Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cultures of Drug-Naïve and Drug-Resistant Cell Lines
2.2. DNA Extraction, Whole-Exome Sequencing (WES) and WES Data Analysis
2.3. Acid Phosphatase Activity (APA) Assay
2.4. Cell Lysates and Western Blotting
2.5. RNA Isolation, cDNA Synthesis, and Quantitative PCR
2.6. Human Phospho-Kinase Array
2.7. Flow Cytometry
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Analysis of AXL Expression Reported in Data Sets from the GEO Database
2.10. Statistical Analysis
3. Results
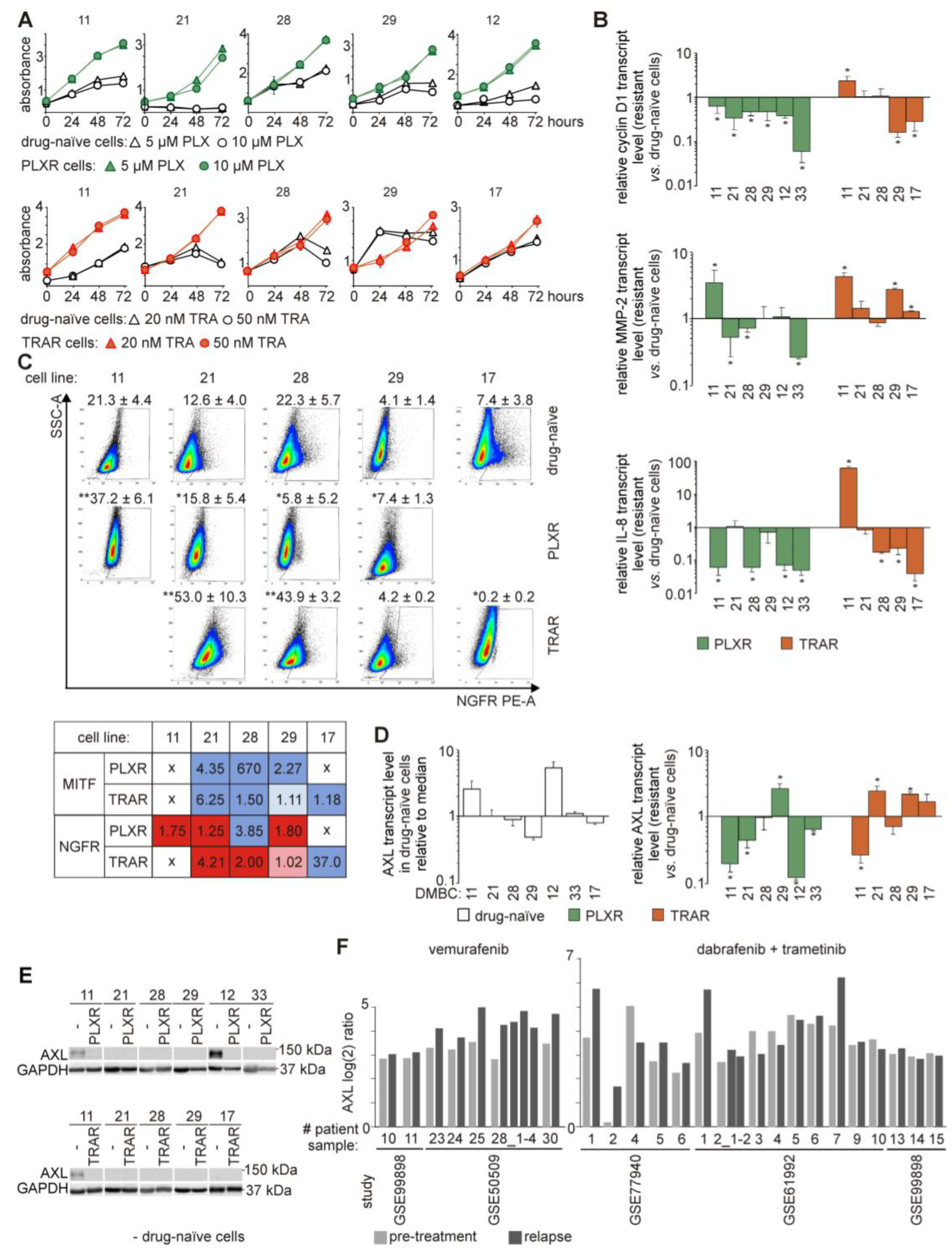
3.1. The Ability of Developing Resistance to Vemurafenib (PLX) or Trametinib (TRA) Differs between Cell Lines Derived from Different Melanoma Specimens
3.2. Drug-Naïve and Drug-Resistant Cells Differ in Transcript Levels of Cyclin D1, MMP-2, and IL-8
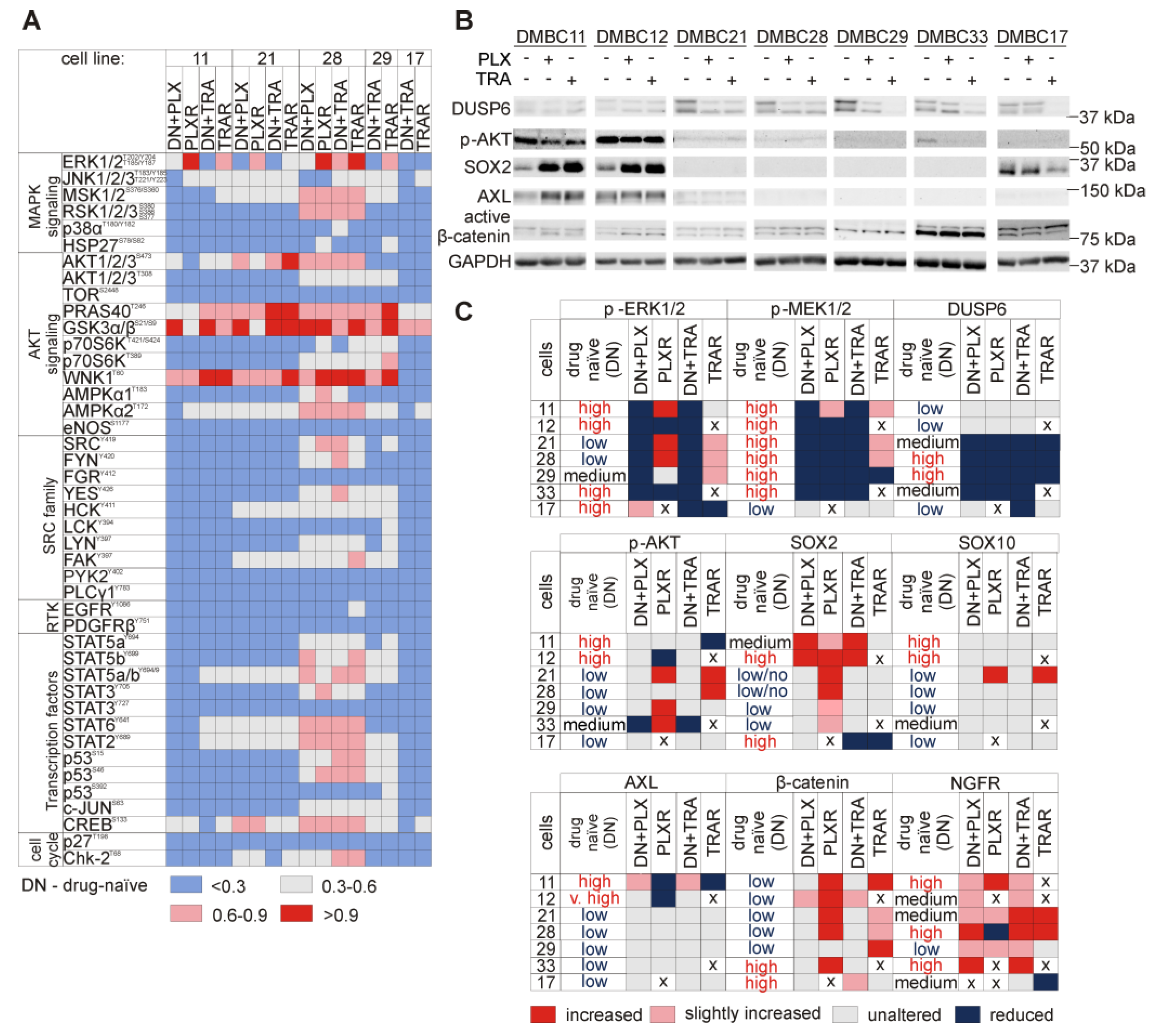
3.3. Increased Frequency of Nerve Growth Factor Receptor (NGFR)-Positive Cells is Accompanied with Suppression of MITF-Dependent Program in the Majority of Melanoma Cell Lines Resistant to Vemurafenib or Trametinib
3.4. AXL is not an Unambiguous Marker of Resistance to Targeted Therapeutics
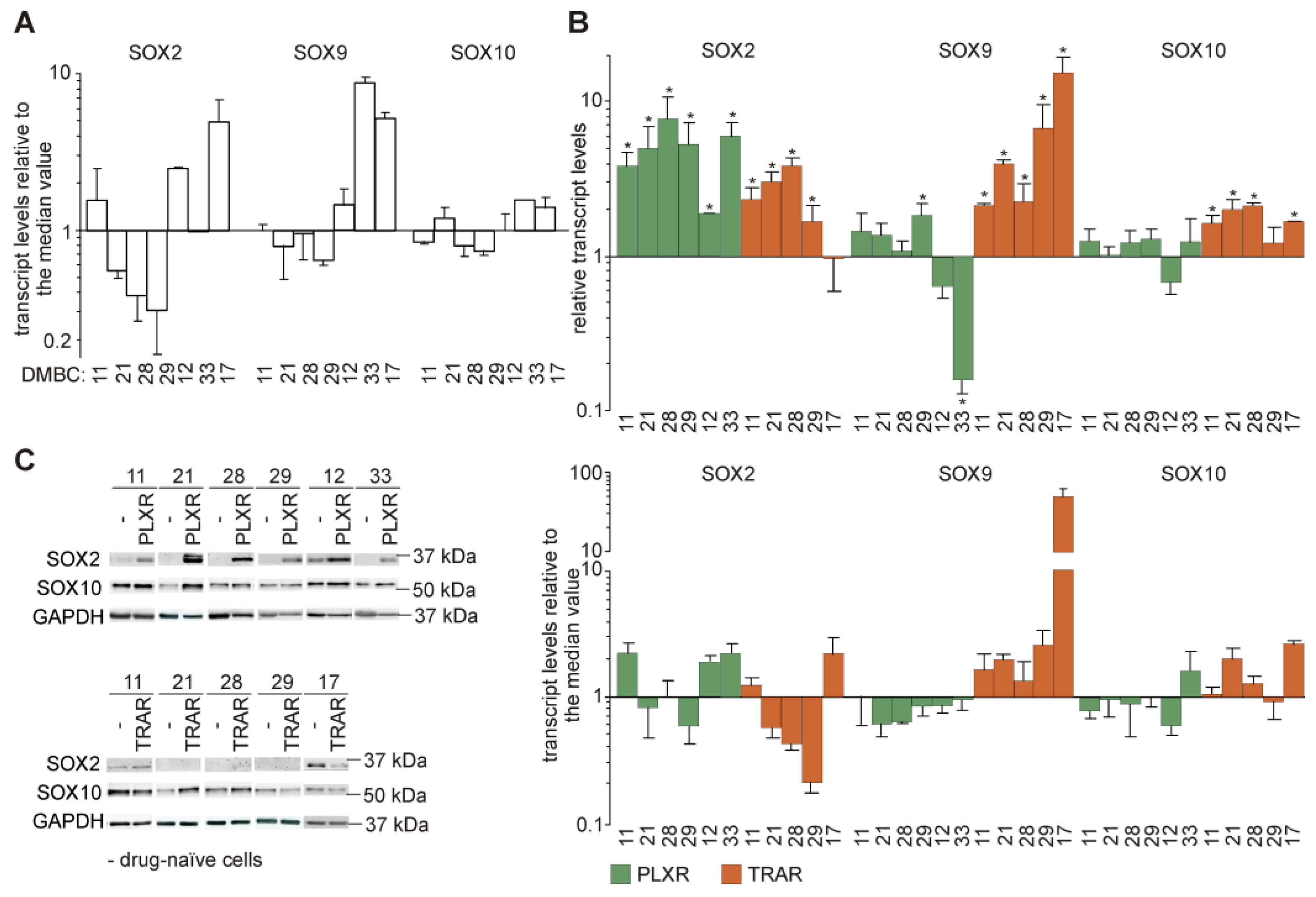
3.5. Genes from the Sex-Determining Region Y-box (SOX) Family are Differentially Expressed in Resistant and Drug-Naïve Melanoma Cell Lines
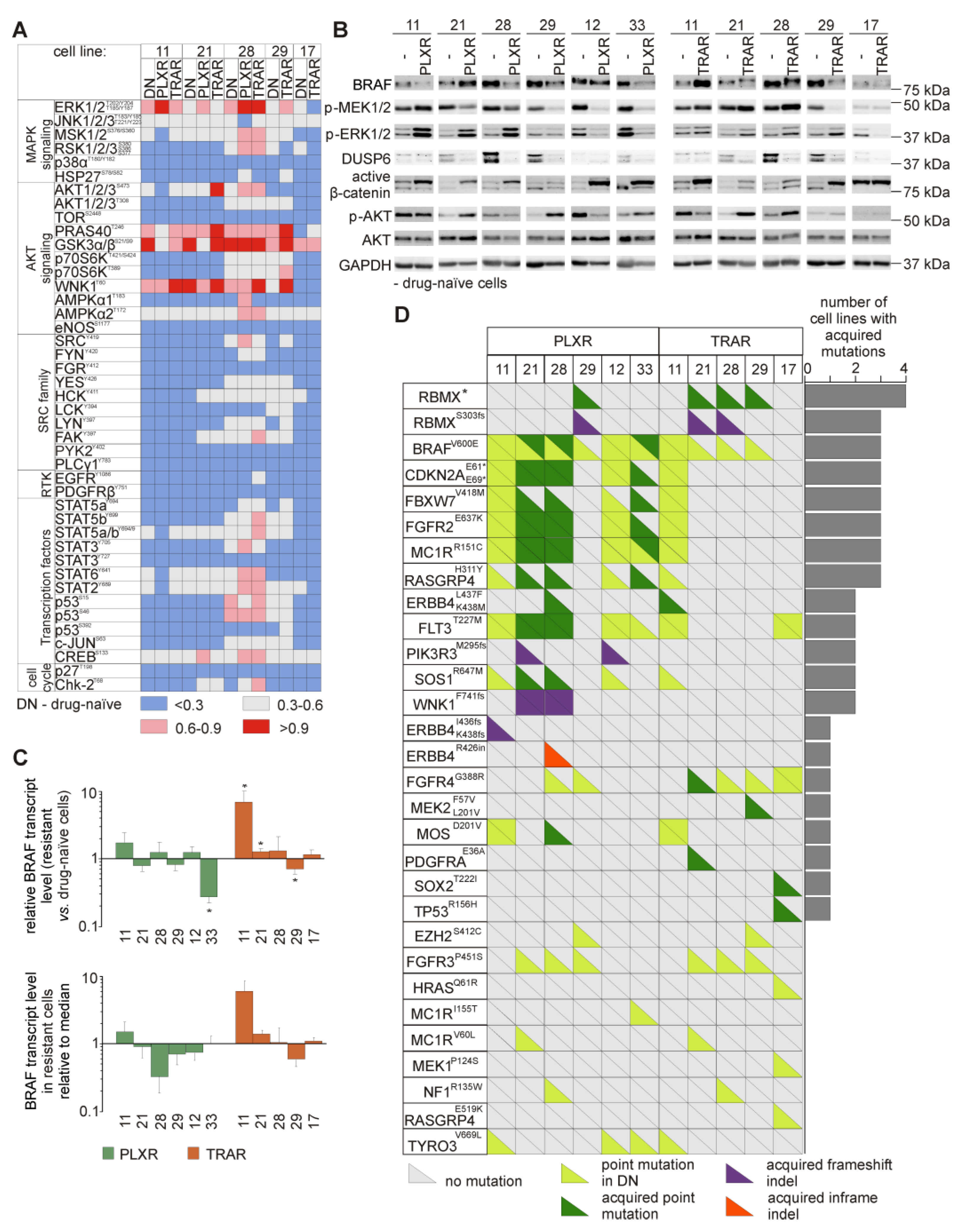
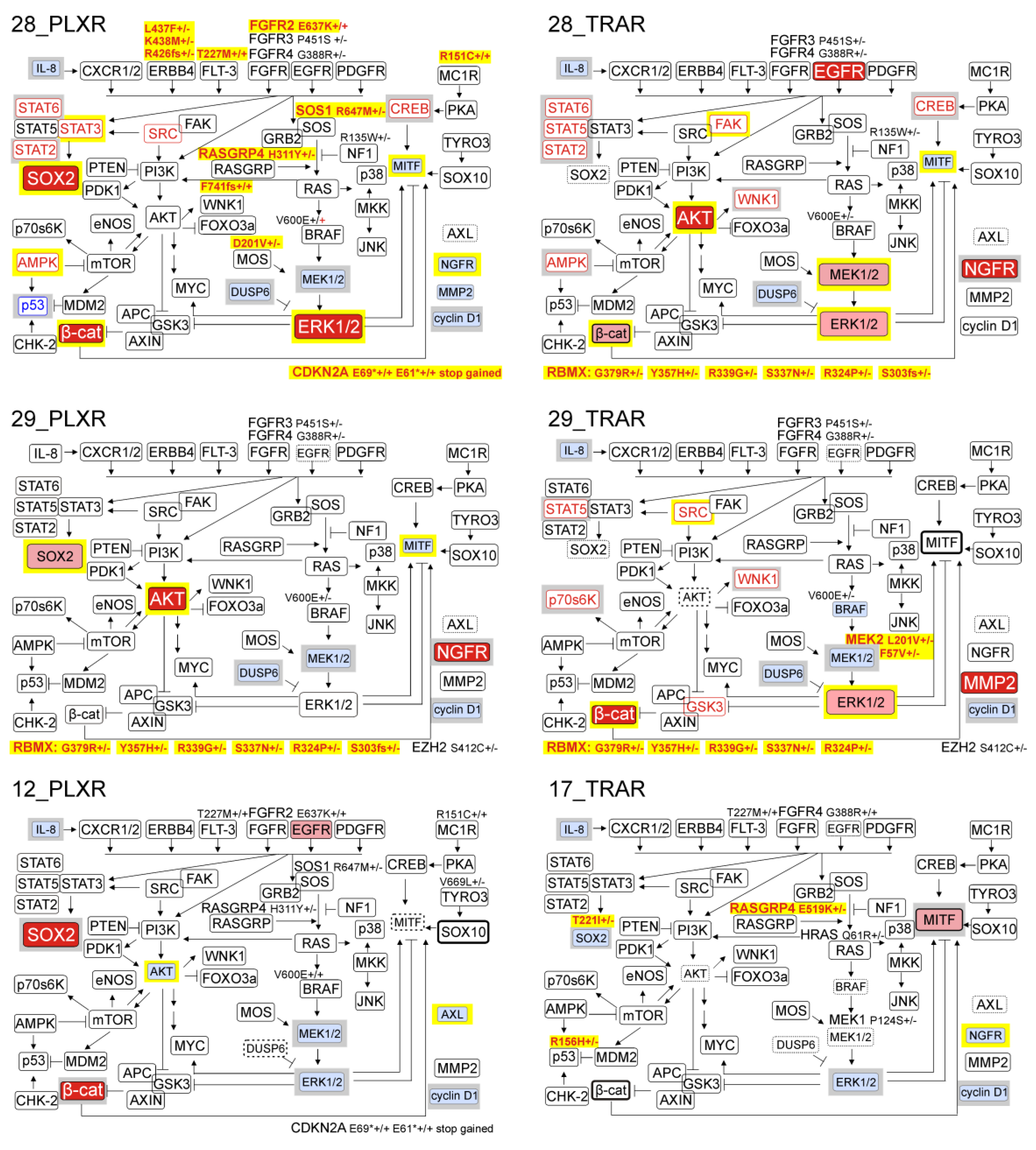
3.6. Resistant Melanoma Cell Lines Reactivate the MAPK Signaling Pathway and/or Trigger the Wingless-Type (WNT) Pathway
3.7. Genetic Analysis Revealed RNA-Binding Motifs X (RBMX) as a Putative Gene Associated with Acquired Resistance of Melanoma Cells with Enhanced Activity of the PI3K/AKT Signaling Pathway
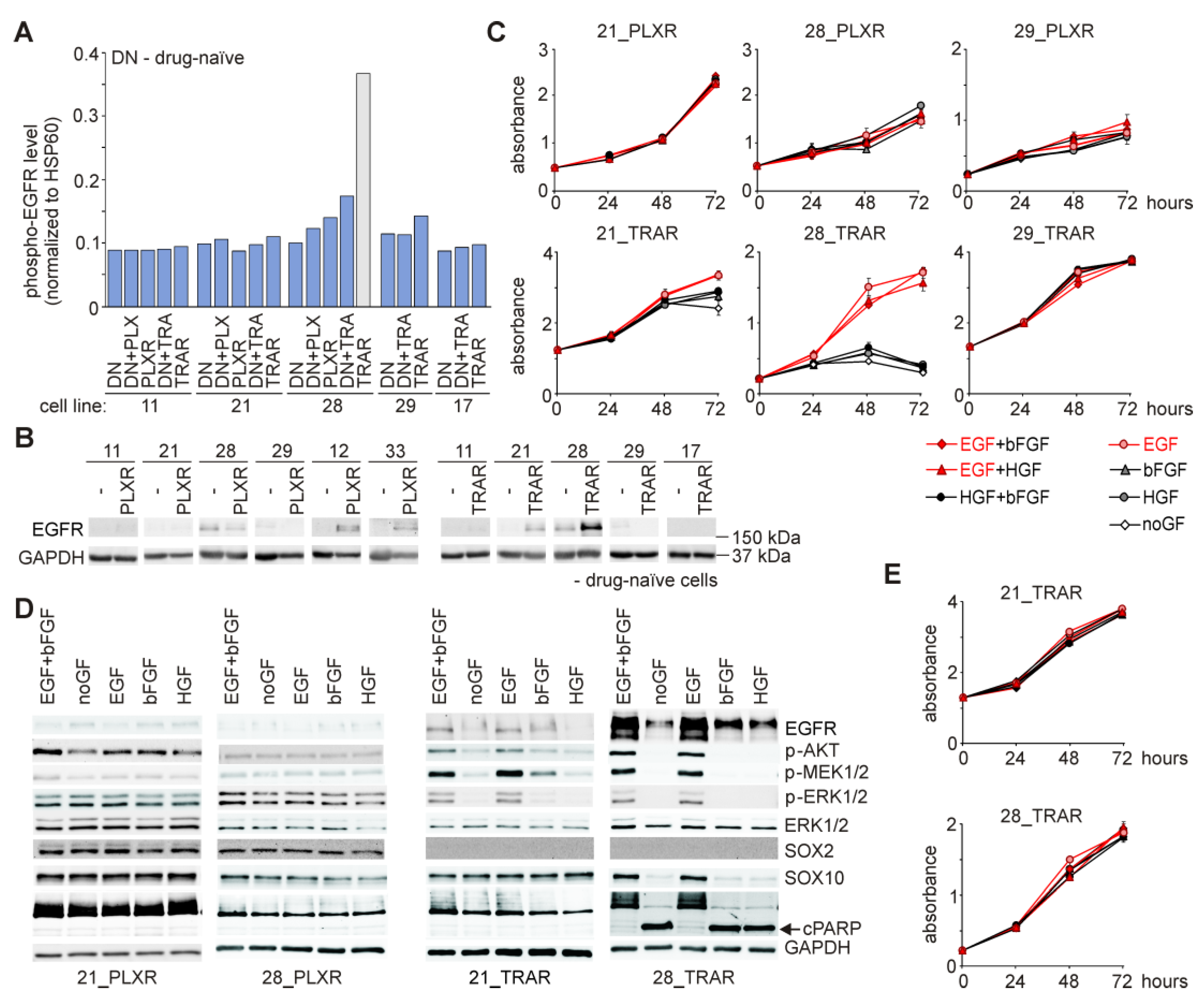
3.8. Resistant Cell Lines do not Frequently Acquire Growth Factor Dependence, and Epidermal Growth Factor (EGF) is Indispensable for Survival of Resistant Cells Only in the Presence of Drug
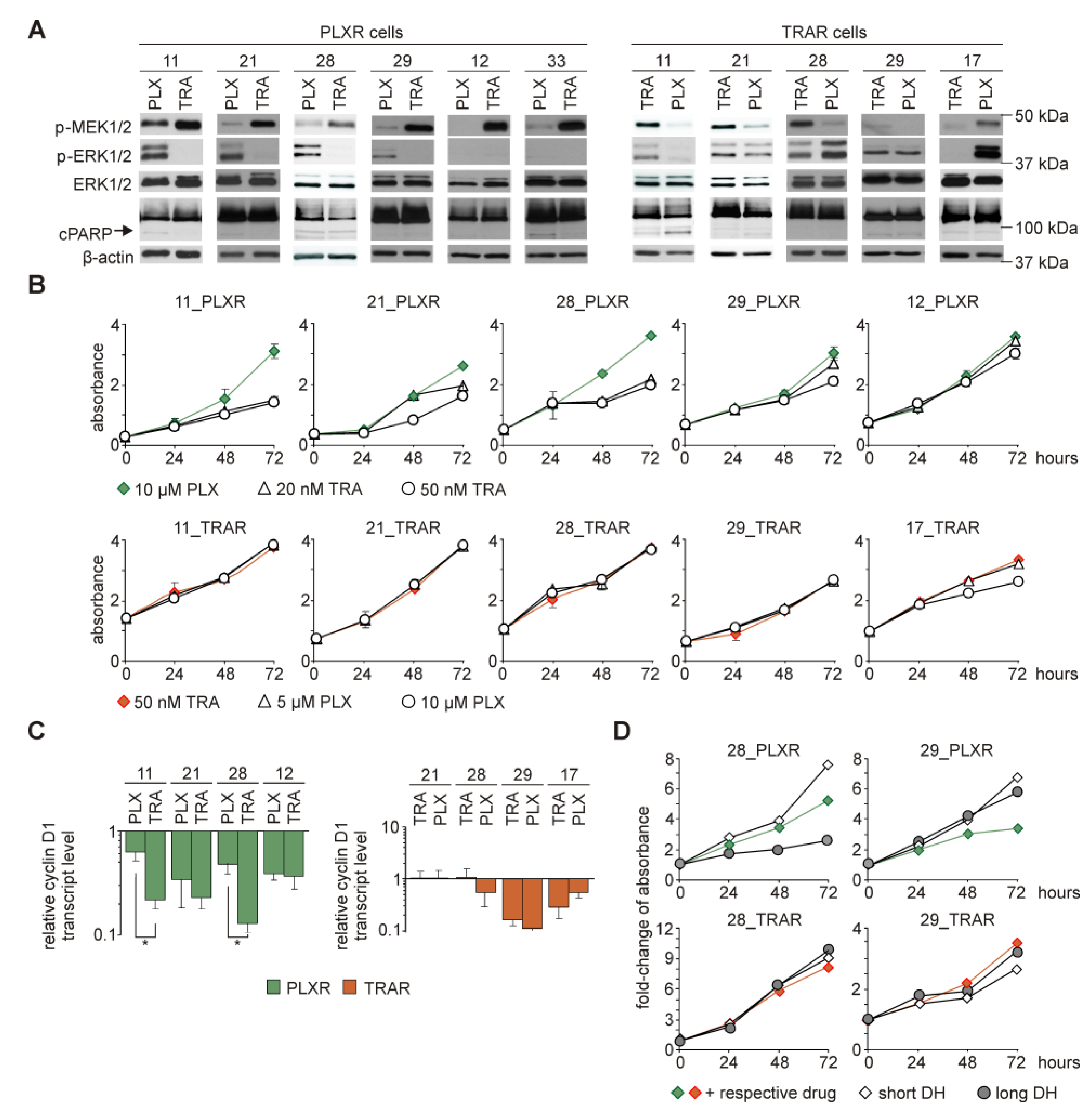
3.9. While Trametinib Inhibits Elevated ERK1/2 Activity and Proliferation of PLXR Cells, Cross-Resistance to Vemurafenib is Developed in TRAR Cell Lines
3.10. Several Alterations in Signaling Pathways Observed in Drug-Resistant Cells are Already Induced during Immediate Response to Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Kim, A.; Cohen, M.S. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [Green Version]
