Mitochondrial Structure and Function in the Metabolic Myopathy Accompanying Patients with Critical Limb Ischemia

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
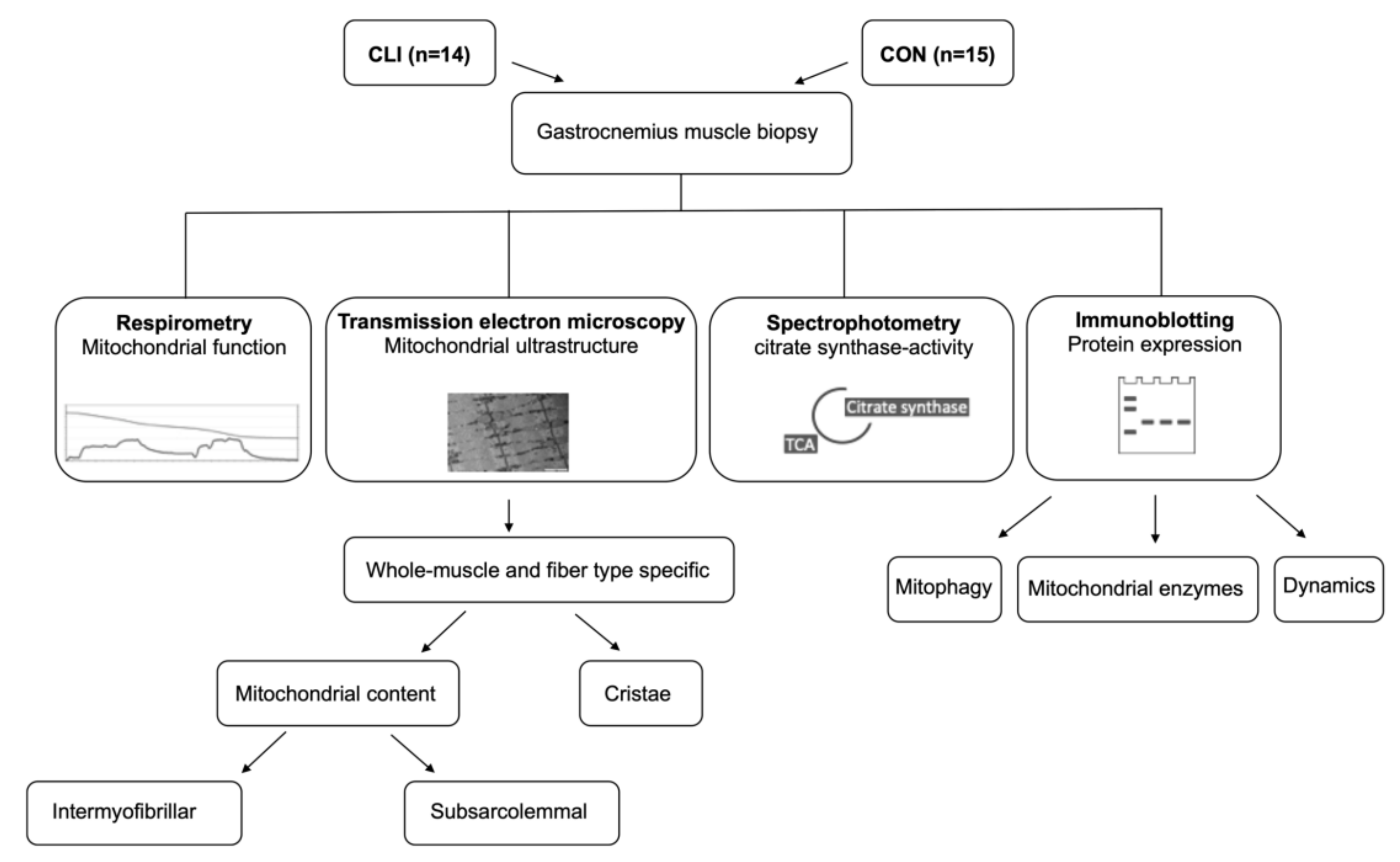
2. Materials and Methods
2.1. Participants
2.2. Ankle-Brachial Index
2.3. Muscle Samples
2.4. Preparation of Permeabilized Muscle Fibers
2.5. High-Resolution Respirometry
2.6. CS Activity
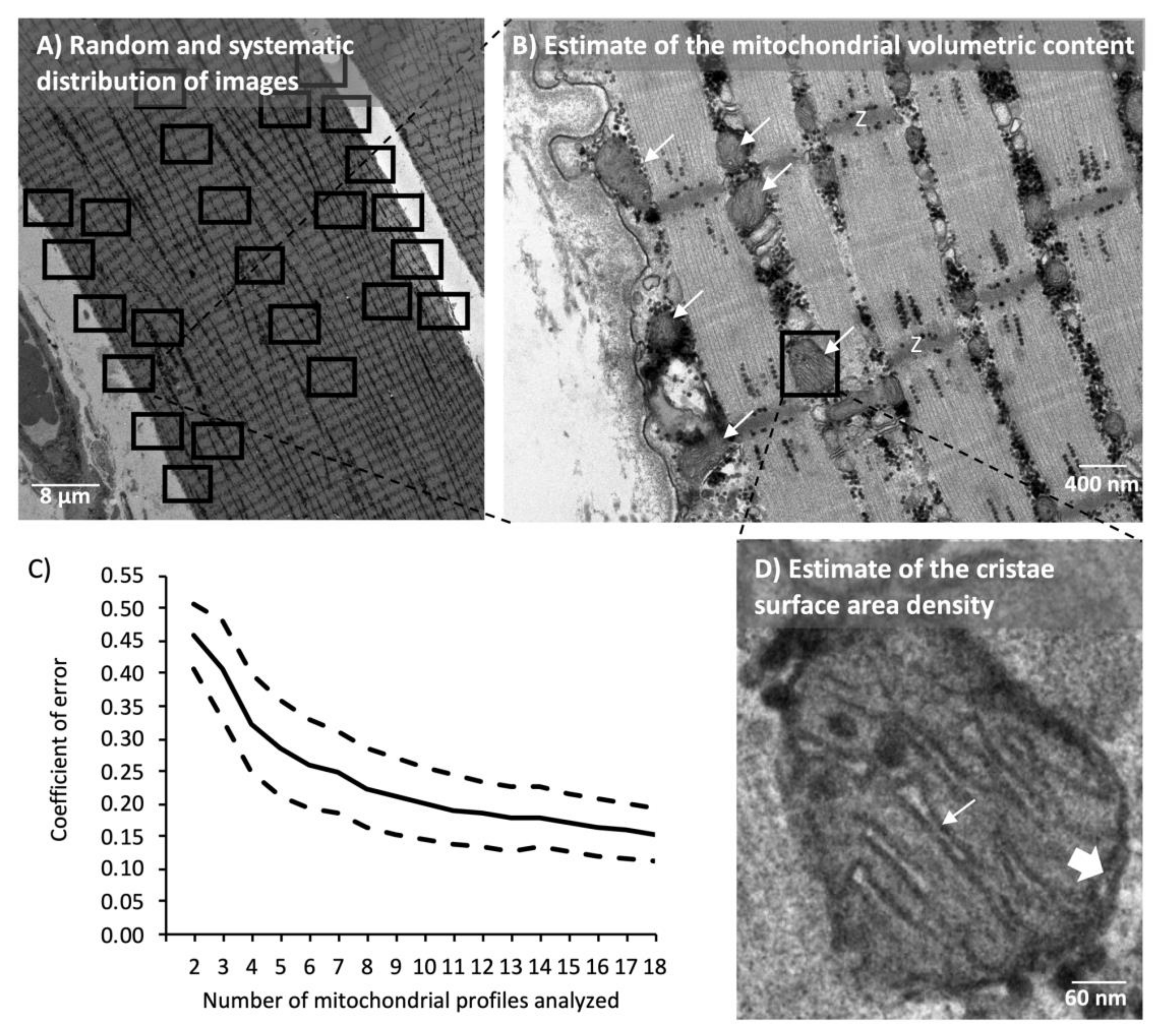
2.7. Transmission Electron Microscopy
2.8. Immunoblotting
2.9. Statistics
3. Results
3.1. Anthropometric and Clinical Characteristics
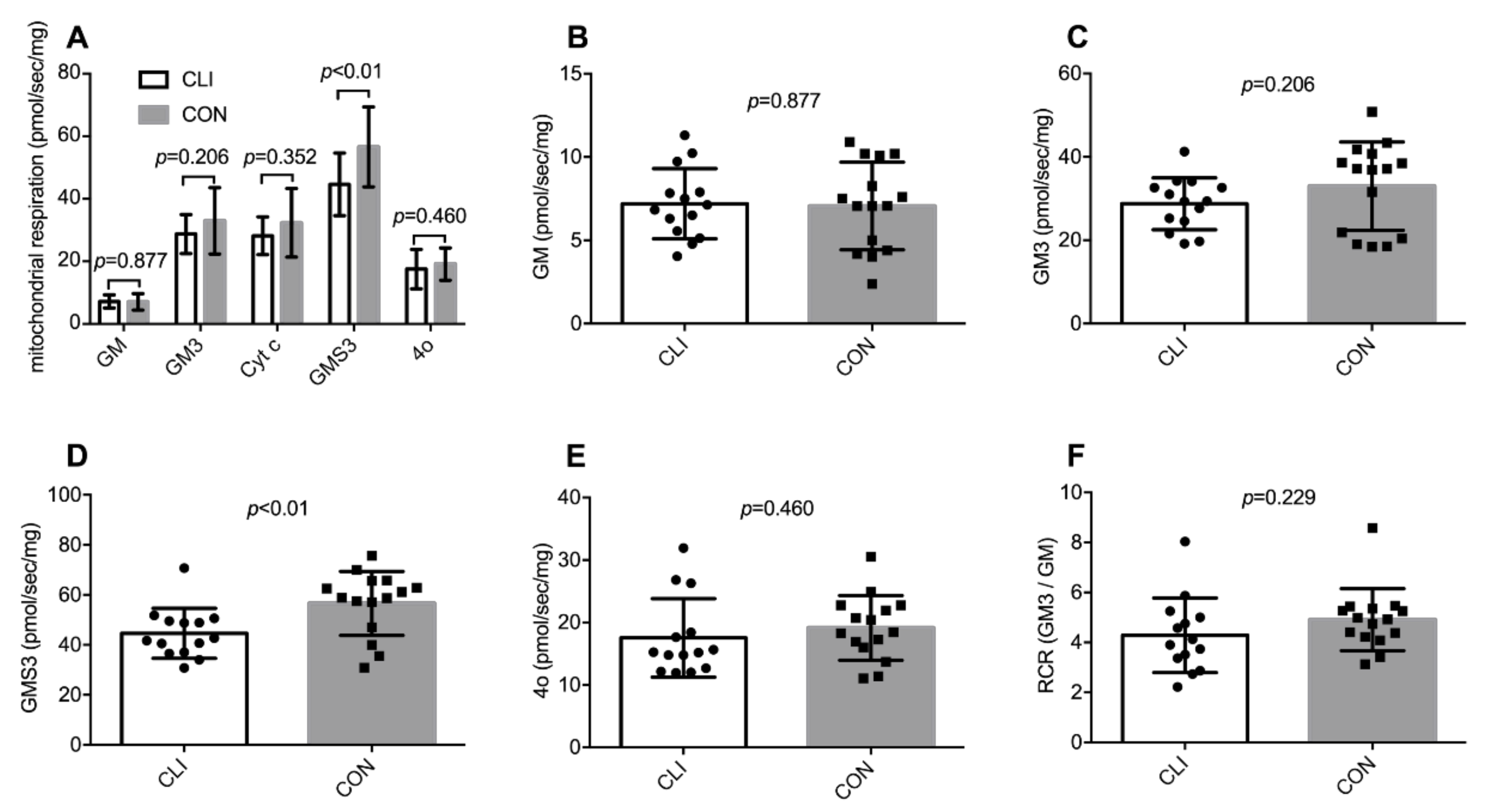
3.2. Global Mitochondrial Respiration
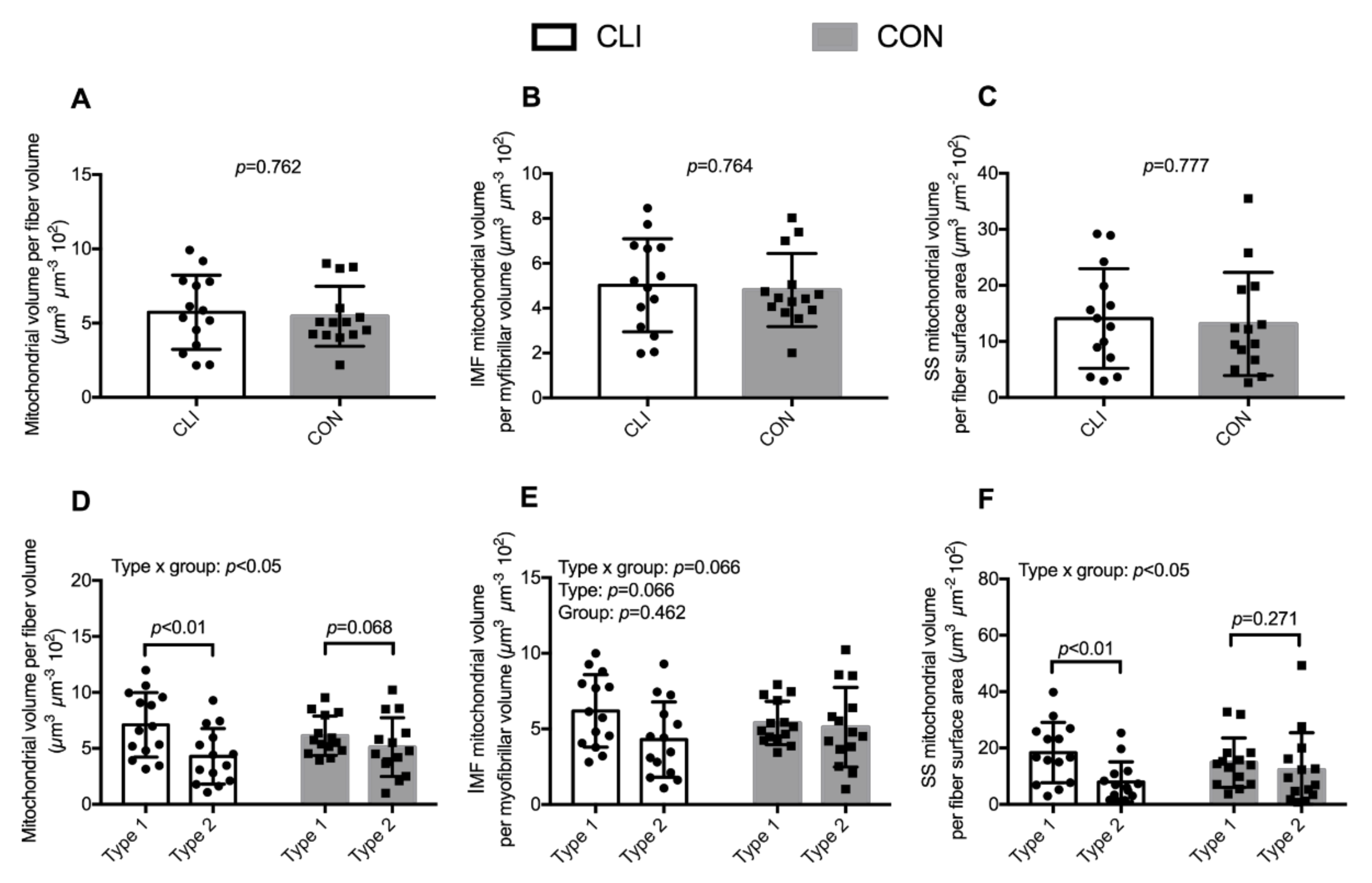
3.3. Mitochondrial Volumetric Content
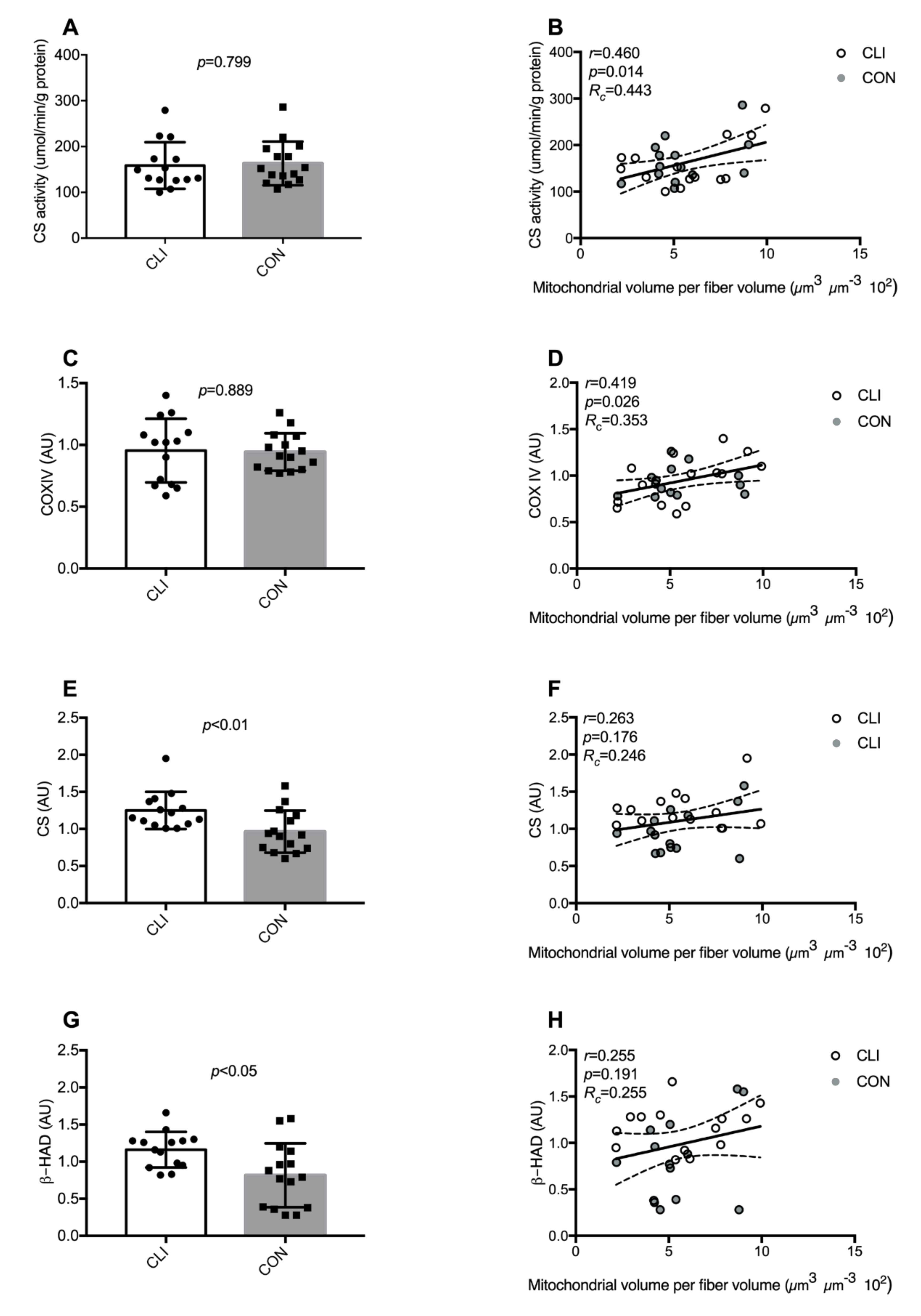
3.4. Common Markers of Mitochondrial Content
3.5. Cristae Content
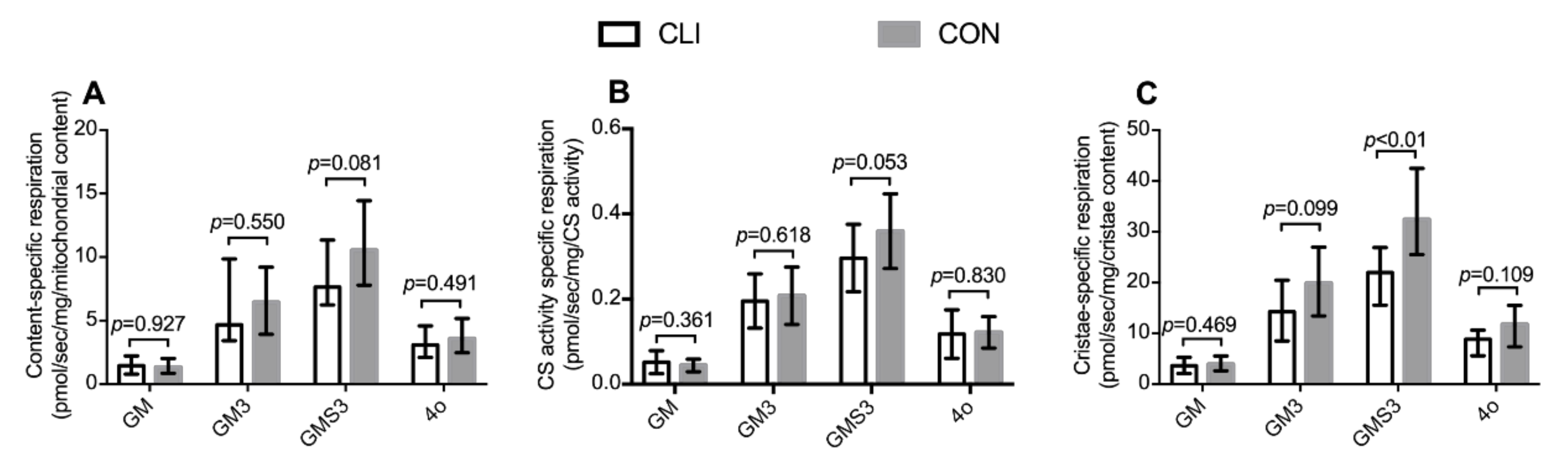
3.6. Mitochondrial Content-Specific Respiration and Cristae-Specific Respiration
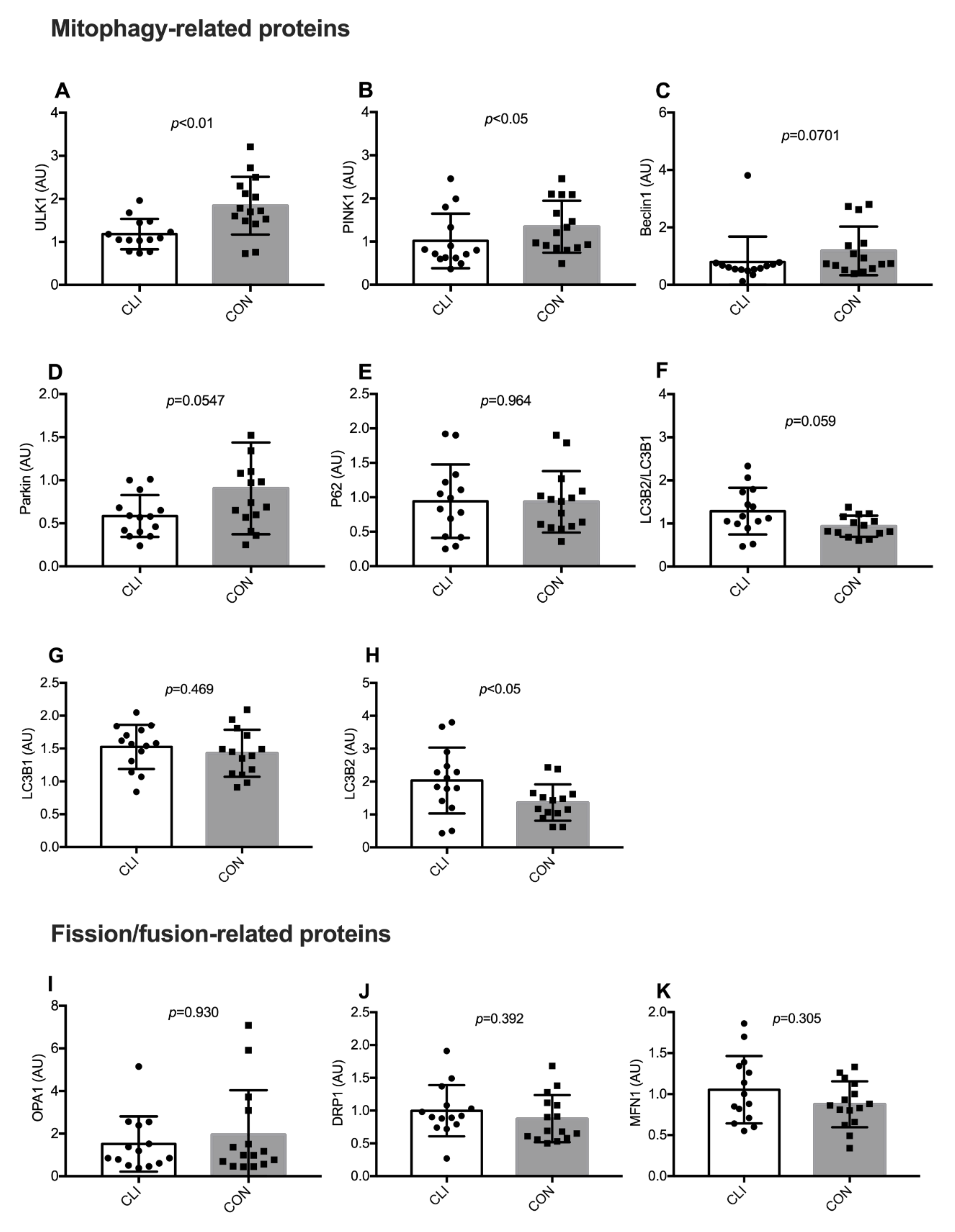
3.7. Expression of Mitophagy-Related and Fission/Fusion Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirsch, A.T.; Haskal, Z.J.; Hertzer, N.R.; Bakal, C.W.; Creager, M.A.; Halperin, J.L.; Hiratzka, L.F.; Murphy, W.R.; Olin, J.W.; Puschett, J.B.; et al. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): A collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): Endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006, 113, e463–e654. [Google Scholar] [CrossRef] [Green Version]
- Klaphake, S.; de Leur, K.; Mulder, P.G.; Ho, G.H.; de Groot, H.G.; Veen, E.J.; Verhagen, H.J.; van der Laan, L. Mortality after major amputation in elderly patients with critical limb ischemia. Clin. Interv. Aging 2017, 12, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Dormandy, J.; Heeck, L.; Vig, S. The fate of patients with critical leg ischemia. Semin. Vasc. Surg. 1999, 12, 142–147. [Google Scholar]
