A Role for Endoplasmic Reticulum Stress in Intracerebral Hemorrhage

 , ,
, , 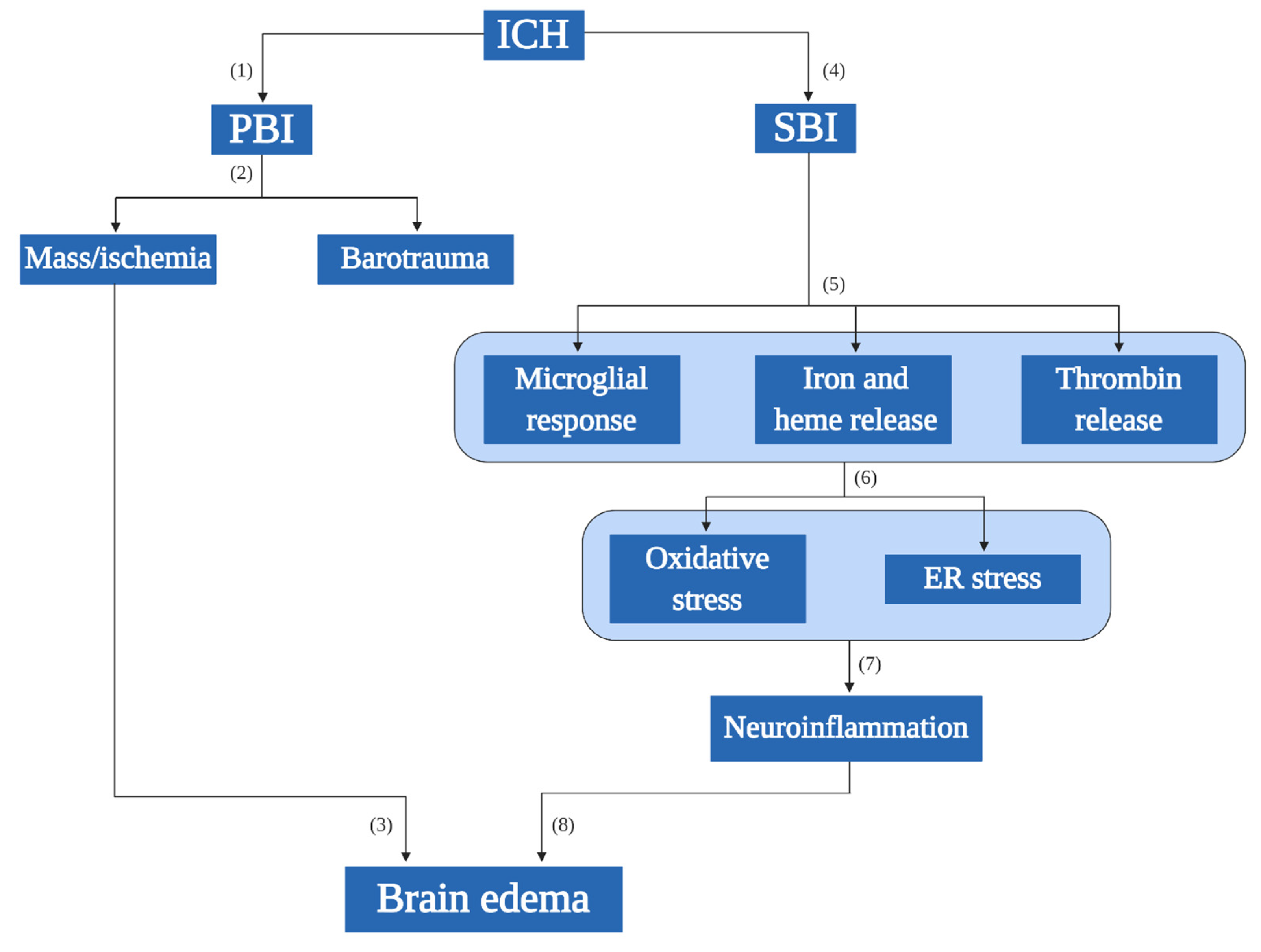{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Therapeutic Interventions for ICH
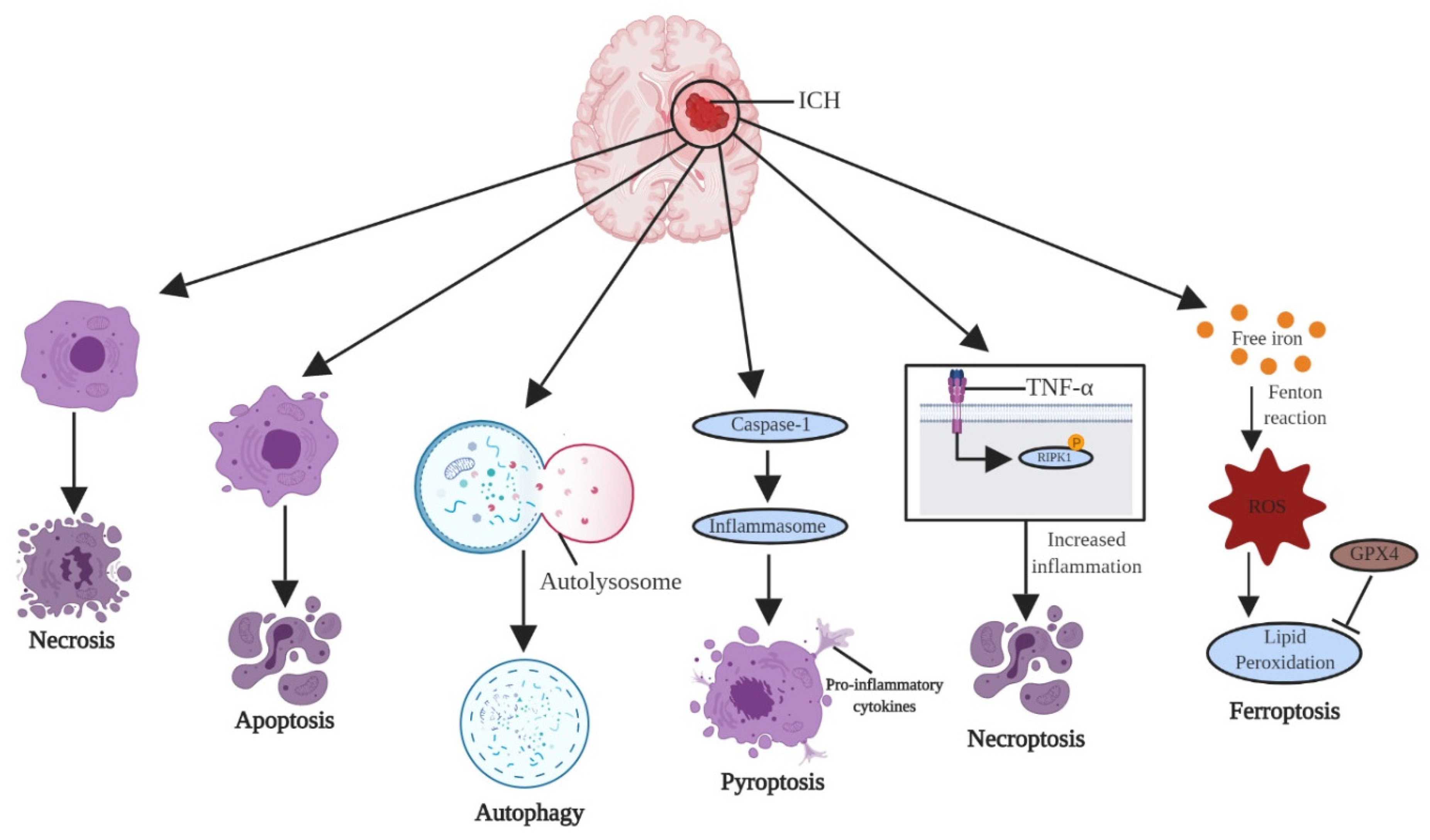
3. Cell Death Pathways after ICH
3.1. Necrosis, Apoptosis, and Necroptosis
3.2. Autophagy
3.3. Pyroptosis
3.4. Ferroptosis
4. Endoplasmic Reticulum Stress
5. Molecular Pathways of ER Stress
5.1. ER Transducer—Inositol-Requiring Protein-1 (IRE1)
5.2. ER Transducer—Activating Transcription Factor 6 (ATF6)
5.3. ER Transducer—Protein Kinase R-Like Endoplasmic Reticulum Kinase (PERK)
5.4. Calcium Homeostasis and ER Stress
6. Crosslinking ER Stress and ICH-Related Cell Death Pathways
6.1. ER Stress in Apoptosis
6.2. ER Stress in Autophagy
6.3. ER Stress in Pyroptosis
6.4. ER Stress in Necroptosis
6.5. ER Stress in Ferroptosis
7. ER Stress Components and ICH Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xi, G.; Keep, R.F.; Hoff, J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- An, S.J.; Kim, T.J.; Yoon, B.W. Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: An update. J. Stroke 2017, 19, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, F.I.; Chiou, H.Y. Stroke: Morbidity, risk factors, and care in Taiwan. J. Stroke 2014, 16, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, W.J. Intracerebral hemorrhage and head trauma: Common effects and common mechanisms of injury. Stroke 2010, 41, S107–S110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Wen, Z.; Shen, H.; Shen, M.; Chen, G. Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid. Med. Cell. Longev. 2016, 2016, 1203285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef]
- Xi, G.; Wagner, K.R.; Keep, R.F.; Hua, Y.; de Courten-Myers, G.M.; Broderick, J.P.; Brott, T.G.; Hoff, J.T. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke 1998, 29, 2580–2586. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 2010, 92, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Dore, S.; Galea, J.; Iihara, K.; et al. Haemoglobin scavenging in intracranial bleeding: biology and clinical implications. Nat. Rev. Neurol. 2018, 14, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Brain injury after intracerebral hemorrhage: The role of thrombin and iron. Stroke 2007, 38, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.A.; Samarasekera, N.; Lerpiniere, C.; Humphreys, C.; McCarron, M.O.; White, P.M.; Nicoll, J.A.R.; Sudlow, C.L.M.; Cordonnier, C.; Wardlaw, J.M.; et al. The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: Model development and diagnostic test accuracy study. Lancet Neurol. 