- Manzano, J.L.; Layos, L.; Bugés, C.; de los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [Green Version]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. MAPK pathway in melanoma part II-secondary and adaptive resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 93–101. [Google Scholar] [CrossRef]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, B.; Delyon, J.; De Moura, C.R.; Battistella, M.; Jouenne, F.; Golmard, L.; Sadoux, A.; Podgorniak, M.-P.; Chami, I.; Marco, O.; et al. A targeted genomic alteration analysis predicts survival of melanoma patients under BRAF inhibitors. Oncotarget 2019, 10, 1669–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2019, in press. [Google Scholar] [CrossRef]
- Cayrefourcq, L.; De Roeck, A.; Garcia, C.; Stoebner, P.E.; Fichel, F.; Garima, F.; Perriad, F.; Daures, J.P.; Meunier, L.; Alix-Panabieres, C. S100-EPISPOT: A new tool to detect viable circulating melanoma cells. Cells 2019, 8, 755. [Google Scholar] [CrossRef] [Green Version]
- Torres-Collado, A.X.; Knott, J.; Jazirehi, A.R. Reversal of Resistance in Targeted Therapy of Metastatic Melanoma: Lessons Learned from Vemurafenib (BRAFV600E-Specific Inhibitor). Cancers 2018, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Filipp, F.V. Precision medicine driven by cancer systems biology. Cancer Metastasis Rev. 2017, 36, 91–108. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Goldberg, R.M.; Lenz, H.J.; Shields, A.F.; Gibney, G.T.; Tan, A.R.; Brown, J.; Eisenberg, B.; Heath, E.I.; Phuphanich, S.; et al. The current state of molecular testing in the treatment of patients with solid tumors, 2019. CA Cancer J. Clin. 2019, 69, 305–343. [Google Scholar] [CrossRef]
- Sztiller-Sikorska, M.; Hartman, M.L.; Talar, B.; Jakubowska, J.; Zalesna, I.; Czyz, M. Phenotypic diversity of patient-derived melanoma populations in stem cell medium. Lab. Investig. 2015, 95, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Rozanski, M.; Osrodek, M.; Zalesna, I.; Czyz, M. Vemurafenib and trametinib reduce expression of CTGF and IL-8 in V600EBRAF melanoma cells. Lab. Investig. 2017, 97, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Mielczarek-Lewandowska, A.; Sztiller-Sikorska, M.; Osrodek, M.; Czyz, M.; Hartman, M.L. 17-Aminogeldanamycin selectively diminishes IRE1α-XBP1s pathway activity and cooperatively induces apoptosis with MEK1/2 and BRAFV600E inhibitors in melanoma cells of different genetic subtypes. Apoptosis 2019, 24, 596–611. [Google Scholar] [CrossRef] [Green Version]
- Zalesna, I.; Osrodek, M.; Hartman, M.L.; Rozanski, M.; Sztiller-Sikorska, M.; Niewinna, K.; Nejc, D.; Czyz, M. Exogenous growth factors bFGF, EGF and HGF do not influence viability and phenotype of V600EBRAF melanoma cells and their response to vemurafenib and trametinib in vitro. PLoS ONE 2017, 12, e0183498. [Google Scholar] [CrossRef]
