Signaling Pathways and Key Genes Involved in Regulation of foam Cell Formation in Atherosclerosis

 ,
,  ,
,  and
and
Abstract
:1. Introduction
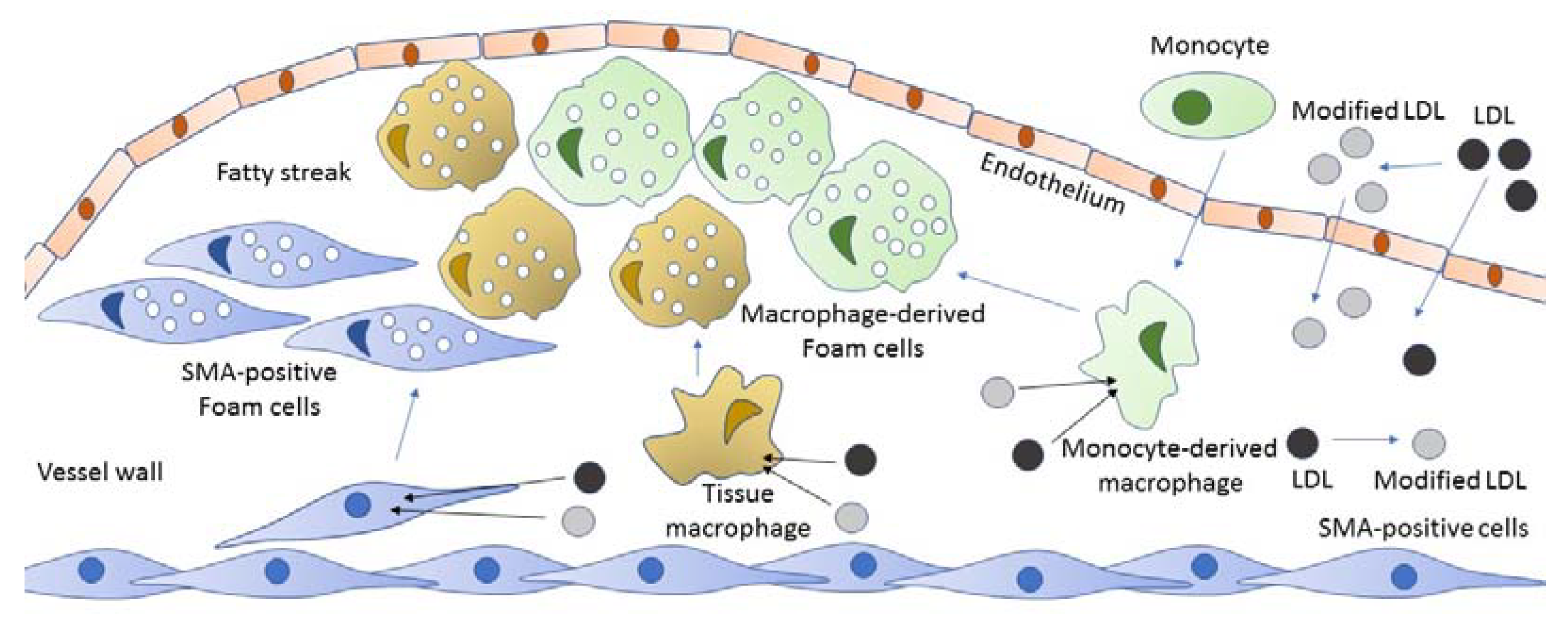
2. The Origin of Foam Cells
3. Foam Cells in Atherosclerosis
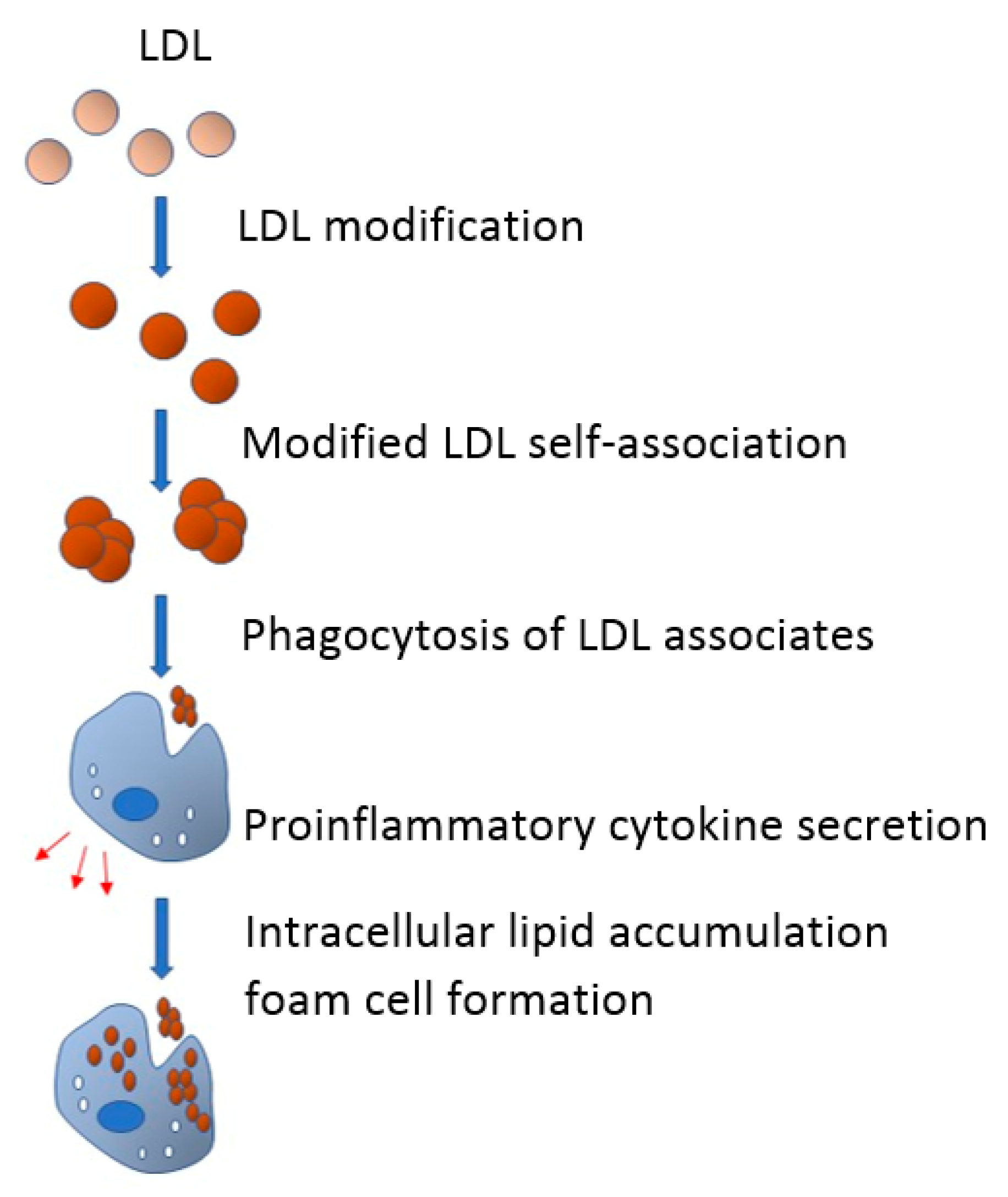
4. Role of Modified LDL in Foam Cells Formation
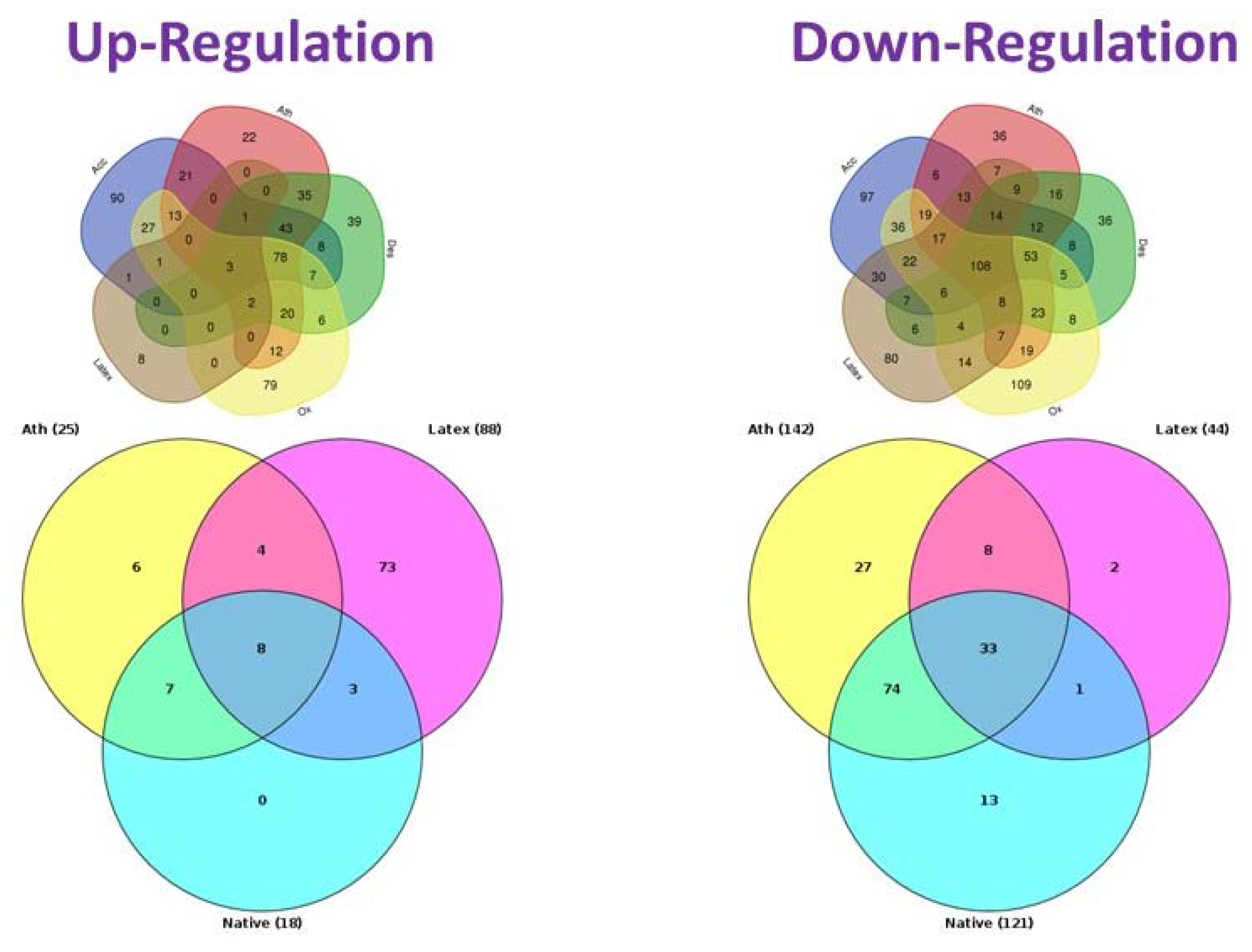
5. Key Genes and Signaling Pathways Involved in Foam Cells Formation
6. Proinflammatory Response as an Inductor of Intracellular Lipid Accumulation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [Green Version]
- Padarti, A.; Zhang, J. Recent advances in cerebral cavernous malformation research. Vessel Plus 2018, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Dis. Stroke Stat. 2018, 137, e67–e492. [Google Scholar]
- Martínez, M.S.; García, A.; Luzardo, E.; Chávez-Castillo, M.; Olivar, L.C.; Salazar, J.; Velasco, M.L.; Rojas Quintero, J.J.; Bermúdez, V. Energetic metabolism in cardiomyocytes: Molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus 2017, 1, 130–141. [Google Scholar] [CrossRef]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. CIRT Investigators. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Hennekens, C.H.; Buring, J.E.; Manson, J.E.; Stampfer, M.; Rosner, B.; Cook, N.R.; Belanger, C.; LaMotte, F.; Gaziano, J.M.; Ridker, P.M.; et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1145–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomized placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar] [CrossRef]
- Babaev, V.R.; Li, L.; Shah, S.; Fazio, S.; Linton, M.F.; May, J.M. Combined vitamin C and vitamin E deficiency worsens early atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1751–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The CANTOS Trial: One Important Step for Clinical Cardiology but a Giant Leap for Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e174–e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, N.; Guevara-Cruz, M.; Velázquez-Villegas, L.A.; Tovar, A.R. Nutrition and Atherosclerosis. Arch. Med. Res. 2015, 46, 408–426. [Google Scholar] [CrossRef] [PubMed]
