miRNAs as Key Players in the Management of Cutaneous Melanoma


Molecular Diagnostics and Pharmacogenetics Unit, IRCCS-Istituto Tumori “Giovanni Paolo II”, 70124 Bari, Italy
*
Author to whom correspondence should be addressed.
†
Equally contributed to this paper.
‡
Equally contributed to this paper.
Cells 2020, 9(2), 415; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020415
Submission received: 4 December 2019
/
Revised: 6 February 2020
/
Accepted: 7 February 2020
/
Published: 11 February 2020
(This article belongs to the Special Issue microRNA as Biomarker)
Abstract
:The number of treatment options for melanoma patients has grown in the past few years, leading to considerable improvements in both overall and progression-free survival. Targeted therapies and immune checkpoint inhibitors have opened a new era in the management of melanoma patients. Despite the clinical advances, further research efforts are needed to identify other “druggable” targets and new biomarkers to improve the stratification of melanoma patients who could really benefit from targeted and immunotherapies. To this end, many studies have focused on the role of microRNAs (miRNAs) that are small non-coding RNAs (18-25 nucleotides in length), which post-transcriptionally regulate the expression of their targets. In cancer, they can behave either as oncogenes or oncosuppressive genes and play a central role in many intracellular pathways involved in proliferation and invasion. Given their modulating activity on the transcriptional landscape, their biological role is under investigation to study resistance mechanisms. They are able to mediate the communication between tumor cells and their microenvironment and regulate tumor immunity through direct regulation of the genes involved in immune activation or suppression. To date, a very promising miRNA-based strategy is to use them as prognosis and diagnosis biomarkers both as cell-free miRNAs and extracellular-vesicle miRNAs. However, miRNAs have a complex role since they target different genes in different cellular conditions. Thus, the ultimate aim of studies has been to recapitulate their role in melanoma in biological networks that account for miRNA/gene expression and mutational state. In this review, we will provide an overview of current scientific knowledge regarding the oncogenic or oncosuppressive role of miRNAs in melanoma and their use as biomarkers, with respect to approved therapies for melanoma treatment.
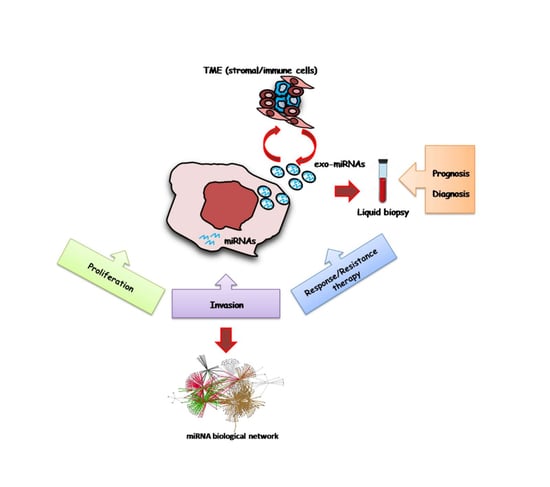
1. Introduction
Cutaneous melanoma is one of the most aggressive types of cancer worldwide, with an incidence that has been increasing over the past 50 years [1].
It occurs from the malignant transformation of melanocytes induced mainly by exposure to intense and prolonged ultraviolet radiation. Unfortunately, this disease can progress rapidly to the advanced aggressive metastatic stage. Depending on the stage and the dissemination of the tumor, different therapeutic options are relied on, such as surgical resection, chemotherapy, radiotherapy, immunotherapy, or targeted therapy. During the past decade, systemic treatment for melanoma has enormously changed as knowledge of the key driver mutations and pathways of tumor cells have led to the development of new therapeutic options. Targeted molecular therapies against specific mutations [2,3] and systemic immunotherapies including immune checkpoint inhibitors [4,5,6], have recently emerged, replacing conventional chemotherapy [7].Notwithstanding the success of the new therapeutic approaches, many research groups have been working to enhance our knowledge of melanoma biology and to identify further reliable biomarkers. Recently, many studies have focused on microRNAs (miRNAs) as important factors in the development, metastasis, and prognosis of melanoma. miRNAs are short (20–25 nucleotides) and single-stranded non-coding RNAs, which target the 3′-untranslated region of an estimated 30% of all human genes at its 5′end domain (position 2 to 8), called the “seed region,” and thus play a role as oncogenes or oncosuppressors [8]. They regulate gene expression at the post-transcriptional and translational levels through degradation of mRNA or a translation block. This review discusses the state of the art of studies on miRNAs and their role in response to therapies, their relationship with other “druggable” pathways, and their potential use as clinical biomarkers.
2. State of the Art of Targeted Therapy Options
Novel treatments for cutaneous metastatic melanoma (CMM) have been proposed based on the central role of the mitogen activated-protein kinase (MAPK) pathway in this disease. The MAPK pathway is activated by extracellular binding of receptor tyrosine kinases (RTK), leading to the activation of the rat sarcoma (RAS) family protein, which subsequently activates intracellular serine-threonine protein kinases of the rapidly accelerated fibrosarcoma (RAF) family (ARAF, BRAF, CRAF). RAF activation triggers the phosphorylation of MAPK extracellular receptor kinase (MEK), which in turn, phosphorylates extracellular signal-regulated kinase (ERK). ERK regulates both cytosolic targets and nuclear transcription factors, thus leading to cellular proliferation, cellular differentiation, survival, and apoptosis. Activated ERK also provides negative feedback at various levels of the pathway [9]. BRAF mutations are more frequent in melanomas that develop in sun-exposed skin. About 50% of CMM harbor an activating mutation of the BRAF gene that consists in the substitution of a single nucleotide in codon 600. The most common mutation is the result of a substitution of glutamic acid for valine in codon 600 (BRAFV600E), which occurs in approximately 90% of BRAF-mutant melanomas. The second most common mutation is BRAFV600K (substituting valine for lysine), which accounts for 5%–6% of BRAF-mutant melanomas [10].
Different clinical characteristics (i.e., gender, age) have been reported between patients with BRAF p.V600E and p.V600K mutation. Vemurafenib and Dabrafenib are selective oral BRAF inhibitors (BRAFi) that have been licensed by the Food and Drug Administration (FDA, Hampton, VA, USA) for the treatment of unresectable or metastatic melanomas harboring activating BRAFV600 mutations [2,11,12]. The initial efficacy of BRAFi is followed by multiple resistance mechanisms caused by inter-tumor (differences between primary and metastatic tumors) or intra-tumor (differences in features of subclones within a tumor) heterogeneity. These mechanisms usually depend on the recovery of the MAPK pathway or the activation of other pathways such as the PI3K/AKT/mTOR pathway through IGF1R and PDGFRb upregulation [13,14].
For this reason, a valid strategy is to target downstream signaling effectors like MEK 1/2. Cobimetinib and Trametinib are oral selective MEK inhibitors (MEKi) approved by the FDA for the treatment of unresectable or metastatic melanomas [11,12]. Clinical trials have shown that BRAFi/MEKi combination therapy improves survival compared to single-agent treatment [15].
MEKi has proven to be effective also in NRAS-mutant melanomas [16]. NRAS and BRAF mutations are usually mutually exclusive in melanoma. The most common RAS mutations occur in codons 12, 61, or 13; 15% of cases have point mutations. RAS proteins function as small GTPases with low intrinsic catalytic activity. Cycling of the RAS protein between a guanosine-5’-triphosphate (GTP)-bound active state and a guanosine diphosphate (GDP)-bound inactive state is catalyzed, respectively, by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). Mutations in NRAS favor the formation of GTP-bound, activating RAS proteins. One of the approaches to inhibit RAS activation has been to directly target RAS by competing for its GTP binding, similar to kinase inhibitors, which compete with ATP.