- Osrodek, M.; Hartman, M.L.; Czyz, M. Physiologically Relevant Oxygen Concentration (6% O2) as an Important Component of the Microenvironment Impacting Melanoma Phenotype and Melanoma Response to Targeted Therapeutics In Vitro. Int. J. Mol. Sci. 2019, 20, E4203. [Google Scholar] [CrossRef] [Green Version]
- Czyz, M.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Osrodek, M.; Hartman, M.L. Plasticity of Drug-Naïve and Vemurafenib- or Trametinib-Resistant Melanoma Cells in Execution of Differentiation/Pigmentation Program. J. Oncol. 2019, 2019, 1697913. [Google Scholar] [CrossRef] [Green Version]
- Sztiller-Sikorska, M.; Koprowska, K.; Jakubowska, J.; Zalesna, I.; Stasiak, M.; Duechler, M.; Czyz, M.E. Sphere formation and self-renewal capacity of melanoma cells is affected by the microenvironment. Melanoma Res. 2012, 22, 215–224. [Google Scholar] [CrossRef]
- Sztiller-Sikorska, M.; Koprowska, K.; Majchrzak, K.; Hartman, M.; Czyz, M. Natural compounds’ activity against cancer stem-like or fast-cycling melanoma cells. PLoS ONE 2014, 9, e90783. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Talar, B.; Noman, M.Z.; Gajos-Michniewicz, A.; Chouaib, S.; Czyz, M. Gene expression profiling identifies microphthalmia-associated transcription factor (MITF) and Dickkopf-1 (DKK1) as regulators of microenvironment-driven alterations in melanoma phenotype. PLoS ONE 2014, 9, e95157. [Google Scholar] [CrossRef] [Green Version]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; de Lima Vazquez, V.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Möller, A.; Algermissen, B.; Worm, M.; Sticherling, M.; Czarnetzki, B.M. IL-8 produced by human malignant melanoma cells In Vitro is an essential autocrine growth factor. J. Immunol. 1993, 151, 2667–2675. [Google Scholar] [PubMed]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-Induced iron-dependent oxidative stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [Green Version]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Almeida, F.V.; Douglass, S.M.; Fane, M.E.; Weeraratna, A.T. Bad company: Microenvironmentally mediated resistance to targeted therapy in melanoma. Pigment Cell Melanoma Res. 2019, 32, 237–247. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L.; Malpicci, D.; Marra, E.; Belleudi, F.; Noto, A.; De Vitis, C.; Pisanu, M.E.; Coluccia, P.; Camerlingo, R.; Roscilli, G.; et al. Combination of antibodies directed against different ErbB3 surface epitopes prevents the establishment of resistance to BRAF/MEK inhibitors in melanoma. Oncotarget 2015, 6, 24823–24841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajos-Michniewicz, A.; Czyz, M. Role of miRNAs in Melanoma Metastasis. Cancers 2019, 11, E326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, M. Fibroblast Growth Factor Receptor Signaling in Skin Cancers. Cells 2019, 8, E540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, F.; Haass, N.K. Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front. Oncol. 2018, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, E3844. [Google Scholar] [CrossRef] [Green Version]
- Kodet, O.; Dvořánková, B.; Bendlová, B.; Sýkorová, V.; Krajsová, I.; Štork, J.; Kučera, J.; Szabo, P.; Strnad, H.; Kolář, M.; et al. Microenvironment‑driven resistance to B‑Raf inhibition in a melanoma patient is accompanied by broad changes of gene methylation and expression in distal fibroblasts. Int. J. Mol. Med. 2018, 41, 2687–2703. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Solal, K.A.; Kaufman, H.L.; Lasfar, A. Transcription factors as critical players in melanoma invasiveness, drug resistance, and opportunities for therapeutic drug development. Pigment Cell Melanoma Res. 2018, 31, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Saei, A.; Eichhorn, P.J.A. Adaptive Responses as Mechanisms of Resistance to BRAF Inhibitors in Melanoma. Cancers 2019, 11, E1176. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnberg, T.; Makino, E.; Krueger, M.A.; Velic, A.; Macek, B.; Rothbauer, U.; Groll, N.; Pötz, O.; Czemmel, S.; Niessner, H.; et al. A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine 2016, 8, 132–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 9, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.R.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Rad, E.B.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4545. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A. Tumor heterogeneity and plasticity as elusive drivers for resistance to MAPK pathway inhibition in melanoma. Oncogene 2015, 34, 2951–2957. [Google Scholar] [CrossRef]
- Lehraiki, A.; Cerezo, M.; Rouaud, F.; Abbe, P.; Allegra, M.; Kluza, J.; Marchetti, P.; Imbert, V.; Cheli, Y.; Bertolotto, C.; et al. Increased CD271 expression by the NF-kB pathway promotes melanoma cell survival and drives acquired resistance to BRAF inhibitor vemurafenib. Cell Discov. 2015, 1, 15030. [Google Scholar] [CrossRef]
- Redmer, T.; Walz, I.; Klinger, B.; Khouja, S.; Welte, Y.; Schäfer, R.; Regenbrecht, C. The role of the cancer stem cell marker CD271 in DNA damage response and drug resistance of melanoma cells. Oncogenesis 2017, 6, e291. [Google Scholar] [CrossRef]