- Gardner, A.W.; Killewich, L.A. Lack of functional benefits following infrainguinal bypass in peripheral arterial occlusive disease patients. Vasc. Med. 2001, 6, 9–14. [Google Scholar] [CrossRef]
- Martinez, R.A.; Shnayder, M.; Parreco, J.; Gaffney, L.; Eby, M.; Cortolillo, N.; Lopez, M.; Zeltzer, J. Nationally Representative Readmission Factors in Patients with Claudication and Critical Limb Ischemia. Ann. Vasc. Surg. 2018, 52, 96–107. [Google Scholar] [CrossRef]
- Roos, S.; Fyhr, I.M.; Sunnerhagen, K.S.; Moslemi, A.R.; Oldfors, A.; Ullman, M. Histopathological changes in skeletal muscle associated with chronic ischaemia. Apmis 2016, 124, 935–941. [Google Scholar] [CrossRef]
- Hiatt, W.R.; Armstrong, E.J.; Larson, C.J.; Brass, E.P. Pathogenesis of the limb manifestations and exercise limitations in peripheral artery disease. Circ. Res. 2015, 116, 1527–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rontoyanni, V.G.; Nunez Lopez, O.; Fankhauser, G.T.; Cheema, Z.F.; Rasmussen, B.B.; Porter, C. Mitochondrial bioenergetics in the metabolic myopathy accompanying peripheral artery disease. Front. Physiol. 2017, 8, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutakis, P.; Miserlis, D.; Myers, S.A.; Kim, J.K.; Zhu, Z.; Papoutsi, E.; Swanson, S.A.; Haynatzki, G.; Ha, D.M.; Carpenter, L.A.; et al. Abnormal accumulation of desmin in gastrocnemius myofibers of patients with peripheral artery disease: Associations with altered myofiber morphology and density, mitochondrial dysfunction and impaired limb function. J. Histochem. Cytochem. 