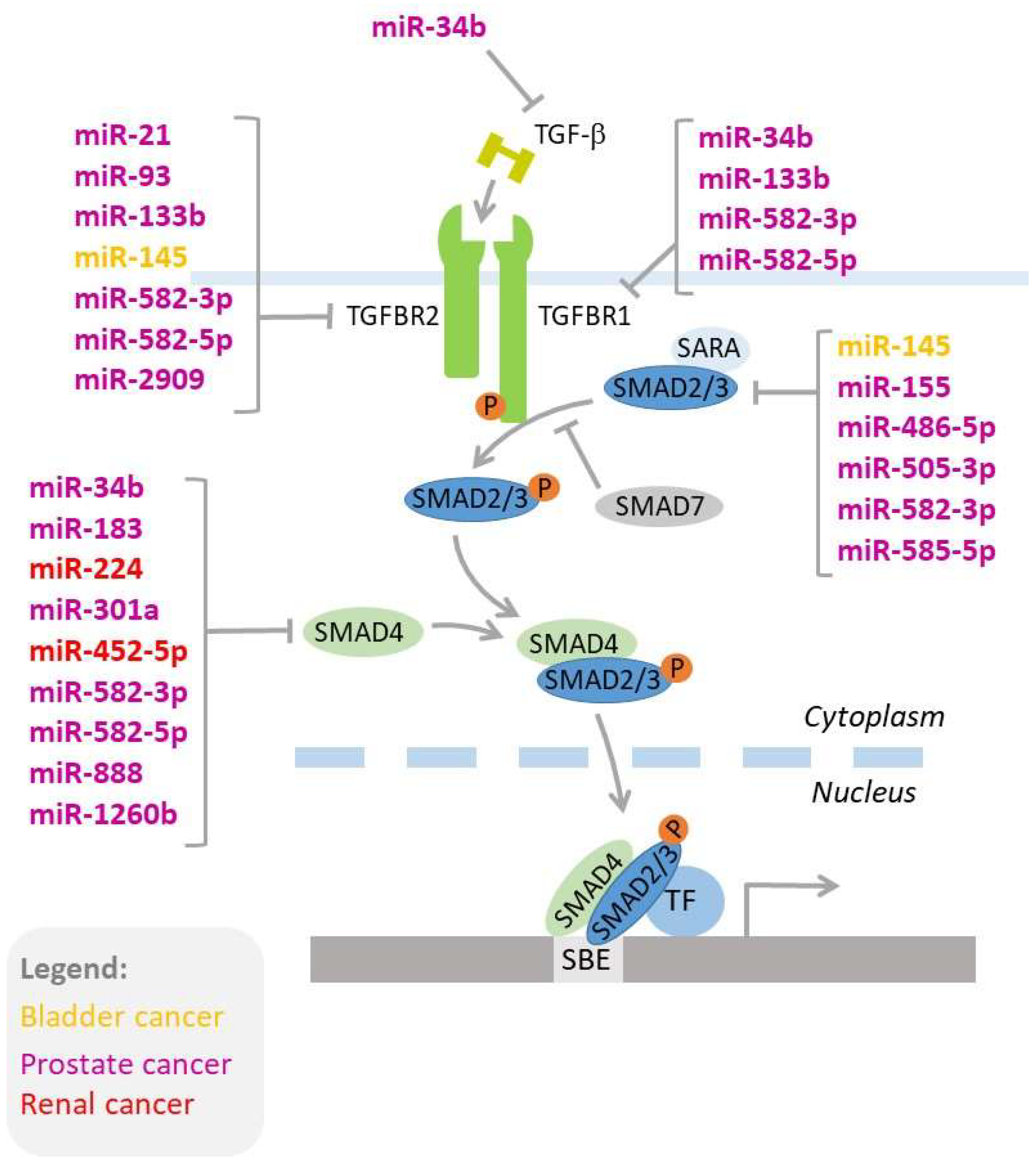{kind=link}
Table 1.
Disturbances of TGF-β1 expression in genitourinary cancers.
| Tumor Type | TGF-β Change (↓/↑) 1 | Ref. |
|---|---|---|
| Renal Cancer | ↑ in serum of RCC patients (vs. healthy donors) | [114] |
| ↑ in plasma of RCC patients (vs. healthy donors) | [115] | |
| ↑ in plasma of metastatic RCC patients (vs. healthy donors) | [116] | |
| ↑ in the peripheral blood of RCC patients (vs. healthy donors) | [117] | |
| ↑ in tumor tissue (vs. normal tissue) | [108] | |
| ↑ in tumor tissue (vs. normal tissue) | [118] | |
| ↑ in tumor tissue (vs. normal tissue) | [110] | |
| Penile Cancer | No data | No data |
| Testicular Cancer | ↑ in tumor tissue (vs. normal tissue) | [119] |
| Bladder Cancer | ↑ in tumor tissue (vs. normal tissue) | [120] |
| ↑ in tumor tissue (vs. normal tissue) | [121] | |
| ↑ in plasma of metastatic patients (vs. healthy donors and vs. patients without metastasis) | [122] | |
| ↑ in urine of bladder cancer patients (vs. healthy donors) | [123] | |
| ↑ in high-grade tumor tissue (vs. low-grade tumors) | [124] | |
| ↑ in serum of patients: i) with invasive tumors (vs. healthy donors); ii) with high-grade tumors (vs. low-grade tumors) | [125] | |
| ↑ in tumor tissue of recurrent patients (v.s non-recurrent patients) | [126] | |
| ↑ in tumor tissue (vs. normal tissue); ↑ in low-grade tumor tissue (vs. high grade); ↑ in superficial BC (vs. invasive BC) | [127] | |
| ↑ in tumor tissue (vs. chronic cystitis) | [128] | |
| ↓ in serum of BC patients (vs. healthy donors) | [129] | |
| Prostate Cancer | ↑ in tumor tissue (vs. normal tissue) | [130] |
| ↑ in tumor tissue (vs. normal tissue) | [131] | |
| ↑ in tumor tissue (vs. normal tissue) | [132] | |
| ↑ in tumor tissue (vs. normal tissue) | [133] | |
| ↑ in tumor tissue (vs. normal tissue) | [134] | |
| ↑ in tumor tissue (vs. normal tissue) | [135] | |
| ↑ in serum of patients with PCa lymph node and/or distant metastases | [136] | |
| ↑ in urine of PCa patients | [133] |
1 ↑ increased expression/↓ decreased expression.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Boguslawska, J.; Kryst, P.; Poletajew, S.; Piekielko-Witkowska, A. TGF-β and microRNA Interplay in Genitourinary Cancers. Cells 2019, 8, 1619. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121619
AMA Style
Boguslawska J, Kryst P, Poletajew S, Piekielko-Witkowska A. TGF-β and microRNA Interplay in Genitourinary Cancers. Cells. 2019; 8(12):1619. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121619
Chicago/Turabian StyleBoguslawska, Joanna, Piotr Kryst, Slawomir Poletajew, and Agnieszka Piekielko-Witkowska. 2019. "TGF-β and microRNA Interplay in Genitourinary Cancers" Cells 8, no. 12: 1619. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121619
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.