Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Transfection Procedures
2.3. RNA Extraction and Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
2.4. Gene Expression Analyses
2.5. Gene Set Enrichment Analyses
2.6. Western Blot Analyses
2.7. Incorporation of 14C Acetate
2.8. Microscopy
2.9. Cell Treatments and Apoptosis and Viability Assays
2.10. Statistics
3. Results
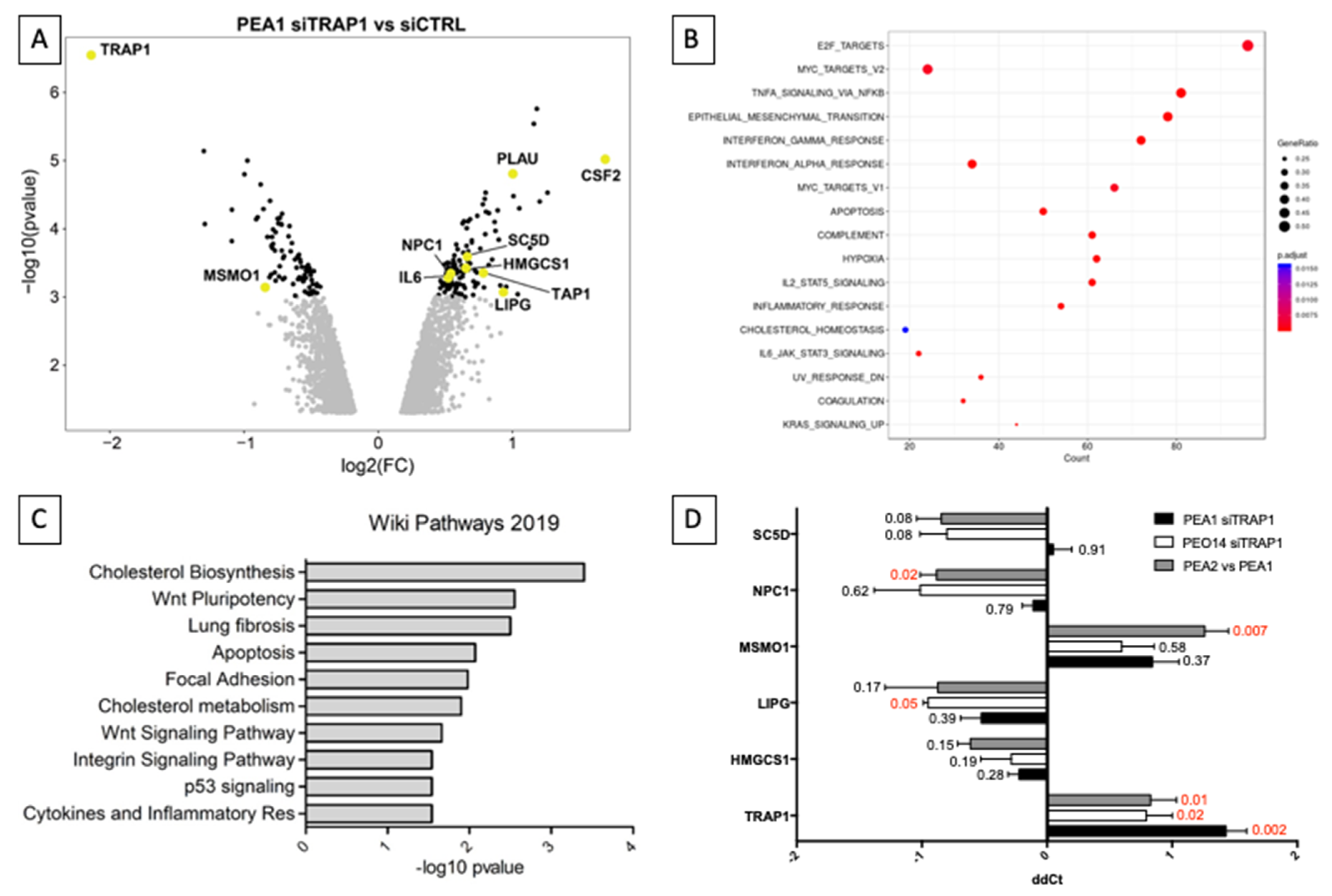