2018, 17, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Aviv, R.I.; Fox, A.J.; Sahlas, D.J.; Gladstone, D.J.; Tomlinson, G.; Symons, S.P. CT angiography “spot sign” predicts hematoma expansion in acute intracerebral hemorrhage. Stroke 2007, 38, 1257–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orito, K.; Hirohata, M.; Nakamura, Y.; Takeshige, N.; Aoki, T.; Hattori, G.; Sakata, K.; Abe, T.; Uchiyama, Y.; Sakamoto, T.; et al. Leakage sign for primary intracerebral hemorrhage: A novel predictor of hematoma growth. Stroke 2016, 47, 958–963. [Google Scholar] [CrossRef]
- Hemphill, J.C., III; Bonovich, D.C.; Besmertis, L.; Manley, G.T.; Johnston, S.C. The ICH score: A simple, reliable grading scale for intracerebral hemorrhage. Stroke 2001, 32, 891–897. [Google Scholar] [CrossRef] [Green Version]
- Cordonnier, C.; Demchuk, A.; Ziai, W.; Anderson, C.S. Intracerebral haemorrhage: current approaches to acute management. Lancet 2018, 392, 1257–1268. [Google Scholar] [CrossRef]
- Matsushita, K.; Meng, W.; Wang, X.; Asahi, M.; Asahi, K.; Moskowitz, M.A.; Lo, E.H. Evidence for apoptosis after intercerebral hemorrhage in rat striatum. J. Cereb. Blood Flow Metab. 2000, 20, 396–404. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Suri, M.F.K.; Ostrow, P.T.; Kim, S.H.; Ali, Z.; Shatla, A.A.; Guterman, L.R.; Hopkins, L.N. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 2003, 52, 1041–1048. [Google Scholar] [CrossRef]
- Burtnick, L.D.; Wong, M.K.C. Interaction of iodoacetamidofluorescein-labelled tropomyosin with deoxyribonuclease I. Fed. Eur. Biochem. Soc. 1989, 244, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Byun, J.; Roh, S.E.; Kim, S.J.; Mook-Jung, I. Reduced IRE1α mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis. 2014, 5, e1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, N.; Kim, B.; Prestigiacomo, C.J. Necroptosis pathway in treatment of intracerebral hemorrhage: Novel therapeutic target. World Neurosurg. 2016, 89, 716–717. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zille, M.; Karuppagounder, S.S.; Chen, Y.; Gough, P.J.; Bertin, J.; Finger, J.; Milner, T.A.; Jonas, E.A.; Ratan, R.R. Neuronal death after hemorrhagic stroke In Vitro and In Vivo shares features of ferroptosis and necroptosis. Stroke 2017, 48, 1033–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Wan, S.; Hua, Y.; Keep, R.F.; Xi, G. Autophagy after experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 28, 897–905. [Google Scholar] [CrossRef]