However, to date, direct pharmacological inhibition of mutant RAS proteins is difficult because of their very tight binding to GDP/GTP [17]. No therapy has yet been approved for NRAS-mutant melanoma. Two MEKi, Binimetinib, and Pimasertib, have been shown to improve progression-free survival (PFS) vs. Dacarbazine significantly but have not proved to provide an overall survival (OS) benefit. Double inhibition with MEKi in combination with PI3Ki or PI3K/mTORi or AKT inhibitors has been used in clinical trials.
Unfortunately, such a combination was too toxic to allow adequate dosing for an antitumor effect, thus leaving a lack of targeted approaches for NRAS-driven melanomas [18]. There are no treatment options available for wildtype-BRAF/NRAS melanomas, which constitute ~30% of all CMMs. Another potentially actionable target gene is c-KIT, a tyrosine-protein kinase that encodes for a receptor essential for the proliferation and survival of mature melanocytes. KIT is often altered in mucosal malignant melanomas, where it activates intracellular signaling cascades, including the MAPK, PI3K, and JAK-STAT pathways. The most common c-KIT mutations in melanoma are L676P and K642E [19]. Trials conducted with Imatinib mesylate, an inhibitor of KIT and other RTKs, in patients with c-KIT–mutant melanoma, have reported median times to disease progression of approximately 3 months that are significantly lower than the time to progression when Imatinib is used to treat gastrointestinal stromal tumors (GIST) (median time to progression of 18 months). Despite the presence of the same mutation, it is unclear why there is such a difference in response between KIT-mutant melanoma and GIST, suggesting that there may be other pathways involved in this treatment resistance [19]. The neurofibromin 1 (NF1) gene is considered one of the driver genes in melanomas, specifically in chronically sun-exposed or older subjects and in desmoplastic melanoma. It encodes for a GTPase-activating protein that inhibits the activity of RAS proteins and negatively regulates MAPK signaling. NF1 mutations are present in <15% of melanoma cases and may be present together with NRAS/BRAF mutations. Combination therapy with MEKi and mTORi has been observed to produce an antitumor effect in BRAF/NF1-mutant allografts [20].
2.1. miRNAs Acting as Oncosuppressors in Melanoma
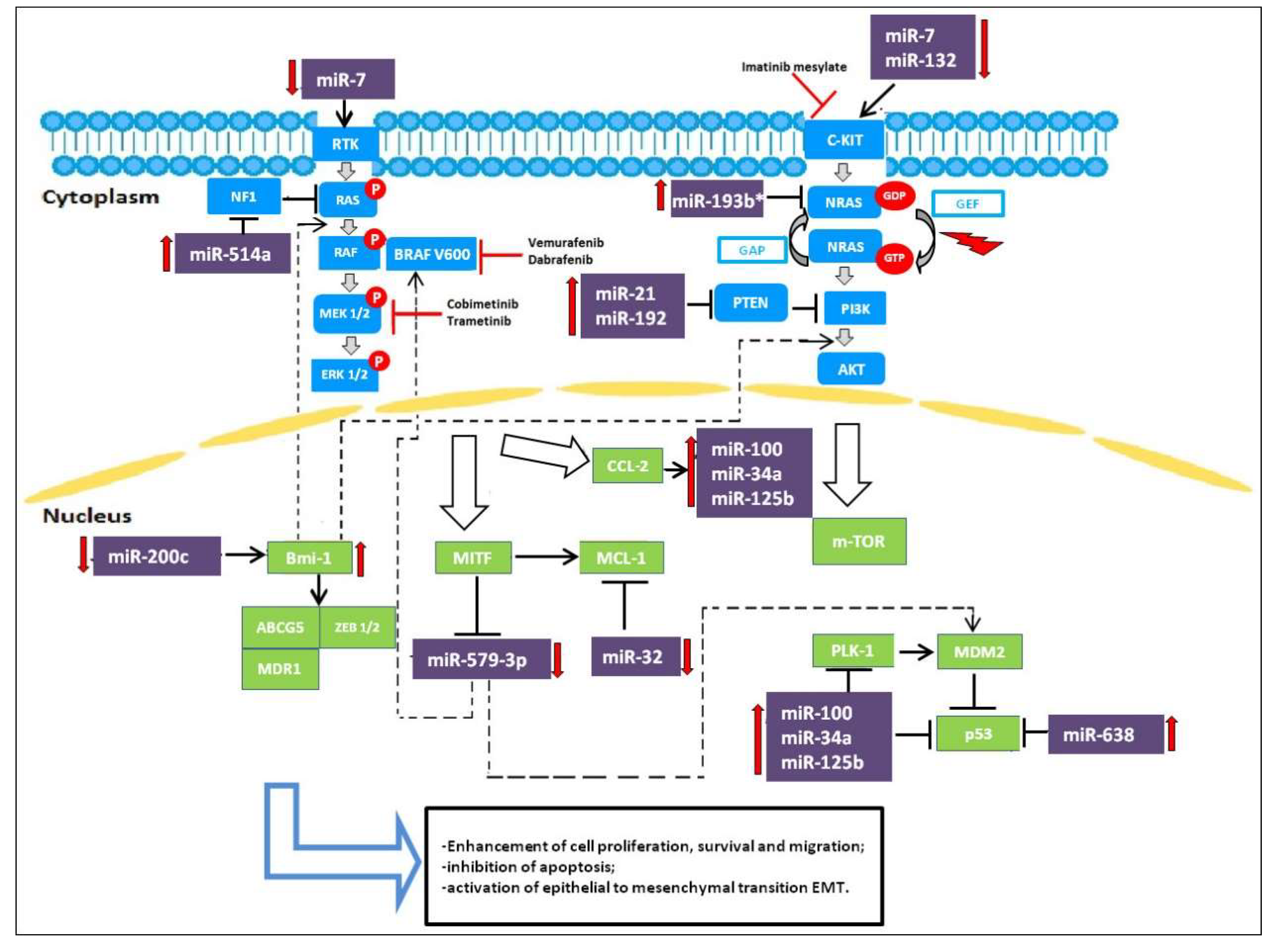
Following the efforts to understand the molecular basis of drug resistance and to establish combination therapies able to revert acquired resistance, studies have focused on miRNAs and their role in controlling and/or reverting drug resistance. In this context, miRNAs act as oncosuppressors. Figure 1 summarizes the involvement of miRNAs in the pathways active in melanoma.
Only a few studies have regarded the impact of miRNAs on BRAFi/MEKi-based targeted therapies. miR-32, a tumor suppressor miRNA, has recently been demonstrated to suppress the growth of melanoma tumors in preclinical models by inhibiting the expression of the myeloid cell leukemia 1 (MCL-1) gene regulating the RAS-RAF-MEK-ERK and the PI3K-AKT-mTOR pathways. Inhibition of MCL-1 through miR-32 may be an effective anti-melanoma strategy, regardless of the status of NRAS, BRAF or PTEN, as MCL-1 inhibition exhibits synergistic effects with Vemurafenib [21]. Low miR-579-3p expression in BRAF-mutated cells is linked to metastatic melanoma progression; expression levels of miR-579-3p decrease from nevi to stage III/IV melanoma samples and even further in cell lines resistant to BRAF/MEK inhibitors. miR-579-3p is able to simultaneously affect both the BRAF/MAPK and the MDM2/p53 pathways. Indeed, in BRAF-mutated cells, further reduction of miR-579-3p expression leads to additional increases in BRAF and MDM2 levels, causing uncontrolled cell growth, enhanced cell proliferation and migration, and inhibition of apoptosis. This condition contributes to the establishment of resistance to targeted therapy. In this regard, it is important to understand the mechanisms responsible for miR-579-3p expression. This miRNA could be regulated by specific transcription factors whose expression is altered during the development of drug resistance. A possible role could be played by a microphthalmia-associated transcription factor (MITF). miR-579-3p is an intronic miR located in intron 11 of the human gene ZFR (Zink-finger recombinase), and it is probably co-expressed with its host gene. Considering that the ZFR gene is supposed to be a target of MITF because its promoter has MITF-consensus binding sites, it has been speculated that MITF downregulation during the development of drug resistance is responsible for decreased expression of downstream miR-579-3p. Hence, miR-579-3p acts not only as an oncosuppressor but also as a factor contributing to the development of drug resistance [22].
