Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma
by
 , ,
, ,
Jason Adhikaree
1,*, 
Julia Moreno-Vicente
2,
Aanchal Preet Kaur
1,
Andrew Mark Jackson
1 and
Poulam M. Patel
1 1
Host-Tumour Interactions Group, Division of Cancer and Stem Cells, BioDiscovery Institute, University of Nottingham, Nottingham NG7 2RD, UK
2
Antibody and Vaccine Group, Centre for Cancer Immunology, University of Southampton, Southampton General Hospital, Southampton, Hants SO16 6YD, UK
*
Author to whom correspondence should be addressed.
Cells 2020, 9(2), 263; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020263
Submission received: 31 December 2019
/
Revised: 15 January 2020
/
Accepted: 16 January 2020
/
Published: 21 January 2020
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Cancers: Glioblastoma)
Abstract
:Glioblastoma (GBM) is inevitably refractory to surgery and chemoradiation. The hope for immunotherapy has yet to be realised in the treatment of GBM. Immune checkpoint blockade antibodies, particularly those targeting the Programme death 1 (PD-1)/PD-1 ligand (PD-L1) pathway, have improved the prognosis in a range of cancers. However, its use in combination with chemoradiation or as monotherapy has proved unsuccessful in treating GBM. This review focuses on our current knowledge of barriers to immunotherapy success in treating GBM, such as diminished pre-existing anti-tumour immunity represented by low levels of PD-L1 expression, low tumour mutational burden and a severely exhausted T-cell tumour infiltrate. Likewise, systemic T-cell immunosuppression is seen driven by tumoural factors and corticosteroid use. Furthermore, unique anatomical differences with primary intracranial tumours such as the blood-brain barrier, the type of antigen-presenting cells and lymphatic drainage contribute to differences in treatment success compared to extracranial tumours. There are, however, shared characteristics with those known in other tumours such as the immunosuppressive tumour microenvironment. We conclude with a summary of ongoing and future immune combination strategies in GBM, which are representative of the next wave in immuno-oncology therapeutics.
1. Introduction
There is a high clinical need for new approaches to combat glioblastoma (GBM) outside of the traditional approaches such as surgery, radiation and chemotherapy. Immunotherapy, and particularly, the inhibition of the programme death 1 (PD-1)/PD-1 ligand (PD-L1) pathway, has arisen as a successful strategy to treat several cancers types [1,2,3,4,5,6,7]. Since the first Food and Drug Agency (FDA) approval in 2014 of Pembrolizumab, a PD-1 inhibitor, to treat metastatic melanoma, there have been a further five drugs developed in this class with almost fifty FDA approvals within the last five years between them. In conjunction with the rapid growth in clinical utility of immune checkpoint inhibitors, there is now increasing knowledge of mechanisms of action, biomarkers, disease response patterns and toxicity in relation to cancer immunotherapy treatment. This has led to a renewed hope that this class of immunotherapies will provide the long-anticipated immunotherapy breakthrough to treat GBM.
Unfortunately in 2019, two-phase III first-line trials recruiting patients with either methylated or unmethylated O6-methylguanine methyltransferase (MGMT) GBM using Nivolumab (a PD-1 inhibitor) concurrently with standard of care radiotherapy or chemoradiotherapy, failed to meet its primary end-point of improved progression-free or overall survival. Likewise, a completed randomised phase III trial using Nivolumab in relapsed patients with GBM, failed to meet its primary outcome of improved overall survival versus Bevacizumab [8]. A disappointing response rate of 8% was seen in this trial, however, those few who responded had a durable response (median 11.1 months with Nivolumab vs. 5.3 months with Bevacizumab) suggesting a subgroup of patients who could derive benefit from such approaches. However, this highlights the current priority area in the immuno-oncology field of how to treat checkpoint blockade unresponsive tumours. This review aims to evaluate the unique challenges of using PD-1/PD-L1 axis inhibiting drugs and other immunotherapies to treat GBM, and barriers that may restrict their success. It also considers what can be learnt from other tumour types and whether this is the beginning or end of PD-1 targeted immunotherapies in GBM.
2. Why is the PD-1/PD-L1 Pathway a Relevant Therapeutic Target to Treat Cancer?
Cytotoxic T-lymphocytes (CTL) can selectively kill cancer and viruses while sparing healthy tissue. However, these tumour-specific T-cells are suppressed in the tumour microenvironment [9]. PD-1 is an inhibitory transmembrane receptor dynamically expressed upon T-cell receptor (TCR) engagement on activated T-lymphocytes. It favours immune evasion in cancer by down-regulating T-cell activation and effector function [10]. Although absent in naïve T-cells, higher levels of PD-1 are found on infiltrating T-lymphocytes, which are thought to be exhausted due to chronic antigen stimulation [11,12]. On binding to its ligand, PD-L1 and PD-L2, SHP-2 phosphatase is recruited to the cytoplasmic immunoreceptor tyrosine-based switch motif (ITSM) domain of PD-1. This and other phosphatases attenuate the co-stimulatory signal predominately through CD28 [13]. Furthermore, signalling through the co-stimulation B7/CD28 complex is required for PD-1 inhibitors to be effective, illustrating the importance of this signal [13,14].
The ligation of PD-1 on T-cells, by tumour or tumour-infiltrating immune cells expressing PD-L1, results in T-cell dysfunction. This includes, but is not limited to, attenuation of activation, decreased cytokine secretion and impaired production of cytotoxic molecules. There is also reduced anti-apoptotic and increased pro-apoptotic gene expression, thereby decreasing T-cell survival. In addition, metabolic reprogramming within T-cells further contributes to T-cell exhaustion and inhibition [10]. Therefore, immune evasion mediated by PD-1/PD-L1 signalling may explain why endogenous or strong vaccine-induced T-cell responses against tumour antigens fail to translate to tumour regression [15]. The relevance of this axis is further supported by the recent explosion of FDA approvals for drugs that target this axis.
It transpires that there are many such receptors that modulate the T-cell response and, therefore, it is somewhat surprising that a single checkpoint has translated to such clinical success [16].
3. Clinical Efficacy of Immune Checkpoint in GBM
The remarkable responses to PD-1 inhibitors seen in lymphoma subtypes, where response rates are high (87% in Hodgkin’s lymphoma), are not recapitulated by solid organ cancers where response rates range from 15 to 40% [17]. This means that, despite significant and durable responses in patients that respond, the majority will progress and, furthermore, be at risk of immunotherapy associated side effects.
Recently, phase III, randomised, multicentre trial, checkmate-143, compared Nivolumab to Bevacizumab in 369 relapsed GBM patients, showed no overall survival advantage for the PD-1 inhibitor above Bevacizumab [8]. In both treatment arms, 40% of patients were on steroids at baseline including approximately 15% in both arms taking ≥4 mg/day. The median overall survival (OS) was 9.8 months with Nivolumab compared to 10.0 months with bevacizumab and the 12-month OS rate was 42% in both treatment arms. The progression-free survival was shorter in the Nivolumab group at 1.5 months, compared to 3.5 months in Bevacizumab group. Likewise, the objective response rate was lower in the Nivolumab group −8% vs. 23%, despite the majority of patients having measurable disease at baseline (83%). The duration of response was, however, longer with Nivolumab −11.1 months compared to 5.3 months and less grade 3–4 treatment-related adverse events (13% vs. 18%). However, on the basis of this phase III trial Nivolumab could not be recommended in the relapsed setting for GBM.
In May 2019, a Bristol Squibb Myers press release stated that the eagerly awaited front line phase III trial, Checkmate-498, had failed to meet its primary endpoint of overall survival (OS) and both secondary endpoints of progression-free survival (PFS), and OS rate at two years. This trial recruited over 500 treatment-naïve patients to receive either Nivolumab plus radiation or Temozolomide plus radiation. After surgery, patients in the experimental arm received Nivolumab every two weeks concurrent with radiation, followed by maintenance with Nivolumab every four weeks until disease progression or unacceptable toxicity. Eligible patients had centrally confirmed MGMT-unmethylated disease and a Karnofsky performance status ≥70. Likewise, the randomised, multicentred phase III frontline trial, Checkmate-548, which recruited MGMT-methylated GBM, had a similar press release in September, stating a failure to meet the primary endpoint of PFS, although OS data is not mature. This trial compared the addition of Nivolumab with Temozolomide and radiation compared to Temozolomide and radiation alone.