3.1. TRAP1 Expression and Chemoresistance Correlate with Metabolism, Inflammation, and Lipid Homeostasis
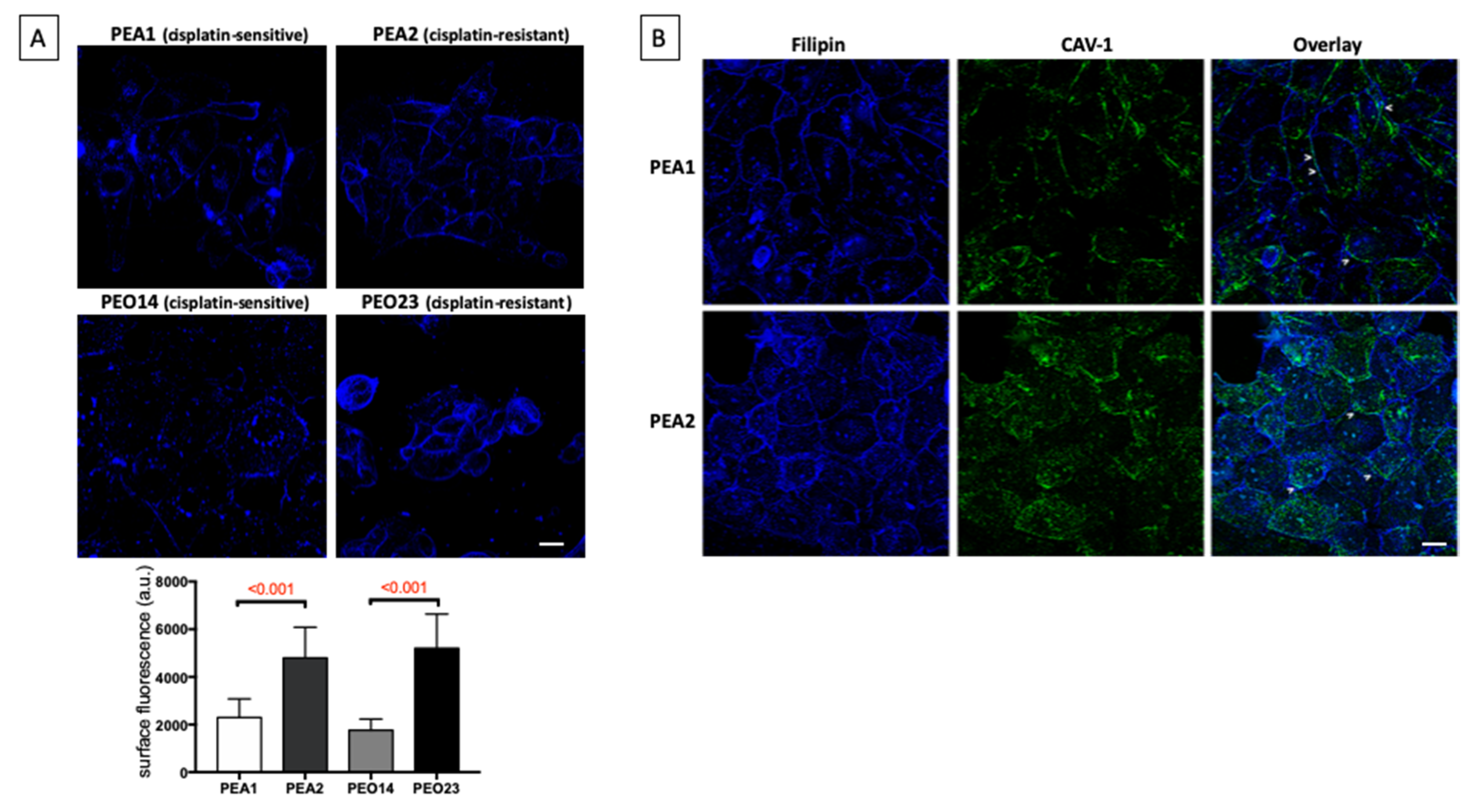
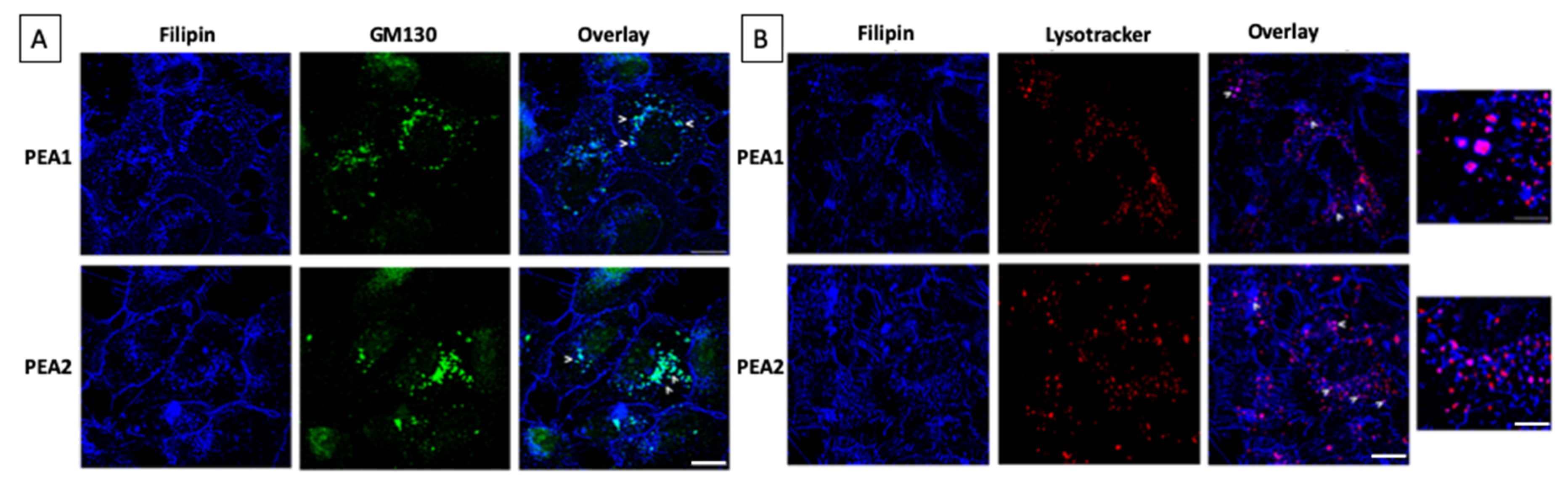