Another potent oncosuppressive miRNA is miR-200c. miR-200c expression is significantly reduced in resistant melanoma cells [23]. miR-200c has been proposed to prevent the establishment of drug resistance in melanoma by targeting Bmi-1, Zeb2, Tubb3, ABCG5, and MDR1, transcriptional repressors that belong to a complex signaling network involved in the epithelial to mesenchymal transition (EMT). Reduction of miR-200c increases Bmi-1 expression, which in turn leads to the activation of the PI3K/AKT and MAPK pathways and the acquisition of features seen in EMT, such as downregulation of E-cadherin and upregulation of N-cadherin, ABCG5, and MDR1 [24]. Low levels of miR-200c have been seen to be correlated with the development of resistance to BRAFi in clinical samples of melanomas and BRAFi-resistant cell lines. Restoration of miR-200c expression or knockdown of Bmi-1 in resistant melanoma cells potentiates the effect of MAPK pathway inhibitory drugs and impairs the establishment of resistance, thus suggesting miR-200c as a potential therapeutic target for overcoming acquired BRAFi resistance [24].
miR-7 expression has been shown to decrease in BRAFi-resistant melanoma cells. Introduction of miR-7 decreases the expression levels of EGFR, IGF-1R, CRAF in vitro as well as in Vemurafenib-resistant cells in melanoma xenograft mice models, which indicates that EGFR, IGF-1R, and CRAF are the target genes of miR-7 that are associated with the acquired resistance to BRAFi. By decreasing the expression levels of EGFR, IGF-1R, CRAF, miR-7 could inhibit the activation of the MAPK and PI3K/AKT pathways and reverse melanoma cell resistance to BRAFi [25].
2.2. miRNAs Acting as Oncogenes
miR-21 has been reported to have a potential role in the treatment of CMM. This miRNA is a well-known modulator of cell proliferation, survival, and migration/invasion. Deregulation of miR-21 has been found in many human cancers. In particular, miR-21 expression is frequently upregulated in human cutaneous melanoma and higher levels correlate with advanced tumor stage, degree of invasion and tumor recurrence. Overexpressed miR-21 may function as an oncogene and promote cancer development by negatively regulating genes that control cell differentiation or apoptosis. Many studies have demonstrated that PTEN is a direct target of miR-21 and levels of PTEN, in turn, modulate the activation status of the PI3K/AKT pathway [26] (Figure 1). Antisense-mediated knockdown of miR-21 has been shown to suppress growth, increase apoptosis, and enhance the chemo- or radio-sensitivity of cutaneous melanoma cells. Such observations indicate that targeting miR-21 will be a potential novel strategy for the treatment of CMM. [27].
Some miRNAs, such as miR- 638 and miR-579-3p, have also been described to affect melanoma cell apoptosis alone or in combination with BRAFi treatments [28].
miR-638 is significantly overexpressed in metastatic melanoma. Indeed, miR-638 overexpression enhances the proliferative, migratory, and clonogenic properties of melanoma cells in vitro and their metastatic capacities in vivo. miR-638 induces its pro-tumorigenic and metastatic effects, protecting melanoma cells from apoptosis and autophagy. Knockdown of miR-638 increases TP53INP2 expression and stimulates expression of p53 and p53 downstream target genes, inducing significant apoptosis and autophagy [29].
The other 3 miRNAs, miR-34a, miR-100, and miR-125b, seem to be involved in the control of cell proliferation and apoptosis. They are upregulated in BRAFi-resistant melanoma cell lines and in the biopsy samples from patients treated with Vemurafenib, decreasing sensitivity to BRAFi therapy. In particular, the adaptative cell response to BRAF inhibitors increases expression of RTK and RTK ligands such as the chemokine monocyte chemoattractant protein-1 (CCL-2), which in turn activates the expression of miR-34a, miR-100, and miR-125b. All this leads to an increase in proliferation and resistance to apoptosis. Inhibition of CCL2 and of these miRNAs restores both cell apoptosis and drug efficacy in resistant melanoma cells [30].
In BRAF-mutated patients treated with Vemurafenib, high expression of miR-192 and miR-193b* and low expression of miR-132 has been associated with short time to progression, indicating a poor prognosis [31].
mir-514a has been reported to be involved in the modulation of BRAFi sensitivity in melanoma cells. In particular, miR-514a plays an important role in initiating melanocyte transformation and promoting melanoma growth by regulating the tumor suppressor NF1 gene. miR-514a overexpression in melanoma cell lines inhibits NF1 expression, which correlates with increased survival of BRAFV600E cells treated with Vemurafenib [32]. Both NF1 direct silencing with siRNA and miR-514a upregulation lead to decreased NF1 levels and thus considerably reduce drug sensitivity in the short term in vitro cell proliferation assays [32].
2.3. miRNA-Based Therapeutic Approaches
Several studies have highlighted the potential of targeting miRNAs and, therefore, it is likely that many new RNA-based therapeutics will be developed within the next few years [33].
Various strategies have been explored to utilize the role of miRNA to develop anticancer therapeutics:
- (a)
- miRNA inhibition therapy with anti-miRNA oligomers, which are 17–22 nucleotides long, single-stranded, chemically modified, competitive inhibitors of miRNAs, causing upregulation of the target mRNA;
- (b)
- Synthetic miRNA mimetic agents used as replacement therapy to replace or substitute lost miRNA;
- (c)
- Small-molecule inhibitors of miRNA (SMIRs) to either inhibit miRNA biogenesis or impede the interaction of miRNA with its target;
- (d)
- miRNAs targeted during transport within the tumor milieu or to distant sites in microvesicles and exosomes [34].
A recent approach to enhance or suppress miRNA function includes delivering synthetic oligoribonucleotides (ORNs) that copy the native miRNA duplex in which high miRNA expression is needed to reproduce single-stranded antisense RNA to use the targeted endogenous miRNA for inhibition studies. Developing modified duplex RNAs that retain their biological activity is a real challenge, and trials to deliver a single-stranded mature miRNA have been unsuccessful [35] probably because of the inability of duplex RNAs to load into the RNA-induced silencing complex. Thus, continuous efforts have been made to improve the stability and cellular uptake of miRNA to be used to treat various diseases.
Despite various studies demonstrating the involvement of miRNAs in melanoma and in specific therapeutic approaches, further systematic research is required to identify miRNAs able to modulate drug resistance. In fact, preclinical studies involving organoids and validation in a large cohort will be key to uncovering the role of miRNA in response to therapies or as molecules to be targeted.
3. The Advent of Immune Checkpoint Inhibitors: Do miRNAs Have a Role?
The advent of immunotherapy approaches has marked a step forward in the treatment of cancer. Generally speaking, immunotherapies, more precisely immune checkpoint inhibitors (ICIs), are based on the interaction between tumor cells and the immune microenvironment, in particular the adaptive immune response and the cytotoxic activity of T lymphocytes.
Antibodies against Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) and Programmed cell Death 1 (PD-1) (Ipilimumab and Nivolumab/Pembrolizumab, respectively) were the first agents to be approved by the FDA. In 1996, the antitumor activity of the CTLA-4 blockade was shown in mice and in 2010, the exceptional results of the phase III MDX010-20 study led to the registration of Ipilimumab for advanced melanoma.