There has also been published case series and early phase trials investigating combination therapy approaches. Part of the checkmate-143 trial comprised multiple phase I safety cohorts of Nivolumab or Nivolumab in combination with Ipilimumab. In the former, the monotherapy arm (n = 10) produced one partial response and no grade 3–4 toxicities. This contrasts to Nivolumab 1 mg and Ipilimumab 3mg, where 9 of the 10 patients experienced grade 3–4 toxicities and 5 discontinued the drug due to adverse events. This is not unexpected, as the combination of Nivolumab with Ipilimumab is known to have a maximum tolerated dose in both melanoma and non-small cell lung cancer (NSCLC), and in combination is too toxic to use at full monotherapy doses [18,19]. However, in contrast to melanoma, increased toxicity was not accompanied by increased objective response rates. In this cohort, 0/10 patients achieved a partial response [20]. Nivolumab at 3 mg and Ipilimumab at 1 mg produced one partial response (n = 20) [21]. The first reports of clinical efficacy using Pembrolizumab in relapsed GBM patients found 1 partial response in the first cohort of 6. The study also extended to combination use with bevacizumab, which was reported as safe [22].
There are also initial reports on multimodal combination strategies. For example, Pembrolizumab at two doses (100 mg and 200 mg every three weeks), combined with hypo-fractionated radiotherapy and bevacizumab was again shown to be safe. The responses of the first three patients treated with 100 mg dose is known and included two stable disease and one complete response at the time of abstract publication [23].
Overall, however, the available data on PD-1 inhibitors in GBM are mainly from published abstracts and case studies, as summarised in Table 1. We can infer that there is a similar safety profile to that known in other solid cancers, but initial response rates are lower (Table 1). More mature trial information will guide objective response rates and, more importantly, survival data. Nevertheless, numerous combination trials are already underway with the aim of improving efficacy.
4. Brain Tumour Immunity and Barrier to Immunotherapy
The brain has historically been seen as an immuno-privileged site, however, it is now clear that this is highly contextual. The lack of traditional lymphatics and known antigen-presenting cells have supported this theory [32]. The challenge of immunotherapy to treat GBM are numerous and includes a limited understanding of basic cellular mechanisms governing anti-tumour responses in the brain, mechanical barriers such as the blood–brain barrier (BBB), in addition to a suppressive tumour microenvironment (TME).
4.1. Antigen Presentation Cell (APC)
Microglia cells are tissue resident macrophages that have entered the CNS in early embryonic life. When activated, microglia express MHC class I and II molecules, as well as adhesion and co-stimulatory molecules, acquiring the ability to act as APCs [33,34,35]. Microglia express toll-like receptors 1–9 and nucleotide-binding oligomerisation domain-like receptors which contributes to their activation and recognition of a range of pathogen-associated molecular patterns [36]. Macrophage and microglial cells have functional plasticity and polarise their phenotype depending on the cytokine milieu and microbial environment. The M1 phenotype is activated by IFN-γ and lipopolysaccharide (LPS) to polarise a macrophage towards a pro-inflammatory IL-12 secreting cell capable of supporting a Th1 response. The M2 or alternatively activated phenotypes are induced by IL-10, glucocorticoids or IL-4 to induce a Th2 or immunoregulatory response [37]. However, in the context of high-grade gliomas, current data suggest that microglia lose their capacity to present antigens due to the highly immunosuppressive TME and resemble alternatively activated macrophages [36,38]. For example, TGF-β inhibits microglial proliferation and when microglial cells are co-cultured with glioma stem cells, they phenotypically revert to an M2 status. These microglial cells have reduced phagocytosis and secrete high levels of IL-10 [39]. The M2 phenotype microglial cells also have lower MHC-class II and surface co-stimulatory molecule expression, reducing antigen-presenting capacity [40,41]. Indeed, microglia have a large regulatory role in the central nervous system (CNS) immunity and plasticity which is utilized by glioma to promote growth. Upwards of 30% of a GBM tumour mass can include these immune cells and higher numbers are seen at higher grades [42]. Microglia release chemoattractants (monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1) when associated with glioma cells promoting further microglial/monocyte recruitment from the peripheral blood and, in combination with the cytokine milieu (IL-10, TGF-β and IL-6), promote further immunosuppression [36,41]. Furthermore, glioma-associated microglia can release matrix metalloproteinase-14, which degrades normal brain parenchyma and promotes tumour invasion [43].
Distinct from the role of microglia, T-cell reactivation can occur in several anatomical niches including the choroid plexus, the meninges and the perivascular space within the CNS parenchyma, where co-localisation with distinct antigen-presenting cells and T-cells are seen [44]. For example, classical myeloid dendritic (cDC) type 1 cells have been identified in the CSF space adjacent to subarachnoid vessels and choroid plexus stroma. cDC type 2 have been identified in post-capillary venules of parenchymal tissue in mouse models [44]. These cells are potent antigen-presenting cells capable of re-stimulating a T-cell response. There are also distinct macrophage populations at these sites, with perivascular macrophages sharing embryonic origins with microglia, while choroid plexus macrophages are replenished by blood-derived monocytes [44]. There is also a consensus that lymphatic drainage of the outer lining and subarachnoid space is via dural sinuses and into deep cervical lymph nodes, where MHC class II-expressing cells localize and can present antigen [45,46]. Hence, this route may indeed prove the pivotal source of antigen presentation within the CNS. Interestingly, recent single-cell mass and fluorescence cytometry in parallel with genetic fate mapping systems, have shown key differences in the dendritic cell, microglia and macrophage distribution and abundance in disease and ageing [47]. It is known that microglial cells appear to be the only leukocyte in the brain parenchyma in the steady-state. However, outside the parenchyma, in the choroid plexus, perivascular space and lining the meninges they found 4 distinct subsets of macrophages which they named border associated macrophages (BAM). These subsets may have different roles in disease, for example the CCR2+ subset was predominately found near the choroid plexus and have a high turnover from bone-marrow. This has implications for disease, for example, in an experimental autoimmune encephalitis (EAE) mouse model, the BAM decreased in frequency, replaced by peripheral monocytes and a homogenous BAM MHCII+CD38+ population was seen [47]. They also found that during EAE, microglia skewed to an inflammatory phenotype, which was also seen in ageing and Alzheimer disease mouse models, suggesting a common activation programme [47]. Additionally, they confirmed that the cDC2, cDC1 and plasmacytoid DC exist intracranially and, consistent with recent descriptions in the periphery, cDC2 are a heterogenous cell group as defined by surface marker expression. Such studies identifying the heterogeneity of innate cells and dynamic infiltration into the brain and will guide future immunotherapy combinations for targeting GBM.
Clinical support of antigen detection in the CNS and extracranial de-novo T-cell responses are seen from reports of the abscopal effect following CNS radiotherapy [48,49]. In a series of 13 patients whom received CNS radiotherapy for metastatic melanoma and had disease progression in the brain following Ipilimumab, 7 experienced a partial response at extracranial sites including liver, lung, pelvic and cutaneous [49]. This provides support to the theory that the presentation of glioma antigens can trigger a peripheral immune response, likely via priming and activation in the deep cervical lymph nodes [46].
4.2. Lymphatics
The lack of traditional lymphatics has led to controversies in regard to the CNS communication with peripheral lymph nodes and therefore the site of antigen presentation and T cell priming. However, the cervical lymph nodes seem to be important with in-vivo tracers following CNS antigens draining through CSF, across the cribriform plate and into the nasal mucosa. This subsequently drains into the cervical lymph nodes [50]. Recently, dissection of mouse meninges and immunohistochemistry staining has co-localised endothelial cells, T-cells and MHC-II expressing cells with high concentration of immune cells near the dural sinuses. Indeed, intravenous and intracerebral injection of lectin dyes and anti-CD45 fluorescent antibodies have revealed alignment of T-cells and antigen-presenting cells along perisinusal vessels [46]. Alternatively, soluble antigens have been shown to travel down a separate pathway in the perivascular spaces within the wall of the cerebral arteries and into the cervical lymph nodes [51].
Further support to the importance of the cervical lymph nodes in T-cell priming comes from a mouse model of MS, EAE. This condition occurs in mice after intracranial injection of purified myelin antigen and is exacerbated by an intracranial cryo-lesion. The removal of the cervical lymph nodes, prior to inflicting the cryo-lesion ameliorates this phenotype [50].