- Bobinger, T.; Burkardt, P.; B. Huttner, H.; Manaenko, A. Programmed cell death after intracerebral hemorrhage. Curr. Neuropharmacol. 2018, 16, 1267–1281. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Y.; Feng, H. P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr. Neuropharmacol. 2018, 16, 1282–1295. [Google Scholar] [CrossRef]
- Lin, X.; Ye, H.; Siaw-Debrah, F.; Pan, S.; He, Z.; Ni, H.; Xu, Z.; Jin, K.; Zhuge, Q.; Huang, L. AC-YVAD-CMK inhibits pyroptosis and improves functional outcome after intracerebral hemorrhage. Biomed. Res. Int. 2018, 2018, 3706047. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Ren, H.; Wang, J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 2019, 4, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Science 2012, 2012, 857516. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Smith, H.L.; Mallucci, G.R. The unfolded protein response: Mechanisms and therapy of neurodegeneration. Brain 2016, 139, 2113–2121. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Merksamer, P.I.; Papa, F.R. The UPR and cell fate at a glance. J. Cell Sci. 2010, 123, 1003–1006. [Google Scholar] [CrossRef] [Green Version]
- Karagoz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of er stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 2017, 171, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the isoform-specific characteristics of ATF6α and ATF6β on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Dussmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6α (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalk, R.; Lehnart, S.E.; Marks, A.R. Modulation of the ryanodine receptor and intracellular calcium. Annu. Rev. Biochem. 2007, 76, 367–385. [Google Scholar] [CrossRef]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.C. Store-Operated calcium channels and pro-inflammatory signals. Acta Pharm. Sin. 2006, 27, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.T.; Hwang, S.Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 2010, 14, 2337–2349. [Google Scholar] [CrossRef]
- Gaut, J.R.; Hendershot, L.M. The modification and assembly of proteins in the endoplasmic reticulum. Curr. Opin. Cell Biol. 1993, 5, 589–595. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, G.; Brostrom, M.A.; Brostrom, C.O. Demonstration of a calcium requirement for secretory protein processing and export. Differential effects of calcium and dithiothreitol. J. Biol. Chem. 1992, 267, 3932–3939. [Google Scholar]
- Lodish, H.F.; Kong, N.; Wikstrom, L. Calcium is required for folding of newly made subunits of the asialoglycoprotein receptor within the endoplasmic reticulum. J. Biol. Chem. 1992, 267, 12753–12760. [Google Scholar] [PubMed]