2015, 63, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipinos, I.I.; Judge, A.R.; Zhu, Z.; Selsby, J.T.; Swanson, S.A.; Johanning, J.M.; Baxter, B.T.; Lynch, T.G.; Dodd, S.L. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med. 2006, 41, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Signolet, I.; Abraham, P.; Chupin, S.; Ammi, M.; Gueguen, N.; Letournel, F.; Picquet, J.; Baufreton, C.; Daligault, M.; Procaccio, V.; et al. Mitochondrial complex I defect resulting from exercise-induced lower limb ischemia in patients with peripheral arterial disease. J. Appl. Physiol. 2018, 125, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Swanson, S.A.; Zhu, Z.; Nella, A.A.; Weiss, D.J.; Gutti, T.L.; McComb, R.D.; Baxter, B.T.; Lynch, T.G.; Casale, G.P. Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R290–R296. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Sharov, V.G.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A.; Todor, A.; Filis, K.A.; Sabbah, H.N. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J. Vasc. Surg. 2003, 38, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.A.; Ryan, T.E.; Lin, C.T.; Inigo, M.M.R.; Green, T.D.; Brault, J.J.; Spangenburg, E.E.; McClung, J.M. Diminished force production and mitochondrial respiratory deficits are strain-dependent myopathies of subacute limb ischemia. J. Vasc. Surg. 2017, 65, 1504–1514.e1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.E.; Yamaguchi, D.J.; Schmidt, C.A.; Zeczycki, T.N.; Shaikh, S.R.; Brophy, P.; Green, T.D.; Tarpey, M.D.; Karnekar, R.; Goldberg, E.J.; et al. Extensive skeletal muscle cell mitochondriopathy distinguishes critical limb ischemia patients from claudicants. JCI Insight 2018, 3, e123235. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.E.; Schmidt, C.A.; Alleman, R.J.; Tsang, A.M.; Green, T.D.; Neufer, P.D.; Brown, D.A.; McClung, J.M. Mitochondrial therapy improves limb perfusion and myopathy following hindlimb ischemia. J. Mol. Cell. Cardiol. 2016, 97, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Spangenburg, E.E.; Neufer, P.D.; McClung, J.M. Targeted Expression of Catalase to Mitochondria Protects Against Ischemic Myopathy in High-Fat Diet-Fed Mice. Diabetes 2016, 65, 2553–2568. [Google Scholar] [CrossRef] [Green Version]
- Baubeta Fridh, E.; Andersson, M.; Thuresson, M.; Sigvant, B.; Kragsterman, B.; Johansson, S.; Hasvold, P.; Nordanstig, J.; Falkenberg, M. Editor’s Choice—Impact of Comorbidity, Medication, and Gender on Amputation Rate Following Revascularisation for Chronic Limb Threatening Ischaemia. Eur. J. Vasc. Endovasc. Surg. 2018, 56, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Berru, F.N.; Gray, S.E.; Thome, T.; Kumar, R.A.; Salyers, Z.R.; Coleman, M.; Dennis, L.; O’Malley, K.; Ferreira, L.F.; Berceli, S.A.; et al. Chronic kidney disease exacerbates ischemic limb myopathy in mice via altered mitochondrial energetics. Sci. Rep. 2019, 9, 15547. [Google Scholar] [CrossRef] [Green Version]
- Aboyans, V.; Criqui, M.H.; Abraham, P.; Allison, M.A.; Creager, M.A.; Diehm, C.; Fowkes, F.G.; Hiatt, W.R.; Jonsson, B.; Lacroix, P.; et al. Measurement and interpretation of the ankle-brachial index: A scientific statement from the American Heart Association. Circulation 2012, 126, 2890–2909. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, N.R.; Yokota, T.; Stottrup, N.B.; Bergdahl, A.; Paelestik, K.B.; Povlsen, J.A.; Dela, F.; Botker, H.E. Pre-ischaemic mitochondrial substrate constraint by inhibition of malate-aspartate shuttle preserves mitochondrial function after ischaemia-reperfusion. J. Physiol. 2017, 595, 3765–3780. [Google Scholar] [CrossRef] [PubMed]
- Groennebaek, T.; Jespersen, N.R.; Jakobsgaard, J.E.; Sieljacks, P.; Wang, J.; Rindom, E.; Musci, R.V.; Botker, H.E.; Hamilton, K.L.; Miller, B.F.; et al. Skeletal Muscle Mitochondrial Protein Synthesis and Respiration Increase With Low-Load Blood Flow Restricted as Well as High-Load Resistance Training. Front. Physiol. 2018, 9, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjostrom, M.; Angquist, K.A.; Bylund, A.C.; Friden, J.; Gustavsson, L.; Schersten, T. Morphometric analyses of human muscle fiber types. Muscle Nerve 1982, 5, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. Stereological Methods: Theoretical Foundations; Academic Press: London, UK, 1980; Volume 2. [Google Scholar]
- Howard, C.V.; Reed, M.G. Unbiased Stereology: Three-Dimensional Measurement in Microscopy; Bios Scientific Publishers: Oxford, UK, 2005. [Google Scholar]
- Schwerzmann, K.; Hoppeler, H.; Kayar, S.R.; Weibel, E.R. Oxidative capacity of muscle and mitochondria: Correlation of physiological, biochemical, and morphometric characteristics. Proc. Natl. Acad. Sci. USA 1989, 86, 1583–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbek, S.K.; Farup, J.; de Paoli, F.; Vissing, K. No differential effects of divergent isocaloric supplements on signaling for muscle protein turnover during recovery from muscle-damaging eccentric exercise. Amino Acids 2015, 47, 767–778. [Google Scholar] [CrossRef]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to beta-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [Green Version]
- Gurtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free technology as a normalization tool in Western blot analysis. Anal. Biochem. 2013, 433, 105–111. [Google Scholar] [CrossRef]
- Lin, L.I. A concordance correlation coefficient to evaluate reproducibility. Biometrics 1989, 45, 255–268. [Google Scholar] [CrossRef]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Elander, A.; Sjostrom, M.; Lundgren, F.; Schersten, T.; Bylund-Fellenius, A.C. Biochemical and morphometric properties of mitochondrial populations in human muscle fibres. Clin. Sci. 1985, 69, 153–164. [Google Scholar] [CrossRef]
- Angquist, K.A.; Sjostrom, M. Intermittent claudication and muscle fiber fine structure: Morphometric data on mitochondrial volumes. Ultrastruct. Pathol. 1980, 1, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Conde Alonso, S.; Ofori, E.; Auwerx, J.; et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Koutakis, P.; Weiss, D.J.; Miserlis, D.; Shostrom, V.K.; Papoutsi, E.; Ha, D.M.; Carpenter, L.A.; McComb, R.D.; Casale, G.P.; Pipinos, I.I. Oxidative damage in the gastrocnemius of patients with peripheral artery disease is myofiber type selective. Redox Biol. 2014, 2, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regensteiner, J.G.; Wolfel, E.E.; Brass, E.P.; Carry, M.R.; Ringel, S.P.; Hargarten, M.E.; Stamm, E.R.; Hiatt, W.R. Chronic changes in skeletal muscle histology and function in peripheral arterial disease. Circulation 1993, 87, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N. Phospho-ubiquitin: Upending the PINK-Parkin-ubiquitin cascade. J. Biochem. 2016, 159, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Shaha, C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci. Rep. 2018, 8, 615. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372. [Google Scholar] [CrossRef]