Furthermore, in the perivascular space, circulates T-cells which display a memory phenotype, rather than naive [35]. In support of this, homing of CD8+ T-cells towards the brain has been found to occur after the presentation of tumour-specific antigens at the cervical lymph nodes [52]. Under physiological circumstances, activated memory CD4+ T-cells enter the CSF from the bloodstream and monitor perivascular spaces as part of the immunosurveillance machinery in the CNS. Upon encounter of antigen-loaded APCs, CD4+ T-cells differentiate into effector cells and acquire the competence to invade the parenchyma, thereby triggering local immune responses in the brain [53]. In a similar fashion, many CD8+ T-cells that are found in the CNS during neuroinflammation display an effector memory phenotype and are thought to be selectively recruited by α4-integrin-expressing endothelial cells at the BBB [54]. This data supports the notion that there is a constant interaction between the extracranial and intracranial immune responses and suggests that the priming and expansion of T-cells occurs outside the CNS in the cervical lymph nodes.
4.3. Blood–Brain Barrier (BBB) and Immune Privilege
The blood–brain barrier is not a static barrier, but dynamic. The integrity of the BBB across CNS microvessels relies on endothelial cells and intact tight junctions between them. This close association reduces permeability and prevents solute exchange to occur paracellularly [55,56,57]. Nevertheless, the endothelial BBB is a dynamic entity that responds to environmental cues. Inflammation and brain pathologies can compromise the integrity of the BBB, thereby increasing its permeability, and thus allowing the infiltration of circulating monocytes and lymphocytes from the periphery [57,58]. As in other tissues, the extravasation of T-cells across the BBB involves the interaction between specific adhesion molecules and chemokines expressed on immune and endothelial cells. However, trafficking into the brain is known to be less efficient than in other organs and may involve transcellular instead of paracellular extravasation, to preserve endothelial tight junctions [57,59]. For example, there is also evidence that inducible metalloproteinases facilitate penetrance of leukocytes migration following perivascular cuffing across the glia limitans and basement membrane, particularly in inflammation such as a diseased pathology of the brain [60]. In the inflamed blood–brain barrier, monocytes, Th1 and Th17 cells migration across the blood–brain barrier is a multistep process involving E- and P- selectin mediated rolling along the surface of the endothelium, followed by chemokine mediated activation and adhesion to the endothelium. This is proceeded by intracellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) and activated leukocyte adhesion molecule (ALCAM) upregulation by pro-inflammatory cytokines. These bind to affiliated T-cell receptors leukocyte function-associated antigen-1 (LFA-1), very late antigen-4 (VLA-4) and CD6. BBB endothelial cells and glial cells are an important source of the pro-inflammatory chemokines such as CCL2/MCP-1, RANTES and CXCL10/IP-10 and thus facilitate immune cell recruitment. CD8+ T-cells seem to be dependent on α4 integrin, distinct to the aforementioned mechanisms [54,57,61,62]. Immunoglobulins can also cross the BBB via the immunoglobulin receptor, FcRn, by carrier-mediated transportation via the cerebral blood vessel and into the brain parenchyma [63,64,65].
Clinical support of the effective penetration of T-cells into the CNS comes from a phase II trial which included 52 patients with untreated or progressive brain metastases diagnosed with metastatic melanoma or NSCLC. These patients had similar responses intracranially, as to their extracranial disease following treatment with Pembrolizumab, a T-cell immune checkpoint inhibitor, targeting PD-1 [66]. Thus, suggesting that these drugs are effective intracranially if the relevant immune signatures against the tumour are present. This has also been observed in GBM, where a recent case report of two patients with recurrent GBM on a background of paediatric biallelic mismatch deficiency, had a deep and durable response to Nivolumab [27]. These tumours have a high mutational burden, which predicts response to immune checkpoint blockade and is discussed later. Although a rare subtype of GBM, in certain countries, where consanguineous rates are higher, biallelic mismatch repair deficiency is estimated to account for 40% of paediatric GBM cases [67].
4.4. T-cell Dysfunction
GBM patients have been recognised to have severe deficits in cell-mediated immunity, particularly within the lymphocyte population [68,69]. This has been narrowed down to the CD4 compartment. T-regulatory cells (Tregs) are highly diverse and plastic subset of CD4 T-cells and have a universal role in immune tolerance [70]. The thymic derived, natural Tregs (nTregs), characterised by high constitutive expression of FoxP3, cause contact-dependent cytokine independent immunosuppression through CTLA-4, PD-L1, granzyme/perforin and Fas/FasL pathways [32]. Inducible Tregs, have transient or absent FoxP3 expression and induce immunosuppression through IL-10 and TGF-β [71]. Fecci et al. described suppressed absolute CD4 counts in 20 patients with GBM, however, they noted the fraction containing CD4+CD25+FoxP3+CD45RO+ T cells was increased compared to healthy controls [72]. Overall these patients’ T-cells showed anergy or secreted Th2 polarising cytokines on stimulation. Removal of the Treg population reversed this cytokine signature. Furthermore, in the murine immunocompetent model using the mouse strain VM/Dk injected with SMA-560 (mouse glioma cell tumour) intracranially, anti-CD25 was shown to deplete Tregs and increase survival [72]. Similarly, the presence of tumour infiltrating lymphocytes has been described as improving prognosis, although the rate of infiltration is low [73]. In support of an important role of Tregs in glioma is an immunohistochemistry study comprising 62 patients, whose tumours were stained for FoxP3 and CD8 and found Treg accumulation at the tumour site was associated with poorer prognosis, while CD8+ tumour infiltrating lymphocytes (TILs) were not associated with increased survival [74]. Indeed, the nTreg population may predominate in glioma, with high FoxP3 expression also seen by another group in glioma samples and linked with higher grade and high levels of Helios transcription factor, another nTreg marker [75,76]. This group expanded this theory to a mouse orthotopic model with intracranially injected (i.c.) GL261 mouse cell line of GBM and found thymectomy, prior to i.c., significantly decreased Treg levels [76]. This may be through high expression of CCL-2 chemokine by glioma. In 19 GBM patients where CD4/CD25bright cells were isolated, Tregs migrated preferentially to glioma conditioned media and was reversed by using a CCL-2 blocking antibody [77].
A more recent study has quantified the CD4 compartment compromise in newly diagnosed GBM as equivalent to that in HIV patients. They found GBM patients had CD4+ counts of ≤200/µL compared to a healthy adult numbers of >1000 [78]. They proposed a mechanism of sequestration of T-cells in the bone marrow of patients causing a relative lymphopenia through loss of sphingosine-1-phosphate (S1P) receptor on the T-cell surface. In health, a S1P ligand gradient directs T-cell chemotaxis to blood and lymph node areas and maintains circulating lymphocyte frequency [78]. They showed in mice which are S1P1-deficient, T cells accumulated in the bone marrow of glioma-bearing mice. However, S1P1-knockin, glioma-bearing mice did not have T-cell sequestration [78].
It is now clear that evidence of pre-existing anti-tumour immunity predicts response to PD-1 therapy [79]. Immunologically ‘hot’ tumours, which includes those with high PD-L1 expression, adjacent CD8 TILs, enriched interferon gene signature and upregulation of other checkpoints have shown stronger predictive value [79]. This is in contrast to ‘cold’ tumours that have no immune infiltrate. A number of studies have shown that PD-L1 is expressed on GBM tissue albeit at variable levels. A range between 50–90% positive cells was described in a cohort of 10 primary GBM samples, which was later backed by a larger cohort of 135 samples, of which 86% expressed the marker [80,81]. Contrary to this, another study of 92 IHC samples showed that a median of 2.7% of tumour cells expressed PD-L1, despite 61% defined as positive using a threshold of ≥1%. The same study also performed flow cytometry on five primary samples, which was supportive of their IHC analysis [82]. Hence, although a reasonable proportion of samples are positive by the 1% cut-off, these are of low intensity. PD-L1 is often viewed as a signature of pre-existing immunity, where this is a fingerprint of previous IFN-γ release by an antigen-specific cytotoxic T-lymphocyte (CTL). However, oncogenic pathways can also lead to constitutive PD-L1 expression. Mutated/loss of PTEN leads to increased PI3K pathway activation and subsequent PD-L1 up-regulation [83]. Mutations/inactivation of the PTEN gene ranges from 5–40% in GBM [84]. Others have shown that in pre-clinical melanoma models, loss of PTEN in tumour cells inhibits T-cell mediated killing and decreases T-cell trafficking. In patients with melanoma, loss of PTEN correlated with decreased T-cell infiltrate and poorer response to PD-1 therapy [85]. However, in this study loss of PTEN was not associated with increased PD-L1 expression and hence, at least in melanoma, PTEN causes immunosuppression independent of PD-L1 [85]. Oncogene activation, such as MYC and EGFR, also up-regulate PD-L1 expression and attenuates the anti-tumour response [86,87,88]. Therefore, whether the mechanism of PD-L1 expression is oncogene-driven intrinsic constitutive expression, or adaptive upregulation via the STAT pathways in response to IFN, is of unknown importance. Ultimately, the clinical response to PD-1 antibody, in the phase III trial of relapsed GBM, had been disappointing [8].