- Strassheim, D.; Karoor, V.; Stenmark, K.; Verin, A.; Gerasimovskaya, E. A current view of G protein-coupled receptor—Mediated signaling in pulmonary hypertension: Finding opportunities for therapeutic intervention. Vessel Plus 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Silverio, D.; Sourlas, A.; Montan, P.D.; Guzman, E.; Garcia, M.J. Anti-inflammatory therapy for cardiovascular disease. Ann. Transl. Med. 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N. LDL and foam cell formation as the basis of atherogenesis. Curr. Opin. Lipidol. 2018, 29, 279–284. [Google Scholar] [CrossRef]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Schouteden, S.; Geenens, R.; Van Duppen, V.; Herijgers, P.; Holvoet, P.; Van Veldhoven, P.P.; Verfaillie, C.; Fadini, G.P.; Verfaillie, C.M. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS ONE 2012, 7, e47286. [Google Scholar] [CrossRef] [Green Version]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Riksen, N.P.; Stienstra, R. Metabolism of innate immune cells: Impact on atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 359–367. [Google Scholar] [CrossRef]
- Dubland, J.A.; Francis, G.A. So Much Cholesterol: The unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr. Opin. Lipidol. 2016, 27, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owsiany, K.M.; Alencar, G.F.; Owens, G.K. Revealing the Origins of Foam Cells in Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Freeman, M.W. Scavenger receptors in atherosclerosis: Beyond lipid uptake. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1273042. [Google Scholar] [CrossRef]
- Ferrari, E.; Lutgens, E.; Weber, C.; Gerdes, N. Atherosclerosis: Cell biology and lipoproteins focus on epigenetic modification and macrophage biology. Curr. Opin. Lipidol. 2017, 28, 220–221. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.G.; Elizova, N.V.; Korobov, G.A.; Aladinskaya, A.V.; Sobenin, I.A.; Bobryshev, Y.V. Tumor Necrosis Factor-α and C-C Motif Chemokine Ligand 18 Associate with Atherosclerotic Lipid Accumulation In situ and In vitro. Curr. Pharm. Des. 2018, 24, 2883–2889. [Google Scholar] [CrossRef]
- Liu, Q.; Fan, J.; Bai, J.; Peng, L.; Zhang, T.; Deng, L.; Wang, G.; Zhao, Y.; Nong, J.; Zhang, M.; et al. IL-34 promotes foam cell formation by enhancing CD36 expression through p38 MAPK pathway. Sci. Rep. 2018, 8, 17347. [Google Scholar] [CrossRef]