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.E.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2019, 180, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, M.; Charalampopoulos, I.; Kenchappa, R.S.; Reversi, A.; Klos-Applequist, J.M.; Karaca, E.; Simi, A.; Spuch, C.; Choi, S.; Friedman, W.J.; et al. Ligand-independent signaling by disulfide-crosslinked dimers of the p75 neurotrophin receptor. J. Cell. Sci. 2009, 122, 3351–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasia, A.; Barker, P.A.; Chao, M.V.; Hempstead, B.L. Detection of p75NTR Trimers: Implications for Receptor Stoichiometry and Activation. J. Neurosci. 2015, 35, 11911–11920. [Google Scholar] [CrossRef] [Green Version]
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. Low neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat. Commun. 2017, 8, 1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Czyz, M. Pro-survival role of MITF in melanoma. J. Investig. Dermatol. 2015, 135, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.P.; Ferguson, J.; Arozarena, I.; Hayward, R.; Marais, R.; Chapman, A.; Hurlstone, A.; Wellbrock, C. Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J. Natl. Cancer Inst. 2013, 105, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Hüser, L.; Sachindra, S.; Granados, K.; Federico, A.; Larribère, L.; Novak, D.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int. J. Cancer. 2018, 143, 3131–3142. [Google Scholar] [CrossRef] [Green Version]
- Bernhardt, M.; Novak, D.; Assenov, Y.; Orouji, E.; Knappe, N.; Weina, K.; Reith, M.; Larribere, L.; Gebhardt, C.; Plass, C.; et al. Melanoma-Derived iPCCs Show Differential Tumorigenicity and Therapy Response. Stem Cell Rep. 2017, 8, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Doxie, D.B.; Greenplate, A.R.; Gandelman, J.S.; Diggins, K.E.; Roe, C.E.; Dahlman, K.B.; Sosman, J.A.; Kelley, M.C.; Irish, J.M. BRAF and MEK inhibitor therapy eliminates Nestin-expressing melanoma cells in human tumors. Pigment Cell Melanoma Res. 2018, 31, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Vachtenheim, J.; Réda, J.; Horák, P.; Ondrušová, L. Inducibly decreased MITF levels do not affect proliferation and phenotype switching but reduce differentiation of melanoma cells. J. Cell Mol. Med. 2018, 22, 2240–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarini, V.; Guida, E.; Todaro, F.; Di Agostino, S.; Tassinari, V.; Nicolis, S.; Favaro, R.; Caporali, S.; Lacal, P.M.; Botti, E.; et al. Sox2 is not required for melanomagenesis, melanoma growth and melanoma metastasis in vivo. Oncogene 2017, 36, 4508–4515. [Google Scholar] [CrossRef] [PubMed]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [Green Version]