Another important element that differentiates immunologically ‘hot’ tumours, likely to respond to immunotherapy, from ‘cold’ inert tumours, is mutational burden [79]. With higher mutational burden, greater neoantigens are created, which leads to a greater potential for T-cell repertoire against tumour specific antigens [89]. This is supported by estimates of neoantigens using exome sequencing of tumours, where melanoma and NSCLC have the greatest numbers of somatic mutations and neoantigens predicted. This is clinically validated, as these tumours are known to be particularly sensitive to checkpoint inhibitors. In contrast, GBM sits in the lower third of neoantigens burden in the 30 tumours reported in this study [90].
One subgroup of patients, that have a high mutational load regardless of cancer site of origin are mismatch repair (MMR) tumours. A phase II study showed that in 41 patients with mainly colorectal cancer, those whom were MMR proficient were unresponsive to PD-1 inhibitor; however, those MMR deficient tumours had objective response rates >70% to PD-1 inhibitor [91]. This was also seen in the non-colorectal MMR deficient cancers (n = 7) in this study. On average, the MMR tumour had 100-fold greater somatic mutations than the proficient tumours [91]. This has also been observed in GBM, where a recent case report of two patients with recurrent GBM on a background of paediatric biallelic MMR deficiency, had a deep and durable response to Nivolumab [27].
It has also been well described that recurrent GBM has a high frequency of mutations in the MSH6 MMR gene, which is seen as a consequence to previous Temozolomide treatment and induces a hypermutated phenotype [92].
Interestingly, two recent phase I trials in GBM have shown that it is possible to turn cold tumours hot. Kerstin et al. used a vaccine designed to target neoantigens, personalised to 8 patients, 5 of which had surgical resection following disease progression while on the vaccine treatment. In the two patients, not taking corticosteroids, TIL infiltrate targeting the neoantigens was seen and had upregulated a number of immune checkpoints, potentially accounting for the disease progression [93]. Likewise, using a similar protocol another group used personalised vaccines based on mutation analyses of the transcriptomes and immunopeptidomes of the individual tumours. Two vaccines were administered in 15 patients against both unmutated and neo-antigens. They were able to elicit CD8+ T-cell expansion in the unmutated antigen vaccine and CD4 responses against neoantigens. In a patient who had a response, tumour resection showed a favourable CD8+:regulatory T (Treg) cell ratio and CD4+ T-cell reactivity against one immunised peptide [94].
It may be that a particular subtype of GBM is more immunogenic. Using The Cancer Genome Atlas data, the mesenchymal subtype upregulated both proinflammatory and immunosuppressive gene profiles. For example, there was mRNA rich signature suggesting immune checkpoint activation such as PD-L1, CTLA-4 and galectin-3 (ligand to TIM-3), immunosuppressive monocyte and macrophage recruiters (CCL2, CD163 and CD204) and Treg markers [95]. This mixed immune signature suggests combination immune strategies may be successful for this subtype.
A recent publication has revealed that the poor response rates seen to PD-1 inhibitor monotherapy may be due to the upregulation of multiple checkpoints and a more severely exhausted T-cell phenotype [96]. Woroniecka et al. have studied T-cell exhaustion in GBM in more depth. They isolated TIL and peripheral blood lymphocytes (PBL) from 21 GBM patients and assessed for the presence of multiple immune checkpoint markers including PD-1, CTLA-4, TIM-3, LAG-3, CD160, 2B4, TIGIT, CD39, and BTLA [96]. Interestingly, they found PD-1+CD8+ were present in 96% of patient TIL samples. The CD8+ T-cells were of an effector memory phenotype (CD45RA−CD62L−), however, noted LAG-3, TIGIT, TIM-3 and CD39 were all upregulated, in addition to PD-1. They also measured post-stimulation levels of intracellular IFN-γ, TNF-α and IL-2 by flow cytometry and found cytokines levels were severely suppressed in TIL expressing PD-1, TIM-3 and LAG-3 (triple positive), but not PD-1 single positive. Indeed, the CD8+PD-1+ produced more IL-2 than CD8+PD-1− which the authors reflected demonstrates that PD-1 alone is an activation marker rather than purely a marker of exhaustion. They modelled T-cell exhaustion in two mouse models of GBM and found the TIL population mirrored those found in mouse models of chronic lymphocytic choriomeningitis virus, with the loss of TbethiPD-1int T cells, and the accumulation EomeshiPD-1hi exhausted T cells [97]. Furthermore, the GBM mouse model TILs had more CD8+PD-1+TIM-3+LAG-3+ than equivalent models in melanoma, breast and lung cancer [96]. Simultaneously, a second group published supportive findings to these showing that CD8+ TILs isolated from GBM showed a severe exhausted phenotype [98]. This group also compared relapsed and primary GBM patients’ TILs and tumour transcriptomic immune signature which they found were similar. However, the relapsed patients had a restricted TCR repertoire clonality and thereby supports the role of boasting antigen presentation and priming through DC targeted immunotherapies [98].
4.5. Immunosuppressive Tumour Microenvironment
GBM has a wide variety of mechanisms of immunosuppression, most of which are also common in non-CNS tumours [32]. Over 4 decades ago impaired cell-mediated immunity was described in brain cancer patients driven by a sera-mediated factor likely to be a cytokine [69]. Indeed, this was likely to be TGF-β isoform 2, originally described as glioblastoma-derived T cell suppressor factor, given its discovery in GBM cell lines and patient serum, where it was noted to be immunosuppressive to T-cell proliferation through IL-2 dependent and independent pathways [99,100]. TGF-β is pleiotropic and plays a role in both glioma tumorgenicity and immunosuppression. Its receptor is highly expressed on glioma cells and RNA silencing of TGF-β reduces glioma proliferation, migration and invasiveness. TGF-β also inhibits the transcription factors for the activating immunoreceptor NKG2D and its ligand MHC class I polypeptide-related sequence A (MICA), thus suppressing CD8 T-cell and NK function [101]. MHC class II expression on glioma cells, microglia and macrophages is also reduced following TGF-β exposure [102]. Interestingly, TGF-β2 has been measured pre- and post-operatively in GBM patients, with a favourable prognosis seen in those with a greater reduction in the cytokine, implying an important role of the cytokine in progression [103].
IL-10 is a potent anti-inflammatory cytokine. IL-10 induces immature and tolerogenic DC and hinders cytotoxic T cell effector function through sustaining the FoxP3 transcription factor on Treg [104,105]. IL-10 messenger RNA has been seen in gliomas, with higher levels associated with higher grades [106]. IL-10 production by gliomas seems to polarise the tumour associated macrophages and microglia in the tumour microenvironment [107]. IL-10 also promotes expression of the negative checkpoint molecule PD-L1 on glioma-associated macrophages and peripheral monocytes [80].
Another important immunomodulatory mechanism of DCs and other immune cells is through indolamine 2,3-dioxygenase 1 (IDO) [32]. This orchestrates response to the cytokine environment, for example, it is upregulated in response to interferons [108]. IDO is a cytosolic enzyme which controls tryptophan degradation particularly kynurenine and in addition to the above, facilitates Treg expansions and inhibits T effector function. Furthermore, it can also attract and activate circulating Tregs in the TME to mediate immunosuppression [77]. Overall it, therefore, downregulates the immune response [109,110]. It is highly expressed by GBM (but not normal brain tissue) and levels of IDO expressions by malignant tumours has been correlated to poorer prognosis. Supportive of the above, mice implanted with IDO producing glioma cells (GL261), had increased intra-tumoral Treg accumulation and reduced survival [109]. Furthermore, administration of 1-methyl-tryptophan (an IDO inhibitor) in combination with PD-L1 and CTLA-4 inhibitors resulted in 100% glioma mice survival, improved over PD-L1 and CTLA-4 dual inhibitor therapy [111].
Overall the TME in glioma is profoundly immunosuppressive through a number of cell populations and cytokine factors.