3.2. Cholesterol Distribution Is Altered in Chemoresistant Cells
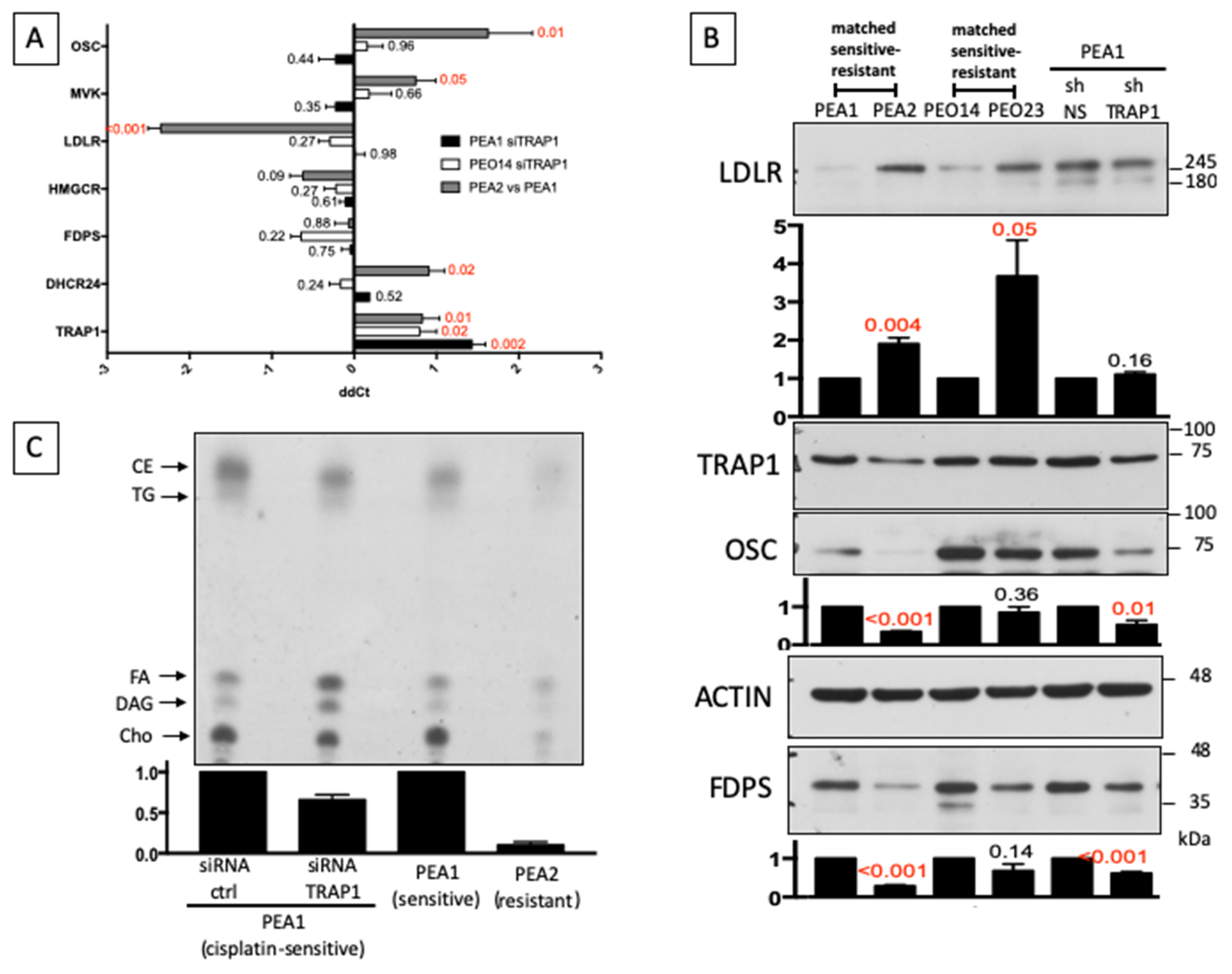
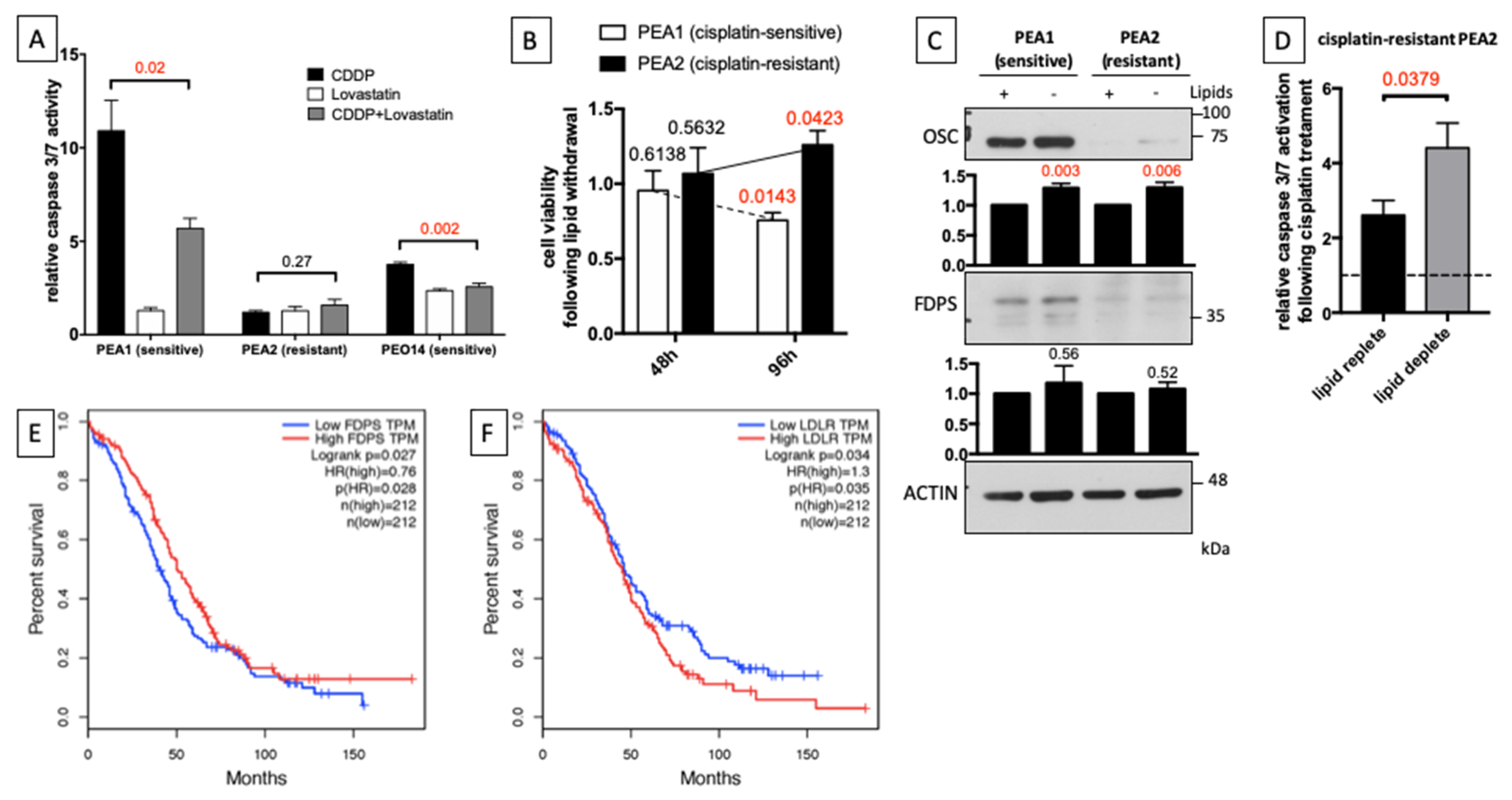
3.3. Reduced Cholesterol Biosynthesis and Increased