- Triolo, M.; Hood, D.A. Mitochondrial breakdown in skeletal muscle and the emerging role of the lysosomes. Arch. Biochem. Biophys. 2019, 661, 66–73. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Miro, O.; Barrientos, A.; Alonso, J.R.; Casademont, J.; Jarreta, D.; Urbano-Marquez, A.; Cardellach, F. Effects of general anaesthetic procedures on mitochondrial function of human skeletal muscle. Eur. J. Clin. Pharmacol. 1999, 55, 35–41. [Google Scholar] [CrossRef] [PubMed]
- La Monaca, E.; Fodale, V. Effects of anesthetics on mitochondrial signaling and function. Curr. Drug Saf. 2012, 7, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Degens, H.; Gayan-Ramirez, G.; van Hees, H.W. Smoking-induced skeletal muscle dysfunction: From evidence to mechanisms. Am. J. Respir. Crit. Care Med. 2015, 191, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Lundby, C. Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J. Appl. Physiol. 2013, 114, 344–350. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON (n = 15) | CLI (n = 14) | p | |
|---|---|---|---|
| Age (y) | 67.4 ± 7.4 | 65.3 ± 7.8 | 0.463 |
| Sex (male/female) | 15/0 | 13/1 | 0.292 |
| Height (cm) | 178.2 ± 8.1 | 178.0 ± 8.4 | 0.948 |
| Weight (kg) | 89.5 ± 15.8 | 89.8 ± 17.7 | 0.973 |
| BMI (kg/m2) | 28.0 ± 3.3 | 28.1 ± 4.3 | 0.958 |
| ABI | 1.11 ± 0.13 | 0.41 ± 0.19 | <0.01 |
| Lower leg rest pain (Y/N) | 0/15 | 7/7 | <0.01 |
| Ischemic wounds (Y/N) | 0/15 | 6/8 | <0.01 |
| Gangrene (Y/N) | 0/15 | 6/8 | <0.01 |