4.6. Corticosteroids
Corticosteroids are universally used to alleviate symptoms of vasogenic oedema in GBM patients. Vasogenic oedema is a major consequence of BBB disruption and increased vascular permeability in GBM, resulting in accumulation of fluid entering the brain extracellular space [112]. Administration of glucocorticoids effectively reduces BBB permeability by reinforcing endothelial tight junctions. In addition, dexamethasone (Dex) can inhibit the effects of tumour-derived VEGF and suppress vascular permeability [113].
This type of oedema is characteristic of brain malignancies that are progressing, but it can also appear after treatment with chemotherapy and radiotherapy due to uncontrolled inflammation of the CNS [114]. Owing to tumour location and volume limitation, cerebral oedema increases intracranial pressure and can compromise neurologic function, causing significant morbidity and mortality [112,115].
Unfortunately, treatment with glucocorticoids also has several negative effects. Of particular relevance is the anti-inflammatory and immunosuppressive action of steroids [116]. Several studies have shown that the use of Dex can lower CD4+ T lymphocytes, as well as exert global immunosuppression by inhibiting the activity of NF-κβ in T-cells [116,117,118]. More recently, studies in GBM animal models and patients indicate that Dex lowers the efficacy of chemotherapy and radiotherapy, resulting in a decreased overall survival [119,120]. Steroids are known to be immunosuppressive and characteristically exert this effect by directing T-cell apoptosis, both at thymic development and in the periphery during activation-induced cell death [121,122]. Indeed clinically, steroids have direct cytotoxic effects on lymphoid origin cancers such as lymphoma and acute lymphoblastic leukaemia whereas have minimal cytotoxic effect on myeloid leukaemias [123]. A group studied the systemic immune suppression in glioma patients (n = 37) including T-cells, myeloid-derived suppressor cells (MDSC) and monocytes and compared patients on and off Dex, to elicit tumour versus corticosteroid-induced immune changes [124]. They found Dex exacerbated lymphopenia in patients compared to healthy controls particularly to the T-cell compartment. In the patients on steroids, although they had similar numbers of monocytes, there was an increase in CD14+HLA-DR−/lo monocytes, which inhibited T cell proliferation and failed to differentiate into MoDC. These cells were also negative for CD80 and had reduced CD86 co-stimulatory molecule expression. Furthermore, these immature cells made up to a third of the monocyte population and were distinct from other CD14+ immune cells such as MDSC [124]. Serum from GBM patients could induce the same changes to healthy monocytes from healthy controls and the authors proposed this to be CCL2 driven. Dex could not induce the loss of HLA-DR+ expression on healthy monocytes, but reduced CCL2 expression in a series of GBM cell lines and, furthermore, inversely correlated with serum CCL2 levels in a dose-dependent manner. Therefore, the authors concluded that tumour induced factors led to the low HLA-DR expression in the monocyte phenotype, however, Dex reduced CCL2 secretion by the tumour, reducing tumour recruitment of monocytes and resulting in increased peripheral CD14+HLA-DR−/lo monocytes [124]. Therefore, these groups suggest the immunosuppressive effects of Dex and tumour-derived factors are additive. Dex is also known to interfere with monocyte differentiation into monocyte-derived DC and production of pro-inflammatory cytokines on maturation [125]. Indeed, measurement of in-vivo circulating DC showed both classical DC and plasmacytoid DC numbers were profoundly suppressed in GBM patients and the numbers were inversely correlated with dexamethasone dose. Furthermore, isolated CD1c+ DC exposed to Dex both in-vivo and in-vitro, showed impaired cytokine secretion and T-cell stimulatory ability [126]. The effect of Dex was recognised as a limiting factor in the success of monocyte-derived DC vaccine trials to treat GBM, particularly with the production of good-quality DCs and hence restricted inclusion criteria to the absence of Dex [127,128]. The Checkmate trial investigating the use of Nivolumab also limited patients to less than 4mg of Dex for trial inclusion.
5. Resistance Mechanisms
Primary resistance to PD-1/PD-L1 blockade has been linked to a transcriptomic signature that was found to be over-represented among non-responding melanoma tumours. This signature was enriched in genes involved in epithelial-to-mesenchymal transition (EMT), angiogenesis, hypoxia and wound healing [129]. These gene signatures were collectively described as signatures of innate anti-PD-1 resistance (IPRES) [129]. There are also higher differentially expressed genes related to immunosuppression (IL-10, VEGFA/C), monocyte and macrophage chemotaxis in non-responders [129]. In a similar fashion, a strong association between EMT and an inflammatory TME was found in lung cancer, where tumours displaying an EMT-like phenotype showed up-regulation of multiple checkpoint molecules (PD-L1/2, Tim-3, CTLA-4) as well as increased Treg infiltration. This suggested that tumours bearing an EMT-like phenotype could trigger immunosuppression through several mechanisms and promote tumour progression [130].
Despite an initial response to therapy, some patients develop secondary resistance and disease progression. In melanoma, 25% of the patients that had responded to PD-1 blockade relapsed and progressed within the first two years of treatment [131]. Genomic analysis of pre-treatment melanoma biopsies and their respective relapsed counterparts identified that alterations in the β-2 microglobulin and JAK1/2 genes were the drivers of the adaptive resistance to PD-1 blockade. Because of their role in the interferon receptor pathway, homozygous loss-of-function mutations in JAK1/2 kinases desensitised cancer cells to IFN signalling, leading to escape from IFN-induced growth inhibition, upregulation of PD-L1 and reduced antigen presentation. On the other hand, the deletion found on the β-2 microglobulin gene prevented the localisation of MHC class I molecules at the cell surface, hence decreasing immunorecognition of cancer cells [132]. Mutations in the JAK1/2 pathway conferring IFN insensitivity and absence of PD-L1 expression has been recently confirmed as a mechanism of primary resistance to PD-1 directed therapies in other studies [133]. Alternate means of adaptive resistance to PD-1 blockade include the expression of alternative checkpoint molecules, such as TIM-3, which appeared to be up-regulated upon tumour progression following an initial response to PD-1 blockade in human and murine samples of lung adenocarcinoma [134]. Indeed, chronic IFN exposure regulates several IFN stimulated genes and T-cell inhibitory receptors, independent of PD-L1 which confers resistance to PD-1 inhibitory agents [135]. For example, epigenetic changes to STAT1 following chronic IFN-γ leads to elevated expression of these genes on resistant tumours [135]. The nature and balance of IFN exposure may regulate when PD-L1-independent adaptive resistance dominates over PD-L1 alone and thus susceptibility to PD-1 therapy (Table 2).
6. Future Immune Strategies and Challenges
Despite the disappointing overall results of the phase III Nivolumab trials in GBM, the information derived from these trials should provide important insights such as favourable subgroups to PD-1 inhibitor treatment. Indeed, the limited success of PD-1 inhibitor monotherapy is not unexpected due to the immunosuppressive barriers in GBM. Furthermore, the cancer immunity cycle reveals the complexity and the multiple steps that are vital for the successful eradication of the tumour [136]. These steps include antigen release and detection, successful priming and activation of the T-cell, followed by successful migration to the tumour site. At the tumour site, the T-cells then must recognise and infiltrate the tumour and subsequently initiate cytotoxicity [136]. Blockade of the PD-1/PD-L1 axis, which predominately dampens cancer immunity in the final steps at the tumour site, is, therefore, targeting one arm of this cycle. This cycle also assumes a T-cell focus for effective immunity against cancer, however natural killer cells, MDSC, macrophages, B-cells may be more than just attenuators in the local TME. Figure 1 summarises some of the approaches already undergoing in clinical trials for GBM, matching them with the process of the cancer immunity cycle that they target.
In C57BL/6 mice implanted with murine glioma cell line GL261-luc2, the use of combination treatment such as radiotherapy, anti-PD-1 and anti-TIM3 has been shown to be synergistic and improve survival above dual therapy [137]. Likewise, in GBM mouse model, the use of toll-like receptor agonist PolyI:C to mature DC combined with anti-PD-1 improved survival than either treatment alone. The combination treatment increased rates of T-effector cells and decreased Treg which reinforces the rationale of targeting two components of the cancer immunity cycle [138].
These approaches only represent a few of over a thousand trial combinations involving a checkpoint blockade backbone that is being tested in oncology trials. Although this is the treatment avenue most likely to achieve success in the management of GBM, it also raises economic challenges for pharmaceutical companies and healthcare systems. Furthermore, the optimum combination may be unique to a tumour type depending on the predominant barrier to immunity. For example, in pancreatic cancer, dense in stromal tissue, a mouse model has shown depletion of carcinoma-associated fibroblast expressing fibroblast activation protein (FAP), restores sensitivity to checkpoint blockade [139]. Whereas, in other cancers, such as renal cell carcinoma, Treg may predominate to attenuate the effect of PD-1/L1 axis inhibition, as supported by higher Treg fraction been a biomarker of poor response in an early clinical trial of Atezolizumab in the treatment of this cancer [140].