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Petrozziello, T.; Tedeschi, V.; Boscia, F.; Vinciguerra, A.; Ciccone, R.; Pannaccione, A.; Molinaro, P.; Pignataro, G.; Annunziato, L. ORAI1/STIM1 interaction intervenes in stroke and in neuroprotection induced by ischemic preconditioning through store-operated calcium entry. Stroke 2019, 50, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Troupes, C.D.; Wallner, M.; Borghetti, G.; Zhang, C.; Mohsin, S.; von Lewinski, D.; Berretta, R.M.; Kubo, H.; Chen, X.; Soboloff, J.; et al. Role of STIM1 (stromal interaction molecule 1) in hypertrophy-related contractile dysfunction. Circ. Res. 2017, 121, 125–136. [Google Scholar] [CrossRef]
- Benard, L.; Oh, J.G.; Cacheux, M.; Lee, A.; Nonnenmacher, M.; Matasic, D.S.; Kohlbrenner, E.; Kho, C.; Pavoine, C.; Hajjar, R.J.; et al. Cardiac Stim1 silencing impairs adaptive hypertrophy and promotes heart failure through inactivation of mTORC2/Akt signaling. Circulation 2016, 133, 1458–1471. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.; Varga-Szabo, D.; Kleinschnitz, C.; Pleines, I.; Bender, M.; Austinat, M.; Bosl, M.; Stoll, G.; Nieswandt, B. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 2009, 113, 2056–2063. [Google Scholar] [CrossRef]
- Ahmad, F.; Boulaftali, Y.; Greene, T.K.; Ouellette, T.D.; Poncz, M.; Feske, S.; Bergmeier, W. Relative contributions of stromal interaction molecule 1 and CalDAG-GEFI to calcium-dependent platelet activation and thrombosis. J. Thromb. Haemost. 2011, 9, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.; Dai, X.; Zou, W.; Yu, X.; Teng, W.; Chen, Q.; Sun, X.; Yu, W.; Ma, H.; Liu, P. Autophagy, endoplasmic reticulum stress and the unfolded protein response in intracerebral hemorrhage. Transl. Neurosci. 2017, 8, 37–48. [Google Scholar] [CrossRef]
- Nakka, V.P.; Prakash-Babu, P.; Vemuganti, R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: Potential therapeutic targets for acute CNS injuries. Mol. Neurobiol. 2016, 53, 532–544. [Google Scholar] [CrossRef]
- He, Z.; Ostrowski, R.P.; Sun, X.; Ma, Q.; Huang, B.; Zhan, Y.; Zhang, J.H. CHOP silencing reduces acute brain injury in the rat model of subarachnoid hemorrhage. Stroke 2012, 43, 484–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, H.K.; Hu, W.F.; Lin, P.B.; Wang, P.K.; Tsai, A.P.; Pang, C.Y.; Chen, T.Y. Over-Activated proteasome mediates neuroinflammation on acute intracerebral hemorrhage in rats. Cells 2019, 8, 1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.E.; Buch, S. Cocaine-Mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos-Moron, E.; Abad-Jimenez, Z.; Maranon, A.M.; Iannantuoni, F.; Escribano-Lopez, I.; Lopez-Domenech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintado, C.; Macias, S.; Dominguez-Martin, H.; Castano, A.; Ruano, D. Neuroinflammation alters cellular proteostasis by producing endoplasmic reticulum stress, autophagy activation and disrupting ERAD activation. Sci. Rep. 2017, 7, 8100. [Google Scholar] [CrossRef] [Green Version]