- Xu, Z.; Dong, A.; Feng, Z.; Li, J. Interleukin-32 promotes lipid accumulation through inhibition of cholesterol efflux. Exp. Ther. Med. 2017, 14, 947–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Ivanova, E.A.; Bobryshev, Y.V. Naturally Occurring Multiple-Modified Low-Density Lipoprotein. In Blood Lipids and Lipoproteins; Ruiz, M., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2015; Chapter 2; pp. 13–54. [Google Scholar]
- Calvo, M.J.; Martínez, M.S.; Torres, W.; Chávez-Castillo, M.; Luzardo, E.; Villasmil, N.; Salazar, J.; Velasco, M.; Bermúdez, V. Omega-3 polyunsaturated fatty acids and cardiovascular health: A molecular view into structure and function. Vessel Plus 2017, 1, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Pentikäinen, M.O.; Oörni, K.; Ala-Korpela, M.; Kovanen, P.T. Modified LDL—Trigger of atherosclerosis and inflammation in the arterial intima. J. Int. Med. 2000, 247, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, N.; Formoso, G.; Pandolfi, A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vasc. Pharmacol. 2016, 84, 1–7. [Google Scholar] [CrossRef]
- Alipov, V.I.; Sukhorukov, V.N.; Karagodin, V.P.; Grechko, A.V.; Orekhov, A.N. Chemical composition of circulating native and desialylated low density lipoprotein: What is the difference? Vessel Plus 2017, 1, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, S.K.; Jialal, I. Biochemistry, Low Density Lipoprotein; In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019; Volume 23, pp. 479–486. [Google Scholar]
- Sánchez-Quesada, J.L.; Villegas, S.; Ordóñez-Llanos, J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr. Opin. Lipidol. 2012, 23, 479–486. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified LDL Particles Activate Inflammatory Pathways in Monocyte-derived Macrophages: Transcriptome Analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef]
- Lopes-Virella, M.F.; Virella, G. Pathogenic role of modified LDL antibodies and immune complexes in atherosclerosis. J. Atheroscler. Thromb. 2013, 20, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Virella, G.; Wilson, K.; Elkes, J.; Hammad, S.M.; Rajab, H.A.; Li, Y.; Chassereau, C.; Huang, Y.; Lopes-Virella, M. Immune complexes containing malondialdehyde (MDA) LDL induce apoptosis in human macrophages. Clin. Immunol. 2018, 187, 1–9. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, X.J.; Cao, L.J.; Liu, X.H.; Liu, Z.H.; Wang, X.Q. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS ONE 2014, 9, e95935. [Google Scholar] [CrossRef]