| Smoking (current/past/non) | 1/5/9 | 10/4/0 | <0.01 |
| Hypertension (Y/N) | 5/10 | 6/8 | 0.597 |
| Diabetes mellitus (Y/N) | 4/11 | 5/9 | 0.599 |
| Lung disease (Y/N) | 2/13 | 5/9 | 0.159 |
| Nephropathy (Y/N) | 0/15 | 1/13 | 0.292 |
| Hypercholesterolemia (Y/N) | 3/12 | 3/11 | 0.924 |
| Statins (Y/N) | 15/0 | 11/3 | 0.058 |
| Metformin (Y/N) | 4/11 | 3/11 | 0.742 |
| Diuretics (Y/N) | 7/8 | 6/8 | 0.837 |
| Antibiotics (Y/N) | 11/4 | 8/6 | 0.359 |
| ACE-inhibitors (Y/N) | 5/10 | 4/10 | 0.782 |
| Insulin (Y/N) | 4/11 | 4/10 | 0.909 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groennebaek, T.; Billeskov, T.B.; Schytz, C.T.; Jespersen, N.R.; Bøtker, H.E.; Olsen, R.K.J.; Eldrup, N.; Nielsen, J.; Farup, J.; de Paoli, F.V.; et al. Mitochondrial Structure and Function in the Metabolic Myopathy Accompanying Patients with Critical Limb Ischemia. Cells 2020, 9, 570. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030570
Groennebaek T, Billeskov TB, Schytz CT, Jespersen NR, Bøtker HE, Olsen RKJ, Eldrup N, Nielsen J, Farup J, de Paoli FV, et al. Mitochondrial Structure and Function in the Metabolic Myopathy Accompanying Patients with Critical Limb Ischemia. Cells. 2020; 9(3):570. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030570
Chicago/Turabian StyleGroennebaek, Thomas, Tine Borum Billeskov, Camilla Tvede Schytz, Nichlas Riise Jespersen, Hans Erik Bøtker, Rikke Kathrine Jentoft Olsen, Nikolaj Eldrup, Joachim Nielsen, Jean Farup, Frank Vincenzo de Paoli, and et al. 2020. "Mitochondrial Structure and Function in the Metabolic Myopathy Accompanying Patients with Critical Limb Ischemia" Cells 9, no. 3: 570. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030570