- Demay, Y.; Perochon, J.; Szuplewski, S.; Mignotte, B.; Gaumer, S. The PERK pathway independently triggers apoptosis and a Rac1/Slpr/JNK/Dilp8 signaling favoring tissue homeostasis in a chronic ER stress Drosophila model. Cell Death Dis. 2014, 5, e1452. [Google Scholar] [CrossRef] [Green Version]
- Adler, H.T.; Chinery, R.; Wu, D.Y.; Kussick, S.J.; Payne, J.M.; Fornace, A.J., Jr.; Tkachuk, D.C. Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol. Cell. Biol. 1999, 19, 7050–7060. [Google Scholar] [CrossRef] [Green Version]
- Morishima, N.; Nakanishi, K.; Nakano, A. Activating transcription factor-6 (ATF6) mediates apoptosis with reduction of myeloid cell leukemia sequence 1 (Mcl-1) protein via induction of WW domain binding protein 1. J. Biol. Chem. 2011, 286, 35227–35235. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Chen, H.; Lin, P.; Wang, A.; Wang, L.; Jin, Y. ATF6 knockdown decreases apoptosis, arrests the S phase of the cell cycle, and increases steroid hormone production in mouse granulosa cells. Am. J. Physiol. Cell Physiol. 2017, 312, C341–C353. [Google Scholar] [CrossRef]
- Eftink, M.R.; Chiron, C.A. Fluorescence quenching studies with proteins. Anal. Biochem. 1981, 114, 199–227. [Google Scholar] [CrossRef]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Harada, C.; Nakamura, K.; Namekata, K.; Okumura, A.; Mitamura, Y.; Iizuka, Y.; Kashiwagi, K.; Yoshida, K.; Ohno, S.; Matsuzawa, A.; et al. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis In Vivo. Am. J. Pathol. 2006, 168, 261–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatai, T.; Matsuzawa, A.; Inoshita, S.; Mochida, Y.; Kuroda, T.; Sakamaki, K.; Kuida, K.; Yonehara, S.; Ichijo, H.; Takeda, K. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 2000, 275, 26576–26581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maundrell, K.; Antonsson, B.; Magnenat, E.; Camps, M.; Muda, M.; Chabert, C.; Gillieron, C.; Boschert, U.; Vial-Knecht, E.; Martinou, J.C.; et al. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J. Biol. Chem. 1997, 272, 25238–25242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deegan, S.; Saveljeva, S.; Gorman, A.M.; Samali, A. Stress-Induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2013, 70, 2425–2441. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef] [Green Version]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Katoh, H.; Imai, T.; Nomura, S.; Watanabe, M. The relationship between inflammasomes and the endoplasmic reticulum stress response in the injured spinal cord. Neurosci. Lett. 2019, 705, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Argon, Y. Stressed-Out endoplasmic reticulum inflames the mitochondria. Immunity 2015, 43, 409–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-Interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-R.; Yao, F.-H.; Zhang, J.-G.; Ji, Z.-Y.; Li, K.-L.; Zhan, J.; Tong, Y.-N.; Lin, L.-R.; He, Y.-N. Ischemia-Reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am. J. Physiol. Ren. Physiol. 2014, 306, F75–F84. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, K.; Li, Z.; Guo, B. ER Stress-Induced inflammasome activation contributes to hepatic inflammation and steatosis. J. Clin. Cell. Immunol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J.M. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-Induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Saha, P.; Gupta, R.; Foley, L.M.; Jiang, T.; Abakumova, O.S.; Hitchens, T.K.; Sen, N. Aberrant ER stress induced neuronal-IFNβ elicits white matter injury due to microglial activation and T-cell infiltration after TBI. J. Neurosci. 2020, 40, 424–446. [Google Scholar] [CrossRef]
- Liu, Y.; Min, J.W.; Feng, S.; Subedi, K.; Qiao, F.; Mammenga, E.; Callegari, E.; Wang, H. Therapeutic role of a cysteine precursor, OTC, in ischemic stroke is mediated by improved proteostasis in mice. Transl. Stroke Res. 2020, 11, 147–160. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, S.; Subedi, K.; Wang, H. Attenuation of ischemic stroke-caused brain injury by a monoamine oxidase inhibitor involves improved proteostasis and reduced neuroinflammation. Mol. Neurobiol. 2020, 57, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ounallah-Saad, H.; Chakraborty, D.; Hleihil, M.; Sood, R.; Barrera, I.; Edry, E.; Kolatt Chandran, S.; Ben Tabou de Leon, S.; Kaphzan, H.; et al. Local inhibition of PERK enhances memory and reverses age-related deterioration of cognitive and neuronal properties. J. Neurosci. 2018, 38, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharibani, P.; Modi, J.; Menzie, J.; Alexandrescu, A.; Ma, Z.; Tao, R.; Prentice, H.; Wu, J.Y. Comparison between single and combined post-treatment with S-Methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015, 300, 460–473. [Google Scholar] [CrossRef]