- Park, Y.M. CD36, A scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackers, I.; Szymanski, C.; Duckett, K.J.; Consitt, L.A.; Silver, M.J.; Malgor, R. Blocking Wnt5a signaling decreases CD36 expression and foam cell formation in atherosclerosis. Cardiovasc. Pathol. 2018, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 1, 3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, M.H.K.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflam. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.; Sinha, A.; Saikia, S.; Gogoi, B.; Rathore, A.K.; Das, A.S.; Pal, D.; Buragohain, A.K.; Dasgupta, S. Inflammation-induced mTORC2-Akt-mTORC1 signaling promotes macrophage foam cell formation. Biochimie 2018, 151, 139–149. [Google Scholar] [CrossRef]
- Singh, R.K.; Haka, A.S.; Asmal, A.; Barbosa-Lorenzi, V.C.; Grosheva, I.; Chin, H.F.; Xiong, Y.; Hla, T.; Maxfield, F.R. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 86–102. [Google Scholar] [CrossRef]
- Sorrentino, R.; Morello, S.; Chen, S.; Bonavita, E.; Pinto, A. The activation of liver X receptors inhibits toll-like receptor-9-induced foam cell formation. J. Cell. Physiol. 2010, 223, 158–167. [Google Scholar] [CrossRef]
- Lee, J.-G.; Lim, E.-J.; Park, D.-W.; Lee, S.-H.; Kim, J.-R.; Baek, S.-H. A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell. Signal. 2008, 20, 2266–2275. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Nègre-Salvayre, A.; Augé, N.; Camaré, C.; Bacchetti, T.; Ferretti, G.; Salvayre, R. Dual signaling evoked by oxidized LDLs in vascular cells. Free Radic. Biol. Med. 2017, 106, 118–133. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.G.; Sukhorukov, V.N.; Kubekina, M.V.; Sobenin, I.A.; Wu, W.K.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; et al. Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; A Transcriptome Analysis. Int. J. Mol. Sci. 2020, 21, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.; Nikiforov, N.; Kubekina, M.; Kashirskikh, D.; Khotina, V.; Kel, A. Genes and Regulatory Pathways Poten-tially Responsible for Foam Cell Formation (Transcriptome Analysis). In Proceedings of the 12th Congress of the Asian-Pacific Society of Atherosclerosis and Vascular Diseases, Taipei, China, 20–22 September 2019. [Google Scholar]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Orekhov, A.N. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem. Biophys. Res. Commun. 1989, 163, 489–494. [Google Scholar] [CrossRef]