In 2014, Nivolumab was approved by the FDA for metastatic melanoma treatment. Based on the results of clinical trials, anti-PD1 antibodies have shown the highest efficacy in melanoma. The CheckMate-066 trial demonstrated improved survival rates and progression-free survival in 210 naïve patients with unresectable/metastatic melanoma treated with Nivolumab [5]. The KEYNOTE-006 trial reported the same results and a durable response after discontinuation of immunotherapy [6,36].
Notwithstanding the remarkable clinical results, no biomarkers have yet been approved for ICI response in melanoma. Elevated serum lactate dehydrogenase (LDH), which is an independent predictor of survival in melanoma according to the American Joint Committee on Cancer (AJCC) guidelines [37], has been shown to be related to a worse outcome in patients treated with Ipilimumab [38] and Pembrolizumab [39,40]. KEYNOTE-006 trial found that LDH is not correlated to the duration of the remission period, even if it could be considered a useful biomarker for monitoring disease, as described by Diem et al. [41]. PD-L1 expression measured by immunohistochemistry was evaluated as a biomarker but the antibodies to be used for detection and cut-off are still under investigation. The results of the Checkmate-066 could not show that PD-L1 could be considered as a predictive biomarker for ICI response because the subset of responding patients included both PDL-1 positive and negative subjects [5]. The introduction of next-generation sequencing approaches has made it possible to explore the genome and transcriptome for every type of tumor. Whole exome sequencing (WES) and RNAseq has been extensively used in several melanoma studies. WES measures the tumor mutational burden (TMB), which is very high in melanoma. Snyder et al. [42] were the first to demonstrate, in a cohort of 64 patients treated with anti-CTLA4, that TMB is associated with a good response to ICIs and they identified a neoantigen signature associated with clinical benefit. Neoantigens derive from somatic mutations, which give rise to mutated proteins presented by antigen-presenting cells (APC). However, Van Allen et al. [43] were not able to confirm the predictive role of the neoantigen signature seen in the study performed by Snyder and colleagues and concluded that the recurrence of neoantigens was a rare event. In another study, Roh et al. [44] did not confirm the association of TMB with an ICI response, even if they found copy number loss was associated with poor outcomes. Several groups have focused on transcriptome analysis to set up a gene expression signature to stratify patients but to date, they have proved discordant [43,45,46]. Identifying biomarkers for ICI response in melanoma represents an unmet clinical need. miRNAs have been evaluated as potential biomarkers, even if, as stated by Dragomir et al. [47], molecular networks could be more helpful in the definition of miRNAs able to influence immune response and thus be considered as biomarkers. Immune checkpoint genes, as any biological process, could be regulated both directly by miRNAs and indirectly through proteins, which in turn can be influenced by miRNAs. Recently, an integrative analysis of deregulated mRNAs, miRNAs, and long noncoding RNAs (lncRNAs) between metastatic and non-metastatic samples included in the TCGA repository led to the definition of a competing endogenous RNA network [48]. Survival analysis of the 3 types of RNAs included in the network demonstrated that 6 lncRNAs, 7 mRNAs, and 5 miRNAs were associated with the prognosis of metastatic melanoma. Such a bioinformatic approach and the use of publicly available data could be applied to define a network of RNAs able to predict response to ICIs.
To date, results regarding miRNAs and ICI responses are sparse, but studies could gain momentum by using the single-cell RNAseq approach to distinguish between the expression of tumor cells and immune components.
miR-29 a/b/c isoforms were studied in 2009 and miR-29a was the first miRNA that was demonstrated to bind to B7-H3 3′UTR and whose expression was found to be inversely related to that of an immune checkpoint inhibitor, in different tumors, B7-H3 [49]. In cutaneous melanoma, an inverse relationship between miR-29c and B7-H3 has been found [50]. Recently, B7-H3 has been proposed as an immune inhibitory protein due to its role in the inhibition of T cell proliferation [51,52]. Promising results have been reported in a clinical trial treating recurrent metastatic neuroblastoma patients with an anti-B7-H3 monoclonal antibody, 8H9 [53]. The metastatic potential of B7-H3 has been dissected in melanoma cells through a silencing technique, and results showed a decrease in the proteins involved in metastasization, namely matrix MMP-2, STAT3 and Il-8 [54].
Moreover, the expression of miR-155 is stimulated by IRF4, which is overexpressed in CD8 T cells from Murine lymphocytic choriomeningitis virus chronic infection [55]. Thus, Martinez-Usatorre and colleagues concluded that miR-155 could be considered a marker of responsiveness of CD8 T cells, as further demonstrated by its upregulation after PD1 blockade [56].
The relationship between T-cell exhaustion and miRNA expression has been investigated. The definition of T-cell exhaustion is controversial [57], but, generally, exhausted T cells are not functional in cancer [58] and overexpress inhibitory receptors like TIM3, PD-1, and BTLA [59,60,61]. Microarray-based profiling was performed in PD1+ and PD1- CD4 T cells sorted from lymph nodes and spleen of tumor-bearing mice. The significantly decreased expression of miR-28, miR-150, and miR-151-5p in PD1+ CD4 T cells was validated but miR-28, in particular, was found to silence PD1 through 3′UTR binding [62]. Moreover, exhausted T cells showed a reduced secretion of IL-2, TNF-α and IFN-γ and the use of miR-28 mimics was able to restore their secretion [62].
PD-L1 overexpressing melanoma have been shown to be resistant BRAFi and MEKi. Audrito et al. [63] demonstrated that miR-17-5p binds to PD-L1, suggesting its role in the resistance to targeted therapies.
Martinez-Usatorre et al. [56] studied the role of miR-155, which had proved to be regulated during CD8 T cells differentiation in previous investigations [64]. They measured miR-155 expression in CD8 T cells isolated from tumor-infiltrating lymph nodes and tumor tissue samples from melanoma patients and murine models. miR-155 expression in CD8 T cells was found to depend on antigen stimulation [56]. Previous investigations had shown that TCR (T Cell Receptor) is responsible for NF-kB and AP1 activation that in turn, upregulates miR-155 expression [65,66]. Moreover, the expression of miR-155 is stimulated by IRF4, which is overexpressed in CD8 T cells from Murine lymphocytic choriomeningitis virus chronic infection [55]. Thus, Martinez-Usatorre and colleagues concluded that miR-155 could be considered a marker of responsiveness of CD8 T cells, as further demonstrated by its upregulation after PD1 blockade [56].
4. miRNAs and the Interplay with Tumor Microenvironment
The tumor microenvironment (TME) is a complex network composed of soluble factors, extracellular matrix, and several types of cells, including endothelial cells, fibroblasts, and immune cells. The interplay between tumor cells and its TME ensures the maintenance of proliferation and, eventually, sprouting of malignancies. Given the significant role of the TME, bioinformatic algorithms based on RNAseq and microarrays have been developed for the deconvolution of bulk gene expression data to infer TME composition computationally (for example, through CIBERSORT [67]).
Several factors, including oxidative stress, pH variation, and acidosis, regulate the dynamics of TME. HIF1α and HIF2α are key players in the response to hypoxic conditions. The survival of melanoma cells in a low oxygen environment has been found to be linked to a low expression of miR-211, which functions as a metabolic regulator through its interaction with PDK4. Downregulation of miR-211 in melanoma cells drive PDK4 overexpression, leading to a decrease in pyruvate dehydrogenase and, in turn, in oxidative phosphorylation [68]. Neo-angiogenesis is another of the mechanisms known to be stimulated by tumor cells [69] to favor proliferation in oxygen/nutrient low environments. ApoE, a suppressor of angiogenesis and cell invasion [70], has been shown to be targeted by miR-1908, miR-199a-5p, and miR-199a-3p in melanoma, thus highlighting the possibility to target them [70,71]. In the previous section, we focused on the relationship between miRNAs and checkpoint inhibitors. miRNAs are able to regulate more generally both innate and adaptive immunity. miR-210 in melanoma inhibits the lysis of tumor cells by T cells, by regulating TNF-α, IL-6, and IFN-β [72]. M1 polarization of macrophages is regulated by miR-29a, and miR-21 is overexpressed in the blood of melanoma patients, targeting COL4A2, SPARC, and TIMP3 [73].Through the interaction between the NKG2D receptor and its ligand NKG2DL, Natural Killer (NK) cells are able to kill tumor cells but this ability is impaired by miR-34a/c and miR-449a/miR-449c [74]. In addition, Myeloid-Derived Suppressor Cells (MDSCs), whose role in cancer is still under evaluation, while its immune suppressive role is well-known [75], could be directly/indirectly regulated by miRNAs. miR-155 is able to induce MDSCs recruitment in TME [76], inhibiting SOCS1. Moreover, MDSC function is influenced by miR-494, whose expression is induced by TGFβ1 [77].