7. Concluding Remarks
PD-1 inhibitors have revolutionised the treatment of cancers such as NSCLC, bladder cancer and metastatic melanoma. These tumours have a high mutational burden and have pre-existing immunity signature on biopsy as represented by high CD8 TILs, PD-L1 expression and IFN-γ enriched signature. In the tumour immunity continuum spanning from pro-inflammatory to immunologically ignorant tumours, GBM would be in the mid-lower end of this spectrum. In addition to targeting immunosuppressive barriers in the TME, the success in the treatment of GBM will likely depend on developing a rational combination therapy able to induce a long-lasting anti-tumour immunity. These combination strategies are not unique to GBM and are now the forefront of questions for the immuno-oncology field to address, particularly in the PD-1/L1 axis inhibition non-responsive and secondary resistant tumours.
Author Contributions
Conceptualization and writing by J.A.; J.M.-V. and P.M.P., supervision by A.M.J. and P.M.P. and review and editing by A.P.K., A.M.J. and P.M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest.
References
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-Positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; de Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs. bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol. 2017, 19, iii21-iii21. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089. [Google Scholar]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug. Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.S.J.; Sahebjam, S. Safety and activity of nivolumab monotherapy and nivolumab in combination with ipilimumab in recurrent glioblastoma: Updated results from CheckMate 143. In Proceedings of the 52nd Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 2–7 June 2016. [Google Scholar]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro. Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Reardon, D.A.; De Groot, J.F.; Colman, H.; Jordan, J.T.; Daras, M.; Clarke, J.L.; Peters, K.B. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Sahebjam, S.J.P.; Forsyth, P.A. Safety and antitumor activity of hypofractionated stereotactic irradiation (HFSRT) with pembrolizumab (Pembro) and bevacizumab (Bev) in patients (pts) with recurrent high grade gliomas: Preliminary results from phase I study. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Blumenthal, D.T. Pembrolizumab: First experience with recurrent primary central nervous system (CNS) tumours. Neuro Oncol. 2015, 17, v107. [Google Scholar] [CrossRef] [Green Version]
- Leibowitz-Amit, R. Initial clinical experience with pembrolizumab in metastatic heavily pre-treated patients with solid cancers in a single institution. Eur. J. Cancer 2015, 51, S111. [Google Scholar] [CrossRef]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of hypermutated glioblastoma: A patient with germline pole deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Carter, T.; Shaw, H.M.; Cohn-Brown, D.; Chester, K.; Mulholland, P.J. Sequential immune checkpoint inhibition with concurrent bevacizumab for relapsed glioblastoma: A single centre experience. J. Clin. Oncol. 2016, 34, e13514-e13514. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: An update. J. Clin. Oncol. 2017, 35, 2042-2042. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Donovan, L. OS09.5 synergistic effect of reirradiation and PD-1 inhibitors in recurrent high-grade gliomas. Neuro Oncol. 2017, 19, iii19-iii19. [Google Scholar] [CrossRef] [Green Version]
- Roth, P.; Valavanis, A.; Weller, M. Long-term control and partial remission after initial pseudoprogression of glioblastoma by anti–PD-1 treatment with nivolumab. Neuro Oncol. 2017, 19, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Perng, P.; Lim, M. Immunosuppressive mechanisms of malignant gliomas: Parallels at Non-CNS sites. Front. Oncol. 2015, 5, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarry, U.; Jeannin, P.; Pineau, L.; Donnou, S.; Delneste, Y.; Couez, D. Efficiently stimulated adult microglia cross-prime naive CD8+ T cells injected in the brain. Eur. J. Immunol. 2013, 43, 1173–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The controversial role of microglia in malignant gliomas. Clin. Dev. Immunol. 2013, 2013, 285246. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Zou, J.P.; Morford, L.A.; Chougnet, C.; Dix, A.R.; Brooks, A.G.; Torres, N.; Shuman, J.D.; Coligan, J.E.; Brooks, W.H.; Roszman, T.L.; et al. Human glioma-induced immunosuppression involves soluble factor(s) that alters monocyte cytokine profile and surface markers. J. Immunol. 1999, 162, 4882–4892. [Google Scholar]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kalin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 2018, 48, 380–395. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, A.M.; Simeone, E.; Giannarelli, D.; Muto, P.; Falivene, S.; Borzillo, V.; Giugliano, F.M.; Sandomenico, F.; Petrillo, A.; Curvietto, M.; et al. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunology 2014, 3, e28780. [Google Scholar] [CrossRef]
- Weller, R.O.; Engelhardt, B.; Phillips, M.J. Lymphocyte targeting of the central nervous system: A review of afferent and efferent CNS-immune pathways. Brain Pathol. 1996, 6, 275–288. [Google Scholar] [CrossRef]
- Laman, J.D.; Weller, R.O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharm. 2013, 8, 840–856. [Google Scholar] [CrossRef]
- Calzascia, T.; Masson, F.; Di Berardino-Besson, W.; Contassot, E.; Wilmotte, R.; Aurrand-Lions, M.; Ruegg, C.; Dietrich, P.Y.; Walker, P.R. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 2005, 22, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivisakk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+ T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ifergan, I.; Kebir, H.; Alvarez, J.I.; Marceau, G.; Bernard, M.; Bourbonniere, L.; Poirier, J.; Duquette, P.; Talbot, P.J.; Arbour, N.; et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 2011, 134, 3560–3577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef]
- Giunti, D.; Borsellino, G.; Benelli, R.; Marchese, M.; Capello, E.; Valle, M.T.; Pedemonte, E.; Noonan, D.; Albini, A.; Bernardi, G.; et al. Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. J. Leukoc. Biol. 2003, 73, 584–590. [Google Scholar] [CrossRef]
- Lossinsky, A.S.; Shivers, R.R. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol. Histopathol. 2004, 19, 535–564. [Google Scholar] [CrossRef]
- Konnecke, H.; Bechmann, I. The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. J. Clin. Dev. Immunol. 2013, 2013, 15. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.; Wang, Y.; Calder, V.L. Lymphocyte adhesion and transendothelial migration in the central nervous system: The role of LFA-1, ICAM-1, VLA-4 and VCAM-1. Immunology 1995, 86, 408–415. [Google Scholar]
- Cayrol, R.; Wosik, K.; Berard, J.L.; Dodelet-Devillers, A.; Ifergan, I.; Kebir, H.; Haqqani, A.S.; Kreymborg, K.; Krug, S.; Moumdjian, R.; et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat. Immunol. 2008, 9, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, F.; Zhu, C.; Pardridge, W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002, 81, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Terasaki, T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm. Res. 2007, 24, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Gettinger, S.N.; Mahajan, A.; Chiang, A.C.; Herbst, R.S.; Sznol, M.; Tsiouris, A.J.; Cohen, J.; Vortmeyer, A.; Jilaveanu, L.; et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: Early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 976–983. [Google Scholar] [CrossRef] [Green Version]
- Amayiri, N.; Tabori, U.; Campbell, B.; Bakry, D.; Aronson, M.; Durno, C.; Rakopoulos, P.; Malkin, D.; Qaddoumi, I.; Musharbash, A.; et al. High frequency of mismatch repair deficiency among pediatric high grade gliomas in Jordan. Int. J. Cancer 2016, 138, 380–385. [Google Scholar] [CrossRef]
- Roszman, T.L.; Brooks, W.H. Immunobiology of primary intracranial tumours. III. Demonstration of a qualitative lymphocyte abnormality in patients with primary brain tumours. Clin. Exp. Immunol. 1980, 39, 395–402. [Google Scholar]