- Girouard, S.D.; Laga, A.C.; Mihm, M.C.; Scolyer, R.A.; Thompson, J.F.; Zhan, Q.; Widlund, H.R.; Lee, C.W.; Murphy, G.F. SOX2 contributes to melanoma cell invasion. Lab. Invest. 2012, 92, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, S.M.; Segalada, C.; Cheng, P.F.; Bonalli, M.; Parfejevs, V.; Levesque, M.P.; Dummer, R.; Nicolis, S.K.; Sommer, L. Sox2 is dispensable for primary melanoma and metastasis formation. Oncogene 2017, 36, 4516–4524. [Google Scholar] [CrossRef] [Green Version]
- Ravindran Menon, D.; Luo, Y.; Arcaroli, J.J.; Liu, S.; KrishnanKutty, L.N.; Osborne, D.G.; Li, Y.; Samson, J.M.; Bagby, S.; Tan, A.C.; et al. CDK1 interacts with Sox2 and promotes tumor initiation in human melanoma. Cancer Res. 2018, 78, 6561–6574. [Google Scholar] [CrossRef] [Green Version]
- Smalley, K.S.; Lioni, M.; Dalla Palma, M.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; Van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef] [Green Version]
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [Green Version]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Rothenberg, S.M.; Hata, A.N.; Faber, A.C.; Piris, A.; Nazarian, R.M.; Brown, R.D.; Godfrey, J.T.; Winokur, D.; Walsh, J.; et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci. Transl. Med. 2013, 5, 196ra98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senawong, T.; Phuchareon, J.; Ohara, O.; McCormick, F.; Rauen, K.A.; Tetsu, O. Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and RAF inhibition: Implications for therapeutic options. Hum. Mol. Genet. 2008, 17, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): A review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 2018, 15, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.E.; Koay, T.S.; Esfandiari, A.; Ho, Y.H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, E3. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Bugide, S.; Wang, B.; Green, M.R.; Johnson, D.B.; Wajapeyee, N. Loss of BOP1 confers resistance to BRAF kinase inhibitors in melanoma by activating MAP kinase pathway. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.H.; Kim, S.H.; Trousil, S.; Frederick, D.T.; Piris, A.; Yuan, P.; Cai, L.; Gu, L.; Li, M.; Lee, J.H.; et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat. Med. 2016, 22, 1056–1061. [Google Scholar] [CrossRef] [Green Version]