- Han, Y.; Yi, W.; Qin, J.; Zhao, Y.; Zhang, J.; Chang, X. Carbon monoxide offers neuroprotection from hippocampal cell damage induced by recurrent febrile seizures through the PERK-activated ER stress pathway. Neurosci. Lett. 2015, 585, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Rubovitch, V.; Barak, S.; Rachmany, L.; Goldstein, R.B.; Zilberstein, Y.; Pick, C.G. The neuroprotective effect of salubrinal in a mouse model of traumatic brain injury. Neuromol. Med. 2015, 17, 58–70. [Google Scholar] [CrossRef]
- Kumar, R.; Azam, S.; Sullivan, J.M.; Owen, C.; Cavener, D.R.; Zhang, P.; Ron, D.; Harding, H.P.; Chen, J.J.; Han, A.; et al. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2α kinase, PERK. J. Neurochem. 2001, 77, 1418–1421. [Google Scholar] [CrossRef] [Green Version]
- Gall, T.; Petho, D.; Nagy, A.; Hendrik, Z.; Mehes, G.; Potor, L.; Gram, M.; Akerstrom, B.; Smith, A.; Nagy, P.; et al. Heme induces endoplasmic reticulum stress (HIER stress) in human aortic smooth muscle cells. Front. Physiol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Meng, C.; Zhang, J.; Dang, B.; Li, H.; Shen, H.; Li, X.; Wang, Z. PERK Pathway activation promotes intracerebral hemorrhage induced secondary brain injury by inducing neuronal apoptosis both In Vivo and In Vitro. Front. Neurosci. 2018, 12, 111. [Google Scholar] [CrossRef]
- Xu, W.; Lu, X.; Zheng, J.; Li, T.; Gao, L.; Lenahan, C.; Shao, A.; Zhang, J.; Yu, J. Melatonin protects against neuronal apoptosis via suppression of the ATF6/CHOP pathway in a rat model of intracerebral hemorrhage. Front. Neurosci. 2018, 12, 638. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Lan, T.; Lu, J.; Zhang, H.; Zhang, D.; Lou, T.; Xu, P.; Ren, J.; Zhao, D.; Sun, L.; et al. DiDang tang inhibits endoplasmic reticulum stress-mediated apoptosis induced by oxygen glucose deprivation and intracerebral hemorrhage through blockade of the GRP78-IRE1/PERK pathways. Front. Pharm. 2018, 9, 1423. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed Thangameeran, S.I.; Tsai, S.-T.; Hung, H.-Y.; Hu, W.-F.; Pang, C.-Y.; Chen, S.-Y.; Liew, H.-K. A Role for Endoplasmic Reticulum Stress in Intracerebral Hemorrhage. Cells 2020, 9, 750. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030750
Mohammed Thangameeran SI, Tsai S-T, Hung H-Y, Hu W-F, Pang C-Y, Chen S-Y, Liew H-K. A Role for Endoplasmic Reticulum Stress in Intracerebral Hemorrhage. Cells. 2020; 9(3):750. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030750
Chicago/Turabian StyleMohammed Thangameeran, Shaik Ismail, Sheng-Tzung Tsai, Hsiang-Yi Hung, Wei-Fen Hu, Cheng-Yoong Pang, Shin-Yuan Chen, and Hock-Kean Liew. 2020. "A Role for Endoplasmic Reticulum Stress in Intracerebral Hemorrhage" Cells 9, no. 3: 750. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030750