- Brooks, W.H.; Netsky, M.G.; Normansell, D.E.; Horwitz, D.A. Depressed cell-meidated immunity in patients with primary intracranial tumours. J. Exp. Med. 1972, 136, 1631–1647. [Google Scholar] [CrossRef]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- Colombo, M.P.; Piconese, S. Regulatory-T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 2007, 7, 880–887. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E., 2nd; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, L.; Di Lorenzo, N.; Guidetti, B. Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. Incidence, fate, and relevance to prognosis in 228 operated cases. J. Neurosurg. 1978, 49, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Zhang, X.; Ye, H.X.; Wang, Y.; Du, Z.G.; Yao, Y.; Mao, Y. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. J. Neurooncol. 2014, 116, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, B.; Xu, X.; Zhang, S.; Yan, X.; Kong, F.; Feng, X.; Wang, J. Clinical significance of FOXP3 expression in human gliomas. Clin. Transl. Oncol. 2014, 16, 36–43. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Sengupta, S.; Han, Y.; Lesniak, M.S. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. 2011, 13, 1308–1323. [Google Scholar] [CrossRef] [Green Version]
- Vasco, C.; Canazza, A.; Rizzo, A.; Mossa, A.; Corsini, E.; Silvani, A.; Fariselli, L.; Salmaggi, A.; Ciusani, E. Circulating T regulatory cells migration and phenotype in glioblastoma patients: An in vitro study. J. Neurooncol. 2013, 115, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Wintterle, S.; Schreiner, B.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Meyermann, R.; Weller, M.; Wiendl, H. Expression of the B7-related molecule B7-H1 by glioma cells: A potential mechanism of immune paralysis. Cancer Res. 2003, 63, 7462–7467. [Google Scholar]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Srividya, M.R.; Thota, B.; Shailaja, B.C.; Arivazhagan, A.; Thennarasu, K.; Chandramouli, B.A.; Hegde, A.S.; Santosh, V. Homozygous 10q23/PTEN deletion and its impact on outcome in glioblastoma: A prospective translational study on a uniformly treated cohort of adult patients. Neuropathology 2011, 31, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227. [Google Scholar] [CrossRef] [Green Version]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Baylot, V.; Felsher, D.W. MYC: Master regulator of immune privilege. Trends Immunol. 2017, 38, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Doucette, T.; Rao, G.; Rao, A.; Shen, L.; Aldape, K.; Wei, J.; Dziurzynski, K.; Gilbert, M.; Heimberger, A.B. Immune heterogeneity of glioblastoma subtypes: Extrapolation from the cancer genome atlas. Cancer Immunol. Res. 2013, 1, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Mohme, M.; Schliffke, S.; Maire, C.L.; Runger, A.; Glau, L.; Mende, K.C.; Matschke, J.; Gehbauer, C.; Akyuz, N.; Zapf, S.; et al. Immunophenotyping of newly diagnosed and recurrent glioblastoma defines distinct immune exhaustion profiles in peripheral and tumor-infiltrating lymphocytes. Clin. Cancer Res. 2018, 24, 4187–4200. [Google Scholar] [CrossRef] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Fontana, A.; Hengartner, H.; de Tribolet, N.; Weber, E. Glioblastoma cells release interleukin 1 and factors inhibiting interleukin 2-mediated effects. J. Immunol. 1984, 132, 1837–1844. [Google Scholar]
- Siepl, C.; Bodmer, S.; Frei, K.; MacDonald, H.R.; De Martin, R.; Hofer, E.; Fontana, A. The glioblastoma-derived T cell suppressor factor/transforming growth factor-beta 2 inhibits T cell growth without affecting the interaction of interleukin 2 with its receptor. Eur. J. Immunol. 1988, 18, 593–600. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Sailer, M.; Ansorge, S.; Firsching, R.; Reinhold, D. Increased concentrations of transforming growth factor beta1 and beta2 in the plasma of patients with glioblastoma. J. Neurooncol. 2006, 79, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Huettner, C.; Paulus, W.; Roggendorf, W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am. J. Pathol. 1995, 146, 317–322. [Google Scholar] [PubMed]
- Wagner, S.; Czub, S.; Greif, M.; Vince, G.H.; Suss, N.; Kerkau, S.; Rieckmann, P.; Roggendorf, W.; Roosen, K.; Tonn, J.C. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int. J. Cancer 1999, 82, 12–16. [Google Scholar] [CrossRef]
- Carlin, J.M.; Borden, E.C.; Sondel, P.M.; Byrne, G.I. Biologic-response-modifier-induced indoleamine 2,3-dioxygenase activity in human peripheral blood mononuclear cell cultures. J. Immunol. 1987, 139, 2414–2418. [Google Scholar]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [Green Version]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [Green Version]
- Kotsarini, C.; Griffiths, P.D.; Wilkinson, I.D.; Hoggard, N. A systematic review of the literature on the effects of dexamethasone on the brain from in vivo human-based studies: Implications for physiological brain imaging of patients with intracranial tumors. Neurosurgery 2010, 67, 1799–1815, discussion 1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, J.D.; Papavassiliou, E.; Merrill, M.J.; Nieman, L.; Knightly, J.J.; Walbridge, S.; Edwards, N.A.; Oldfield, E.H. Mechanism of dexamethasone suppression of brain tumor-associated vascular permeability in rats. Involvement of the glucocorticoid receptor and vascular permeability factor. J. Clin. Investig. 1996, 98, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S.; Consortium, N.C. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, M.C.; Saadoun, S.; Binder, D.K.; Manley, G.T.; Krishna, S.; Verkman, A.S. Molecular mechanisms of brain tumor edema. Neuroscience 2004, 129, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Bourke, E.; Moynagh, P.N. Antiinflammatory Effects of Glucocorticoids in Brain Cells, Independent of NF-KB. J. Immunol. 1999, 163, 2113. [Google Scholar] [PubMed]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-KB activity through induction of IKB synthesis. Science 1995, 270, 286. [Google Scholar] [CrossRef]
- Hughes, M.A.; Parisi, M.; Grossman, S.; Kleinberg, L. Primary brain tumors treated with steroids and radiotherapy: Low CD4 counts and risk of infection. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1423–1426. [Google Scholar] [CrossRef]
- Wong, E.T.; Lok, E.; Gautam, S.; Swanson, K.D. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br. J. Cancer 2015, 113, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef] [Green Version]
- Herold, M.J.; McPherson, K.G.; Reichardt, H.M. Glucocorticoids in T cell apoptosis and function. Cell Mol. Life Sci. 2006, 63, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, J.; Rao, K.; Pastorino, S.; Kesari, S. Corticosteroids in brain cancer patients: Benefits and pitfalls. Expert Rev. Clin. Pharmacol. 2011, 4, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Pufall, M.A. Glucocorticoids and cancer. Adv. Exp. Med. Biol. 2015, 872, 315–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010, 12, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piemonti, L.; Monti, P.; Allavena, P.; Sironi, M.; Soldini, L.; Leone, B.E.; Socci, C.; Di Carlo, V. Glucocorticoids Affect Human Dendritic Cell Differentiation and Maturation. J. Immunol. 1999, 162, 6473. [Google Scholar]
- Adhikaree, J.; Franks, H.A.; Televantos, C.; Vaghela, P.; Kaur, A.P.; Walker, D.; Schmitz, M.; Jackson, A.M.; Patel, P.M. Impaired circulating myeloid CD1c+ dendritic cell function in human glioblastoma is restored by p38 inhibition—Implications for the next generation of DC vaccines. Oncoimmunology 2019, 8, 1593803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gool, S.; Maes, W.; Ardon, H.; Verschuere, T.; Van Cauter, S.; De Vleeschouwer, S. Dendritic cell therapy of high-grade gliomas. Brain Pathol. 2009, 19, 694–712. [Google Scholar] [CrossRef]
- Rozkova, D.; Horvath, R.; Bartunkova, J.; Spisek, R. Glucocorticoids severely impair differentiation and antigen presenting function of dendritic cells despite upregulation of Toll-like receptors. Clin. Immunol. 2006, 120, 260–271. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.; Diao, L.; Cuentas, E.R.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of Pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.; Pauken, K.E.; Huang, A.C.; et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination therapy with Anti-PD-1, Anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fasso, M.; Wang, Y.V.; et al. Atezolizumab, an Anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: Long-term safety, clinical activity, and immune correlates from a phase Ia study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic of combination therapies with PD-1/L1 inhibitors (outer circle) targeting different aspects of the cancer-immunity cycle (inner circle).
Figure 1.
Schematic of combination therapies with PD-1/L1 inhibitors (outer circle) targeting different aspects of the cancer-immunity cycle (inner circle).