The EMT constitutes one of the hallmarks of cancer because of its role in resistance to treatments due to the acquisition of invasiveness. The crosstalk with the EMT (e.g., metabolic modification, stroma/immune cells, growth factor, and hypoxia) is responsible for the switch of tumor cells [78]. Melanoma cells are not epithelial but EMT markers have been identified, which are negatively correlated with the state of differentiation of melanocytes [79,80]. miR-205-5p, miR-542-3p, miR-9, and miR-31 target different pathways and regulate EMT components [81,82,83,84,85].
5. Circulating MicroRNA Biomarkers in Melanoma
Although therapeutic options for melanoma have substantially changed in the past few years, the development of non-invasive methods for monitoring disease progression or treatment resistance continues to be a major challenge. Recently, the use of liquid biopsy results has proved to be useful as a source of non-invasive biomarkers that provide the entire genetic panorama of the tumoral landscape, allowing for earlier intervention and melanoma therapeutic decisions. Liquid biopsy is defined as the detection of circulating tumor cells (CTCs) or tumor-derived nucleic acids such as tumor DNA (ctDNA), mRNA, or miRNA that are released into circulation by cancer cells [86]. It is not an invasive method thus it can be used repetitively, unlike classic biopsy.
In the past decade, circulating miRNAs have emerged as powerful novel tools for the diagnosis and monitoring of patients with melanoma [87]. Most miRNAs are found within the cells, however, low levels are also detectable in the extracellular space including the bloodstream where miRNAs, referred to as circulating miRNAs, are attached to lipoproteins, proteins, or loaded inside exosomes. Exosomes are little vesicles derived from endosomes and are released from cells by fusion of the multivesicular endosome with the plasma membrane. Like other extracellular vesicles, exosomes contain proteins, RNA, miRNA, DNA, and lipids [88] that are delivered to the intercellular environment playing a pivotal role in cell–cell communication. Loading of circulating miRNAs into exosomes prevents degradation by serum and plasma RNases [89,90]. As such, circulating miRNAs are very stable under the harsh conditions of the blood [91], a feature that points to their potential use as easily accessible markers to help clinicians monitor cutaneous melanoma progression and treatment response [92].Since 2010, numerous efforts have been made to prove that circulating miRNAs are useful as melanoma diagnostic biomarkers. Table 1 summarizes the circulating miRNAs described in the literature as potential biomarkers in melanoma. Leidinger et al. [93] were the first researchers to use a microarray-based approach to screen ~866 human miRNAs in blood cells and the qRT-PCR technique to validate a 16-miRNA signature able to distinguish melanoma patients from healthy controls.
More recently, Fogli et al. [94] studied 30 patients with different stages of melanoma showing upregulation of plasma miR-15b-5p, miR-149-3p, miR-150-5p, and downregulation of miR-193a-3p and miR-524-5p in patients with melanoma compared to healthy subjects, suggesting that these 5 miRNAs may be new potential biomarkers in human cutaneous melanoma. Following their investigations, Li P. et al. [95] indicated that circulating miR-221-3p was a useful biomarker for staging. Lower serum levels of this miRNA were observed in stage I–II patients than in stage III–IV patients. A recent study by Stark et al. [96] measured the expression of a panel of 17 miRNAs (MELmiR-17) in melanoma tissue and serum samples from 255 melanoma patients and 130 controls. The authors also indicated that a seven-miRNA panel (MELmiR-7) made up of miR-16, miR-211-5p, miR-4487, miR-4706, miR-4731, miR-509-3p, and miR-509-5p was a useful tool to predict melanoma progression and recurrence, with a good relevance also in melanoma diagnosis and prognosis.
In addition to studies on the identification of circulating miRNAs with diagnostic power, several investigations regarded miRNAs able to distinguish drug responders from non-responders and predict melanoma recurrence and progression [97]. Kanemaru and co-workers [98] indicated circulating miR-221-3p as a biomarker for melanoma exhibiting with significantly different levels between stage I/IV melanoma patients and healthy controls. They described how miR-221-3p levels decreased after surgical removal of the primary tumor and increased upon disease recurrence, suggesting that circulating miRNA-221-3p could have a role as a new tumor marker.
Fleming et al. [99] identified a serum miRNA signature, including miR-150, miR-15b, miR-425, and miR-30d, which distinguished recurrent from non-recurrent cases and stratified patients into groups at high and low risk of recurrence. Friedman et al. [100] screened 355 miRNAs in sera from 80 melanoma patients at primary diagnosis and identified a 5-miRNA signature, which comprised miR-150, miR-15b, miR-199a-5p, miR-33a, and miR-424 and was able to distinguish patients with a high risk of recurrence from those with a low risk. In agreement with the study of Fleming et al. [99], upregulation of miR-150-5p and downregulation of miR-15b-5p were observed in the serum of melanoma patients at high risk of recurrence. Friedman et al. [100] also reported a signature of 5 miRNAs able to classify melanoma patients into high and low recurrence risk. A longitudinal evaluation of circulating miRNA expression in pre- and post-recurrence serum samples of 17 melanoma stage II patients highlighted a significant increase in the expression levels of miR-103a-3p and miR-221-3p at the time of primary diagnosis and upon recurrence.
Similarly, Tian et al. [101] suggested that miR-206 could be used as a potential prognostic and predictive biomarker, demonstrating that its levels in the serum of melanoma patients were associated with disease progression, poor prognosis, and response to treatment. Margue et al. [102] identified a set of 8 miRNAs that were profoundly deregulated in late-stage melanoma patients. Among them, miR-193b-3p and miR-720 discriminated melanoma patients from healthy controls and non-metastatic from metastatic melanoma patient groups. The first study that identified a circulating miRNA panel useful to detect the presence of metastasis in melanoma patients was performed by Shiiyama and co-workers [103]. It suggested that serum miR-9-5p, miR-145-5p, miR-150-5p, miR-155-5p and miR-205-5p could be used as prognostic biomarkers to discriminate between primary and metastatic melanoma patients. Some years later, Van Laar et al. [104] identified a 38-miRNA signature (MEL38) able to discriminate melanoma from normal plasma samples and an 18-miRNA signature (MEL18) able to distinguish between non-metastatic (stage I/II) and metastatic (stage III/IV) melanoma patients.