- van Herpen, C.M.L.; Agarwala, S.S.; Hauschild, A.; Berking, C.; Beck, J.T.; Schadendorf, D.; Jansen, R.; Queirolo, P.; Ascierto, P.A.; Blank, C.U.; et al. Biomarker results from a phase II study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS- or BRAF-mutated melanoma. Oncotarget 2019, 10, 1850–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, D.J.; Dalgliesh, C.; Hysenaj, G.; Ehrmann, I. RBMX family proteins connect the fields of nuclear RNA processing, disease and sex chromosome biology. Int. J. Biochem. Cell Biol. 2019, 108, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell. Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Antonello, Z.A.; Hsu, N.; Bhasin, M.; Roti, G.; Joshi, M.; Van Hummelen, P.; Ye, E.; Lo, A.S.; Karumanchi, S.A.; Bryke, C.R.; et al. Vemurafenib-resistance via de novo RBM genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with BRAFV600E. Oncotarget 2017, 8, 84743–84760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef]
- Zheng, Y.; Sun, Y.; Liu, Y.; Zhang, X.; Li, F.; Li, L.; Wang, J. The miR-31-SOX10 axis regulates tumor growth and chemotherapy resistance of melanoma via PI3K/AKT pathway. Biochem. Biophys. Res. Commun. 2018, 503, 2451–2458. [Google Scholar] [CrossRef]
- Villanueva, J.; Infante, J.R.; Krepler, C.; Reyes-Uribe, P.; Samanta, M.; Chen, H.Y.; Li, B.; Swoboda, R.K.; Wilson, M.; Vultur, A.; et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013, 4, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.C.; Lai, F.; Thorne, R.F.; Yang, F.; Liu, H.; Hersey, P.; Zhang, X.D. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin. Cancer. Res. 2011, 17, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, K.; Snoyman, S.; Pupo, G.M.; Becker, T.M.; Kefford, R.F.; Rizos, H. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J. Investig. Dermatol. 2012, 132, 1850–1859. [Google Scholar] [CrossRef] [Green Version]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Erin Chen, Y.; Kumar, R.; Taylor, M.; Jenny Njauw, C.N.; Miao, B.; Frederick, D.T.; Wargo, J.A.; Flaherty, K.T.; Jönsson, G.; et al. MITF modulates therapeutic resistance through EGFR signaling. J. Investig. Dermatol. 2015, 135, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Dermatol. 2015, 135, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.; Niemetz-Rahn, A.; Nonnenmacher, A.; Tucholski, J.; Keilholz, U.; Fusi, A. Expression and activity of EGFR in human cutaneous melanoma cell lines and influence of vemurafenib on the EGFR pathway. Target. Oncol. 2015, 10, 77–84. [Google Scholar] [CrossRef]