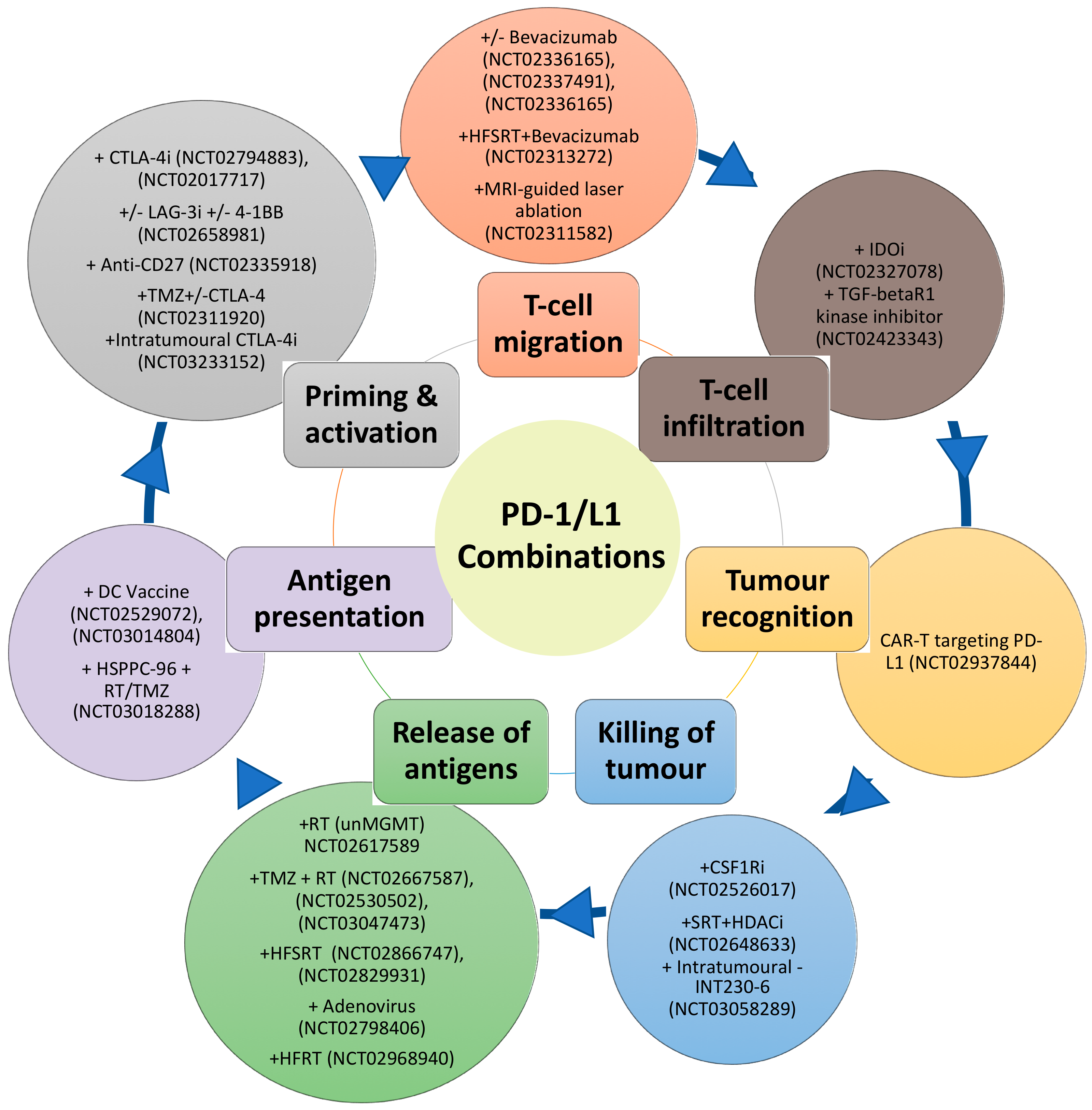{kind=link}
Table 1.
Clinical efficacy of programme death 1 (PD-1) therapy in Glioblastoma.
| Drug | Patient Cohort | Study Type | Toxicity | Efficacy | Reference |
|---|---|---|---|---|---|
| Pembrolizumab | Recurrent GBM (n = 9), AA (n = 2), BSG (n = 1), Paeds (n = 5) | Retrospective | Safe | No responses | Blumenthal et al. 2015 [24] |
| Pembrolizumab + Bevacizumab | Recurrent GBM N = 6 | Phase I | Safe | 1 PD 2 SD 3 PD | Reardon et al. 2016 [22] |
| Pembrolizumab + hypo-fractionated RT + Bevacizumab | Recurrent GBM or AA N = 3 | Phase I | Safe | 1 CR 2 SD | Sahebjam et al. 2016 [23] |
| Pembrolizumab | Recurrent solid tumours, Glioma (n = 3) | Case series | Safe | 1 MR | Leibowitz-amit et al. 2015 [25] |
| Pembrolizumab | Hypermutated GBM (POLE germline deficiency) | Case study | Safe | 1 PR | Johanns et al. 2016 [26] |
| Nivolumab +/− Ipilimumab | Recurrent GBM Monotherapy (3 mg/kg) (n = 10) | Phase I | 0 grade 3–4 AE 1 discontinued due to AE | 1 PR 5 SD 3 PD | Reardon et al. 2016 [20] |
| Nivolumab 1 mg/Ipilimumab 3 mg (n = 10) | Phase I | 9/10 grade 3–4 AE 5 discontinued | 0 PR 4 SD 6 PD | ||
| Nivolumab 3 mg/Ipilimumab 1 mg (n = 20) | Phase I | 5/20 grade 3–4 AE 2 discontinued | 0 PR 10 SD 9 PD | ||
| Nivolumab | Paediatric GBM with biallelic mismatch repair deficiency (n = 2) | Case study | 1 seizure at initiation | 2 PR | Bouffet et al. 2016 [27] |
| Pembrolizumab + Bevacizumab + GMCSF | Recurrent GBM (n = 4) | Case series | Safe | 1 PR 3 SD | Brown et al. 2016 [28] |
| Durvalumab | Recurrent GBM (n = 31) | Phase II, cohort B | Safe 9.7% Grade 3–4 AE | 4 PR 14 SD 12mOS −44.4% | Reardon et al. 2017 [29] |
| PD-1 inhibitors + RT | Recurrent HGG (n = 20) | Case series | Safe | 7 PR 5 SD 8PD | Iwamoto et al. 2017 [30] |
| Nivolumab | Recurrent GBM | Case report | Safe | PR | Roth et al. 2017 [31] |
| Nivolumab vs. Bevacizumab | Recurrent GBM (n = 369) | Phase III | 13% grade 3–4 toxicity Nivolumab | RR 8% vs. 23% (Nivolumab vs. Bevacizumab | Reardon et al. 2017 [8] |
| Nivolumab + RT vs. TMZ + RT | 1st line (n = 550) unmethylated-MGMT | Phase III | not published | failed to extend OS and PFS | BMS Press release |
| Nivolumab+TMZ + RT vs. TMZ + RT | 1st line (n = 693) methylated MGMT | Phase III | not published | failed to extend PFS | BMS Press release |
RT = radiotherapy; TMZ = Temozolomide; PR = partial response; SD = stable disease; MR = mixed response; PD = progressive disease; HGG = high grade glioma; AA = anaplastic astrocytoma; BSG = brain stem glioma; Paeds = paediatric; POLE = DNA polymerase epsilon deficiency; OS = overall survival; PFS = progression-free survival; GMCSF = granulocyte-macrophage colony-stimulating factor.
Table 2.
Restriction to PD-1 inhibitor response.
| GBM Specific Barriers | Primary Resistance * | Secondary Resistance * |
|---|---|---|
| Low-intermediate PD-L1 expression | Enriched genes epithelial-mesenchymal transition | Alterations β2 microglobulin |
| Low-Intermediate mutational burden | Angiogenesis, wound healing, hypoxia, IL-10, VEGF-A/C gene signature | Alteration JAK1/2 genes |
| Low-intermediate CD8 TIL infiltrate | High monocyte, macrophage chemotaxis genes | Upregulation of alternate checkpoints e.g., TIM-3 |
| Blood–brain barrier | ||
| High corticosteroid use | ||
| Low dendritic cell populations | ||
| High IL-10, TGF-β, CCL2 and IDO suppressive humoral factors |
* in all tumours.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020263
AMA Style
Adhikaree J, Moreno-Vicente J, Kaur AP, Jackson AM, Patel PM. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells. 2020; 9(2):263. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020263
Chicago/Turabian StyleAdhikaree, Jason, Julia Moreno-Vicente, Aanchal Preet Kaur, Andrew Mark Jackson, and Poulam M. Patel. 2020. "Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma" Cells 9, no. 2: 263. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020263
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.