Exo-miRNAs
Tumor-derived exosomes also play a role in epigenetic regulation. They contain various enzymes that are involved in the synthesis and regulation of miRNAs, the most abundant RNAs in exosomes (exo-miRNA) [105]. In the past few years, miRNA profiles of tumor exosomes were found to be correlated with tumor burden or disease risk and thus, the detection of exo-miRNA has been indicated as a promising, non-invasive method for cancer diagnosis and a new tool for drug delivery [106]. Using miRNA profiling, Rappa et al. [107] revealed 49 different miRNAs with higher concentrations in metastatic-melanoma derived microvesicles, named prom1-exo, than in parental cells. Among these deregulated miRNAs, 20 proved to have a specific cancer-related function. Later, Alegre and co-workers [108] examined the exosome-associated miRNA pool of melanoma patients and controls. Significantly lower levels of miR-125b were observed in the serum exosomes of patients with advanced melanoma than in those of disease-free patients and healthy controls. However, no significant difference was observed between the miRNAs from the whole serum of melanoma patients and from that of healthy controls. More recently, Tengda L et al. [109] detected the levels of 5 miRNAs, namely miRNA-532-5p, miRNA-106b, miRNA-200c, miRNA-199a-5p, and miRNA-210, in serum exosomes isolated from 30 melanoma patients and 30 healthy individuals. Serum exo-miRNA-532-5p and exo-miRNA-106b proved to have the potential to be used as biomarkers for the diagnosis and monitoring of melanoma in a clinical setting. The authors also developed and subsequently validated an exo-miRNA panel in 95 serum samples from melanoma patients and healthy controls and concluded that it was a powerful diagnostic tool to distinguish patients with metastasis from those without metastasis, stage I-II patients from stage IV-V patients, and patients who had received Pembrolizumab treatment from those who were untreated.
Several studies have suggested using circulating miRNAs for melanoma staging and recurrence prediction. However, the lack of reproducibility among the results reported by different research groups constitutes a substantial obstacle for the future use of circulating miRNAs in clinical practice. To date, there have been very few multi-center studies, and cohorts have often been insufficiently powered. The lack of standardized analytical methods and pre-analytical procedures, together with the use of different technological platforms and statistical methodologies, has contributed to these discrepancies. Adequate standardization of methods is required before circulating miRNAs can be used in clinical trials to investigate their potential as diagnostic and prognostic biomarkers for melanoma management.
6. Conclusions
This review clearly highlights the urgent need to identify novel biomarkers of response/resistance to therapies for melanoma treatment. miRNAs regulate the expression of genes involved in the pathways affected by targeted therapies and immune checkpoint inhibitors. miRNAs have been found to stratify patients in terms of diagnosis and prognosis. The interplay with TME and exo-miRNAs modulates the states of the cytotypes interacting with melanoma cells, favoring proliferation, and invasion.
Author Contributions
Conceptualization, S.D.S. and S.T.; resources, C.L. and K.D.; writing—original draft preparation, C.L. and S.D.S.; writing—review and editing, K.D., R.P. and S.T.; supervision, S.T. All authors have read and agree to the published version of the manuscript.
Funding
Partially funded by Ricerca Corrente 2019 (Italian Ministry of Health).
Acknowledgments
The AA. thanks A. Papa for language revision.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Austrilia, 2017. [Google Scholar]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Buder-Bakhaya, K.; Machiraju, D.; Hassel, J.C. Liquid biopsy: Value for melanoma therapy. Oncol. Res. Treat. 2017, 40, 430–434. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Flaherty, K.T. Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 2013, 49, 1297–1304. [Google Scholar] [CrossRef]
- Bello, D.M.; Ariyan, C.E.; Carvajal, R.D. Melanoma mutagenesis and aberrant cell signaling. Cancer Control 2013, 20, 261–281. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma treatment in review. Immuno Targets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T. Targeting Metastatic Melanoma. Annu. Rev. Med. 2012, 63, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Falkenbach, E.; Villanueva, C.A.; Garrido, M.C.; Ruano, Y.; Garcìa-Martìn, R.M.; Godoy, E.; Ortiz-Romero, P.L.; Rìos-Martìn, J.J.; Santos-Briz, A.; Rodrìguez-Peralto, J.L. Intra- and inter-tumoral homogeneity BRAFV600E mutations in melanoma tumors. J. Investig. Dermatol. 2015, 135, 3078–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco. Targets. Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.L.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Babaei, M.A.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Mishra, P.J.; Merlino, G. Integrated Genomics Identifies miR-32/MCL-1 Pathway as a Critical Driver of Melanomagenesis: Implications for miR-Replacement and Combination Therapy. PLoS ONE 2016, 11, e0165102. [Google Scholar] [CrossRef]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Laganà, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Caponea, M.; et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, E5005–E5013. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Cui, R.; Xu, X. MiR-200c inhibits melanoma progression and drug resistance through down-regulation of Bmi-1. Am. J. Pathol. 2012, 181, 1823–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Wang, T.; Yang, R.; Xie, L.; Zhang, G.; Krepler, C.; Xiao, M.; Beqiri, M.; Xu, W.; et al. miR-200c/Bmi1 axis and epithelial-mesenchymal transition contribute to acquired resistance to BRAF inhibitor treatment. Pigment Cell Melanoma Res. 2015, 28, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, J.; Sun, Y.; Zhang, Y.; Dong, L.; Shen, C.; Yang, L.; Yang, M.; Li, Y.; Shen, G.; et al. miR-7 reverses the resistance to BRAFi in melanoma by targeting EGFR/IGF-1R/CRAF and inhibiting the MAPK and PI3K/AKT signaling pathways. Oncotarget 2016, 7, 53558–53570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Chen, M.; Wang, Y.; Cui, P.; Liu, S.; Xu, Z. MicroRNA-21 regulates the ERK/NF-kB signalingpathway to affect the proliferation, migration, and apoptosis of human melanoma A375 cells by targeting SPRY1, PDCD4, and PTEN. Mol. Carcinog. 2017, 56, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lv, X.; Li, J.; Li, J.; Li, X.; Li, W.; Li, Y. The status of microRNA-21 expression and its clinical significance in human cutaneous malignant melanoma. Acta Histochem. 2012, 114, 582–588. [Google Scholar] [CrossRef]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Schmitz, U.; Raatz, Y.; Schönherr, M.; Kottek, T.; Schauer, M.; Franz, S.; Saalbach, A.; Anderegg, U.; Wolkenhauer, O.; et al. miR-638 promotes melanoma metastasis and protects melanoma cells from apoptosis and autophagy. Oncotarget 2015, 6, 2966–2980. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2016, 7, 4428–4441. [Google Scholar] [CrossRef] [Green Version]
- Pinto, R.; Strippoli, S.; De Summa, S.; Albano, A.; Azzariti, A.; Guida, G.; Popescu, O.; Lorusso, V.; Guida, M.; Tommasi, S. MicroRNA expression in BRAF-mutated and wild-type metastatic melanoma and its correlation with response duration to BRAF inhibitors. Expert Opin. Ther. Targets 2015, 19, 1027–1035. [Google Scholar] [CrossRef]