- Tertov, V.V.; Orekhov, A.N.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Yaroslavov, A.A.; Smirnov, V.N. Three types of naturally occurring modified lipoproteins induce intracellular lipid accumulation due to lipoprotein aggregation. Circ. Res. 1992, 71, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2019, 18, 1. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Direction | Signaling Pathways |
|---|---|
| up | Neurotrophic signaling TLR2-mediated signaling TLR9 pathway VEGF-A pathway |
| down | Aurora-B cell cycle regulation Cdc20 deubiquitination Cdc20 ubiquitination CyclinB1 ubiquitination → anaphase onset Fzr1 → cyclin B1 degradation Metaphase to Anaphase transition Securin degradation Usp44 → Cdc20 |
| Intracellular Cholesterol, % of Control | P | ||
|---|---|---|---|
| Vs. Control | vs LDL | ||
| Control | 100.0 ± 21.0 | N/A | N/A |
| + LDL, 100 µg/mL | 162.5 ± 20.3 | <0.01 | N/A |
| + LDL + IL-6, 50 ng/mL | 199.5 ± 16.5 | <0.01 | <0.01 |
| + IL-6, 50 ng/mL | 129.5 ± 8.2 | 0.01 | 0.01 |
| + LDL + IL-15, 50 ng/mL | 187.0 ± 13.12 | <0.01 | <0.01 |
| + IL-15, 50 ng/mL | 107.9 ± 7.3 | 0,6 | <0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Wu, W.-K.; Melnichenko, A.A.; Wetzker, R.; Sukhorukov, V.; Markin, A.M.; Khotina, V.A.; Orekhov, A.N. Signaling Pathways and Key Genes Involved in Regulation of foam Cell Formation in Atherosclerosis. Cells 2020, 9, 584. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030584
Poznyak AV, Wu W-K, Melnichenko AA, Wetzker R, Sukhorukov V, Markin AM, Khotina VA, Orekhov AN. Signaling Pathways and Key Genes Involved in Regulation of foam Cell Formation in Atherosclerosis. Cells. 2020; 9(3):584. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030584
Chicago/Turabian StylePoznyak, Anastasia V., Wei-Kai Wu, Alexandra A. Melnichenko, Reinhard Wetzker, Vasily Sukhorukov, Alexander M. Markin, Victoria A. Khotina, and Alexander N. Orekhov. 2020. "Signaling Pathways and Key Genes Involved in Regulation of foam Cell Formation in Atherosclerosis" Cells 9, no. 3: 584. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030584