- Rizzolio, S.; Cagnoni, G.; Battistini, C.; Bonelli, S.; Isella, C.; Van Ginderachter, J.A.; Bernards, R.; Di Nicolantonio, F.; Giordano, S.; Tamagnone, L. Neuropilin-1 upregulation elicits adaptive resistance to oncogene-targeted therapies. J. Clin. Investig. 2018, 128, 3976–3990. [Google Scholar] [CrossRef]
- Grimont, A.; Pinho, A.V.; Cowley, M.J.; Augereau, C.; Mawson, A.; Giry-Laterrière, M.; Van den Steen, G.; Waddell, N.; Pajic, M.; Sempoux, C.; et al. SOX9 regulates ERBB signalling in pancreatic cancer development. Gut 2015, 64, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Ishii, S.; Richert, N.; Merlino, G.T.; Pastan, I. Epidermal growth factor regulates the expression of its own receptor. Proc. Natl. Acad. Sci. USA 1985, 82, 8374–8378. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.K.; Lee, J.Y.; Supko, K.M.; Khan, A.; Torres, S.M.; Berwick, M.; Ho, J.; Kirkwood, J.M.; Siegfried, J.M.; Stabile, L.P. Pan-erbB inhibition potentiates BRAF inhibitors for melanoma treatment. Melanoma Res. 2014, 24, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, B.; Jacobs, K.; Ferdinande, L.; Taildeman, J.; Lambert, J.; Peeters, M.; Bracke, M.; Pauwels, P.; Brochez, L. EGFR in melanoma: Clinical significance and potential therapeutic target. J. Cutan. Pathol. 2011, 38, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, H.; Fukushima, S.; Yamashita, J.; Jinnin, M.; Sakai, K.; Masuguchi, S.; Aoi, J.; Makino, T.; Inoue, Y.; Ihn, H. Serum epidermal growth factor receptor levels in patients with malignant melanoma. Clin. Exp. Dermatol. 2013, 38, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Margheri, F.; Biagioni, A.; Chillà, A.; Pimpinelli, N.; Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Calorini, L.; Serratì, S.; et al. EGFR/uPAR interaction as druggable target to overcome vemurafenib acquired resistance in melanoma cells. EBioMedicine 2019, 39, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.N.; Walker, A.L.; Liu, Y.Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, E803. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman, M.L.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Czyz, M. Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity. Cells 2020, 9, 142. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9010142
Hartman ML, Sztiller-Sikorska M, Gajos-Michniewicz A, Czyz M. Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity. Cells. 2020; 9(1):142. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9010142
Chicago/Turabian StyleHartman, Mariusz L., Malgorzata Sztiller-Sikorska, Anna Gajos-Michniewicz, and Malgorzata Czyz. 2020. "Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity" Cells 9, no. 1: 142. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9010142