- Stark, M.S.; Bonazzi, V.F.; Boyle, G.M.; Palmer, J.M.; Symmons, J.; Lanagan, C.M.; Schmidt, C.W.; Herington, A.C.; Ballotti, R.; Pollock, P.M.; et al. miR-514a regulates the tumour suppressor NF1 and modulates BRAFi sensitivity in melanoma. Oncotarget 2015, 6, 17753–17763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Long, G.V.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Long-term outcomes in patients (pts) with ipilimumab (ipi)-naive advanced melanoma in the phase 3 KEYNOTE-006 study who completed pembrolizumab (pembro) treatment. J. Clin. Oncol. 2017, 35, 9504. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Kelderman, S.; Heemskerk, B.; Van Tinteren, H.; Van Den Brom, R.R.H.; Hospers, G.A.P.; Van Den Eertwegh, A.J.M.; Kapiteijn, E.W.; De Groot, J.W.B.; Soetekouw, P.; Jansen, R.L.; et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol. Immunother. 2014, 63, 449–458. [Google Scholar] [CrossRef]
- Nosrati, A.; Tsai, K.K.; Goldinger, S.M.; Tumeh, P.; Grimes, B.; Loo, K.; Algazi, A.P.; Nguyen-Kim, T.D.L.; Levesque, M.; Dummer, R.; et al. Evaluation of clinicopathological factors in PD-1 response: Derivation and validation of a prediction scale for response to PD-1 monotherapy. Br. J. Cancer 2017, 116, 1141–1147. [Google Scholar] [CrossRef] [Green Version]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for response of melanoma patients to immune checkpoint inhibitors: A systematic review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, 3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Kendall, S.M.; Makarov, V.; Walsh, L.A.; Desrichard, A.; Weinhold, N.; Chan, T.A. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat. Genet. 2016, 48, 1327–1329. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.; Chen, B.; Fu, X.; Calin, G.A. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol. Med. 2018, 15, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.X.; Wan, C.; Dong, Z.B.; Wang, B.H.; Liu, H.Y.; Li, Y. Integrative Analysis of Long Noncoding RNA (lncRNA), microRNA (miRNA) and mRNA Expression and Construction of a Competing Endogenous RNA (ceRNA) Network in Metastatic Melanoma. Med. Sci. Monit. 2019, 25, 2896–2907. [Google Scholar] [CrossRef]
- Xu, H.; Cheung, I.Y.; Guo, H.F.; Cheung, N.K.V. MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: Potential implications for immune based therapy of human solid tumors. Cancer Res. 2009, 69, 6275–6281. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chong, K.K.; Nakamura, Y.; Nguyen, L.; Huang, S.K.; Kuo, C.; Zhang, W.; Yu, H.; Morton, D.L.; Hoon, D.S.B. B7-H3 associated with tumor progression and epigenetic regulatory activity in cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 2050–2058. [Google Scholar] [CrossRef] [Green Version]
- Prasad, D.V.R.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 Is a Negative Regulator of T Cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [Green Version]
- Suh, W.K.; Gajewska, B.U.; Okada, H.; Gronski, M.A.; Bertram, E.M.; Dawicki, W.; Duncan, G.S.; Bukczynski, J.; Plyte, S.; Elia, A.; et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 2003, 4, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Kushner, B.H.; Modak, S.; Pandit-Taskar, N.; Smith-Jones, P.; Zanzonico, P.; Humm, J.L.; Xu, H.; Wolden, S.L.; Souweidane, M.M.; et al. Compartmental intrathecal radioimmunotherapy: Results for treatment for metastatic CNS neuroblastoma. J. Neurooncol. 2010, 97, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekle, C.; Nygren, M.K.; Chen, Y.W.; Dybsjord, I.; Nesland, J.M.; Mælandsmo, G.M.; Fodstad, Ø. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int. J. Cancer 2012, 130, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Usatorre, A.; Sempere, L.F.; Carmona, S.J.; Carretero-Iglesia, L.; Monnot, G.; Speiser, D.E.; Rufer, N.; Donda, A.; Zehn, D.; Jandus, C.; et al. MicroRNA-155 expression is enhanced by T-cell receptor stimulation strength and correlates with improved tumor control in Melanoma. Cancer Immunol. Res. 2019, 7, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Olive, D.; Kuchroo, V.; Zarour, H.M. CD8 + T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012, 72, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audrito, V.; Serra, S.; Stingi, A.; Orso, F.; Gaudino, F.; Bologna, C.; Neri, F.; Garaffo, G.; Nassini, R.; Baroni, G.; et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 2017, 8, 15894–15911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaun, B.; Yamamoto, T.; Badran, B.; Tsunetsugu-Yokota, Y.; Roux, A.; Baitsch, L.; Rouas, R.; Fayyad-Kazan, H.; Baumgaertner, P.; Devevre, E.; et al. Differentiation associated regulation of microRNA expression in vivo in human CD8+T cell subsets. J. Transl. Med. 2011, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, P.; Krishna, S.; Wang, J.; Lin, X.; Huang, H.; Xie, D.; Gorentla, B.; Huang, R.; Gao, J.; et al. Unexpected positive control of NFκB and miR-155 by DGKa and ζ ensures effector and memory CD8+ T Cell differentiation. Oncotarget 2016, 7, 33744–33764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Wang, X.; McBride, J.; Fewell, C.; Flemington, E. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 2008, 283, 2654–2662. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Mazar, J.; Qi, F.; Lee, B.; Marchica, J.; Govindarajan, S.; Shelley, J.; Li, J.-L.; Ray, A.; Perera, R.J. MicroRNA 211 Functions as a Metabolic Switch in Human Melanoma Cells. Mol. Cell. Biol. 2016, 36, 1090–1108. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent multi-miRNA targeting of ApoEgrives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathsyaraja, H.; Thies, K.; Taffany, D.A.; Deighan, C.; Liu, T.; Yu, L.; Fernandez, S.A.; Shapiro, C.; Otero, J.; Timmers, C.; et al. CSF1-ETS2-induced microRNA in myeloid cells promote metastatic tumor growth. Oncogene 2015, 34, 3651–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Not only in tumor immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Fan, J.; Ye, C.; Dominguez, D.; Zhang, Y.; Curiel, T.J.; Fang, D.; Kuzel, T.M.; Zhang, B. Host miR155 promotes tumor growth through a myeloid-derived suppressor cell-dependent mechanism. Cancer Res. 2015, 75, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 Is Required for the Accumulation and Functions of Tumor-Expanded Myeloid-Derived Suppressor Cells via Targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Skourti, E.; Logotheti, S.; Kontos, C.K.; Pavlopoulou, A.; Dimoragka, P.T.; Trougakos, I.P.; Gorgoulis, V.; Scorilas, A.; Michalopoulos, I.; Zoumpourlis, V. Progression of mouse skin carcinogenesis is associated with the orchestrated deregulation of mir-200 family members, mir-205 and their common targets. Mol. Carcinog. 2016, 55, 1229–1242. [Google Scholar] [CrossRef]
- Liu, S.; Kumar, S.M.; Lu, H.; Liu, A.; Yang, R.; Pushparajan, A.; Guo, W.; Xu, X. MicroRNA-9 up-regulates E-cadherin through inhibition of NF-κB1-Snail1 pathway in melanoma. J. Pathol. 2012, 226, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2014, 10, 103–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asangani, I.A.; Harms, P.W.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Maher, C.A.; Fullen, D.R.; Johnson, T.M.; Giordano, T.J.; Palanisamy, N.; et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget 2012, 3, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Sole, C.; Arnaiz, E.; Manterola, L.; Otaegui, D.; Lawrie, C.H. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin. Cancer Biol. 2019, 58, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Mumford, S.L.; Towler, B.P.; Pashler, A.L.; Gilleard, O.; Martin, Y.; Newbury, S.F. Circulating microRNA biomarkers in melanoma: Tools and challenges in personalised medicine. Biomolecules 2018, 8, E21. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 8. [Google Scholar] [CrossRef]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release Mechanisms, Functions and Challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Carpi, S.; Polini, B.; Fogli, S.; Nieri, P.; Romanini, A. Circulating MicroRNAs in Cutaneous Melanoma Diagnosis and Prognosis. Manag. Malig. Melanoma. 2016, 12. [Google Scholar]
- Leidinger, P.; Keller, A.; Borries, A.; Reichrath, J.; Rass, K.; Jager, S.U.; Lenhof, H.P.; Meese, E. High-throughput miRNA profiling of human melanoma blood samples. BMC Cancer 2010, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogli, S.; Polini, B.; Carpi, S.; Pardini, B.; Naccarati, A.; Dubbini, N.; Lanza, M.; Breschi, M.C.; Romanini, A.; Nieri, P. Identification of plasma microRNAs as new potential biomarkers with high diagnostic power in human cutaneous melanoma. Tumor Biol. 2017, 39, 1010428317701646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; He, Q.Y.; Luo, C.Q.; Qian, L.Y. Circulating miR-221 expression level and prognosis of cutaneous malignant melanoma. Med. Sci. Monit. 2014, 20, 2472–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBioMedicine 2015, 2, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Polini, B.; Carpi, S.; Romanini, A.; Breschi, M.C.; Nieri, P.; Podestà, A. Circulating cell-free microRNAs in cutaneous melanoma staging and recurrence or survival prognosis. Pigment Cell Melanoma Res. 2019, 32, 486–499. [Google Scholar] [CrossRef]
- Kanemaru, H.; Fukushima, S.; Yamashita, J.; Honda, N.; Oyama, R.; Kakimoto, A.; Masuguchi, S.; Ishihara, T.; Inoue, Y.; Jinnin, M.; et al. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J. Dermatol. Sci. 2011, 61, 187–193. [Google Scholar] [CrossRef]
- Fleming, N.H.; Zhong, J.; Da Silva, I.P.; De Miera, E.V.S.; Brady, B.; Han, S.W.; Hanniford, D.; Wang, J.; Shapiro, R.L.; Hernando, E.; et al. Serum-based miRNAs in the prediction and detection of recurrence in melanoma patients. Cancer 2015, 121, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Friedman, E.B.; Shang, S.; de Miera, E.V.; Fog, J.U.; Teilum, M.W.; Ma, M.W.; Berman, R.S.; Shapiro, R.L.; Pavlick, A.C.; Hernando, E.; et al. Serum microRNAs as biomarkers for recurrence in melanoma. J. Transl. Med. 2012, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Liu, T.; Qiao, L.; Gao, M.; Li, J. Decreased serum microRNA-206 level predicts unfavorable prognosis in patients with melanoma. Int. J. Clin. Exp. Pathol. 2015, 8, 3097–3103. [Google Scholar] [CrossRef]
- Margue, C.; Reinsbach, S.; Philippidou, D.; Beaume, N.; Walters, C.; Schneider, J.G.; Nashan, D.; Behrmann, I.; Kreis, S. Comparison of a healthy miRNome with melanoma patient miRNomes: Are microRNAs suitable serum biomarkers for cancer? Oncotarget 2015, 6, 12110–12127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiiyama, R.; Fukushima, S.; Jinnin, M.; Yamashita, J.; Miyashita, A.; Nakahara, S.; Kogi, A.; Aoi, J.; Masuguchi, S.; Inoue, Y.; et al. Sensitive detection of melanoma metastasis using circulating microRNA expression profiles. Melanoma Res. 2013, 23, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, R.; Lincoln, M.; Van Laar, B. Development and validation of a plasmabased melanoma biomarker suitable for clinical use. Br. J. Cancer 2018, 118, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin-Kujundzic, V.; Sola, I.M.; Predavec, N.; Potkonjak, A.; Somen, E.; Mioc, P.; Serman, A.; Vranic, S.; Serman, L. Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers. Cells 2019, 8, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Shen, Q.; Yang, X.; Qiu, Y.; Zhang, W. The role of extracellular vesicles: An epigenetic view of the cancer microenvironment. Biomed Res. Int. 2015, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Pope, R.M.; Lorico, A. Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol. Cancer 2013, 12, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre, E.; Sanmamed, M.F.; Rodriguez, C.; Carranza, O.; Martín-Algarra, S.; González, Á. Study of circulating MicroRNA-125b levels in serum exosomes in advanced melanoma. Arch. Pathol. Lab. Med. 2014, 138, 828–832. [Google Scholar] [CrossRef]
- Tengda, L.; Shuping, L.; Mingli, G.; Jie, G.; Yun, L.; Weiwei, Z.; Anmei, D. Serum exosomal microRNAs as potent circulating biomarkers for melanoma. Melanoma Res. 2018, 28, 295–303. [Google Scholar] [CrossRef]
Figure 1.
miRNAs involved as oncogenes and oncosuppressors targeting MAPK and PI3K/AKT pathways in melanoma resistant cells to BRAFi.
Figure 1.
miRNAs involved as oncogenes and oncosuppressors targeting MAPK and PI3K/AKT pathways in melanoma resistant cells to BRAFi.


{kind=link}
{kind=link}
Table 1.
Circulating miRNAs with a diagnostic and predictive role in melanoma.
| Main Deregulated miRNA | Body Fluid Type | Role | Detection Method | Reference |
|---|---|---|---|---|
| miR-186, let-7d*, miR-18a*, miR-145, miR-99a, miR-664, miR-501-5p, miR-378*, miR-29c*, miR-1280, miR-365, miR-1249, miR-328, miR-422a, miR-30d, miR-17* | Blood | Prognostic | microarray/qRT-PCR | [93] |
| miR-15b-5p, miR-149-3p, miR-150-5p, miR-193a-3p, miR-524-5p | Plasma | Prognostic | qRT-PCR | [94] |
| miR-221-3p | Serum | Prognostic | qRT-PCR | [95,98] |
| MELmiR-17 panel (hsa-miR-211, miR-508-3p, miR-514a, miR-4731, miR-146a, miR-509-3p, miR-506-3p, miR-509-5p, miR-508-5p, miR-4487, miR-16, miR-204, miR-513c, miR-513b, miR-145, miR-363-3p, miR-4706) and MELmiR-7 panel (miR-16, miR-211-5p, miR-4487, miR-4706, miR-4731, miR-509-3p, miR-509-5p) | FFPE tissue and serum | Prognostic and Predictive | qRT-PCR | [96] |
| miR-150, miR-15b, miR-425, miR-30d | Serum | Prognostic and Predictive | qRT-PCR | [99] |
| miR-150, miR -15b, miR -199a-5p, miR-33a, miR-424 | Serum | Prognostic and Predictive | qRT-PCR | [100] |
| miR-206 | Serum | Prognostic and Predictive | qRT-PCR | [101] |
| miRNome including miR-193b-3p and miR-720 | Serum, whole blood samples, melanoma tissue, primary melanocyte and keratinocyte cell lines | Prognostic and Predictive | qPCR arrays | [102] |
| miR-9, miR-145, miR-150, miR-155, miR-203, and miR-205 | Serum | Prognostic | qRT-PCR | [103] |
| 38-miRNA signature (MEL38) and 18-miRNA signature (MEL18) | Plasma | Prognostic and Predictive | microarray | [104] |
| miR-216b, miR-889, miR-4307, miR-4272, miR-203, miR-4289, miR-3149, miR-203, miR-3145, miR-1911, miR-513a-3p, miR-3916, miR-886-3p, miR-1182, miR-3613-5p, let-7i, miR-3132, miR-3914, miR-3618, miR-1307, miR-3614-3p, miR-3160, miR-519c-3p, miR-3153, miR-4278, miR-3646, miR-3926, miR-515-5p, miR-3169, miR-10a, miR-140-5p, miR-3148, miR-4271, miR-627, miR-548d-3p, miR-3613-3p, miR-481, miR-571, miR-4274, miR-4277, miR-3686, miR-3074, miR-95, miR-590-3p, miR-525-5p, miR-548g, miR-365, miR-525-3p, miR-320d | exosomes | Prognostic | microarray | [107] |
| miR-16, miR-125b | serum exosomes | Prognostic | qRT-PCR | [108] |
| miR-532-5p, miR-106b, miR-200c, miR-199a-5p, miR-210 | serum exosomes | Prognostic and Predictive | qRT-PCR | [109] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lorusso, C.; De Summa, S.; Pinto, R.; Danza, K.; Tommasi, S. miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells 2020, 9, 415. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020415
AMA Style
Lorusso C, De Summa S, Pinto R, Danza K, Tommasi S. miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells. 2020; 9(2):415. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020415
Chicago/Turabian StyleLorusso, Celeste, Simona De Summa, Rosamaria Pinto, Katia Danza, and Stefania Tommasi. 2020. "miRNAs as Key Players in the Management of Cutaneous Melanoma" Cells 9, no. 2: 415. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020415
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.