Cholesterol Uptake from Extracellular Environment Induce Chemoresistance
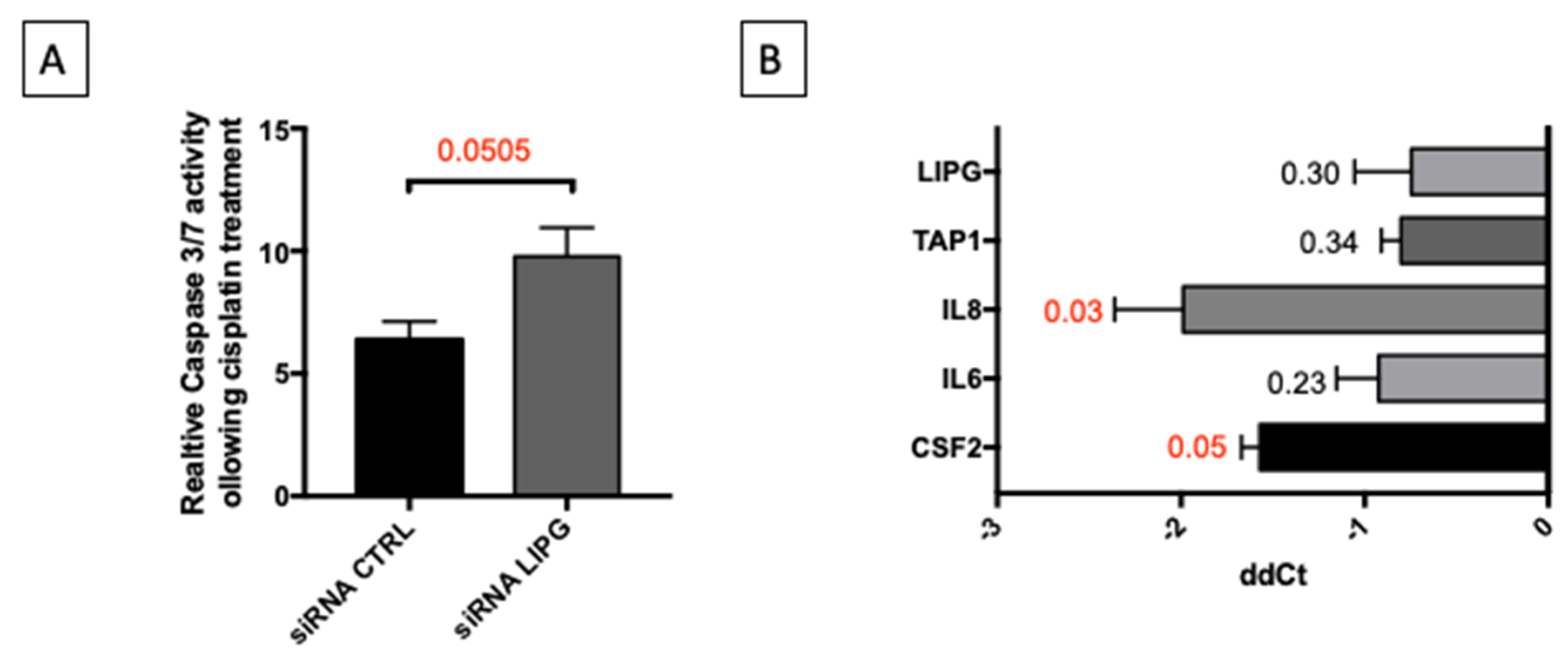
3.4. Oxidative Stress Induces Inflammatory Response and Remodeling of Lipid Metabolism
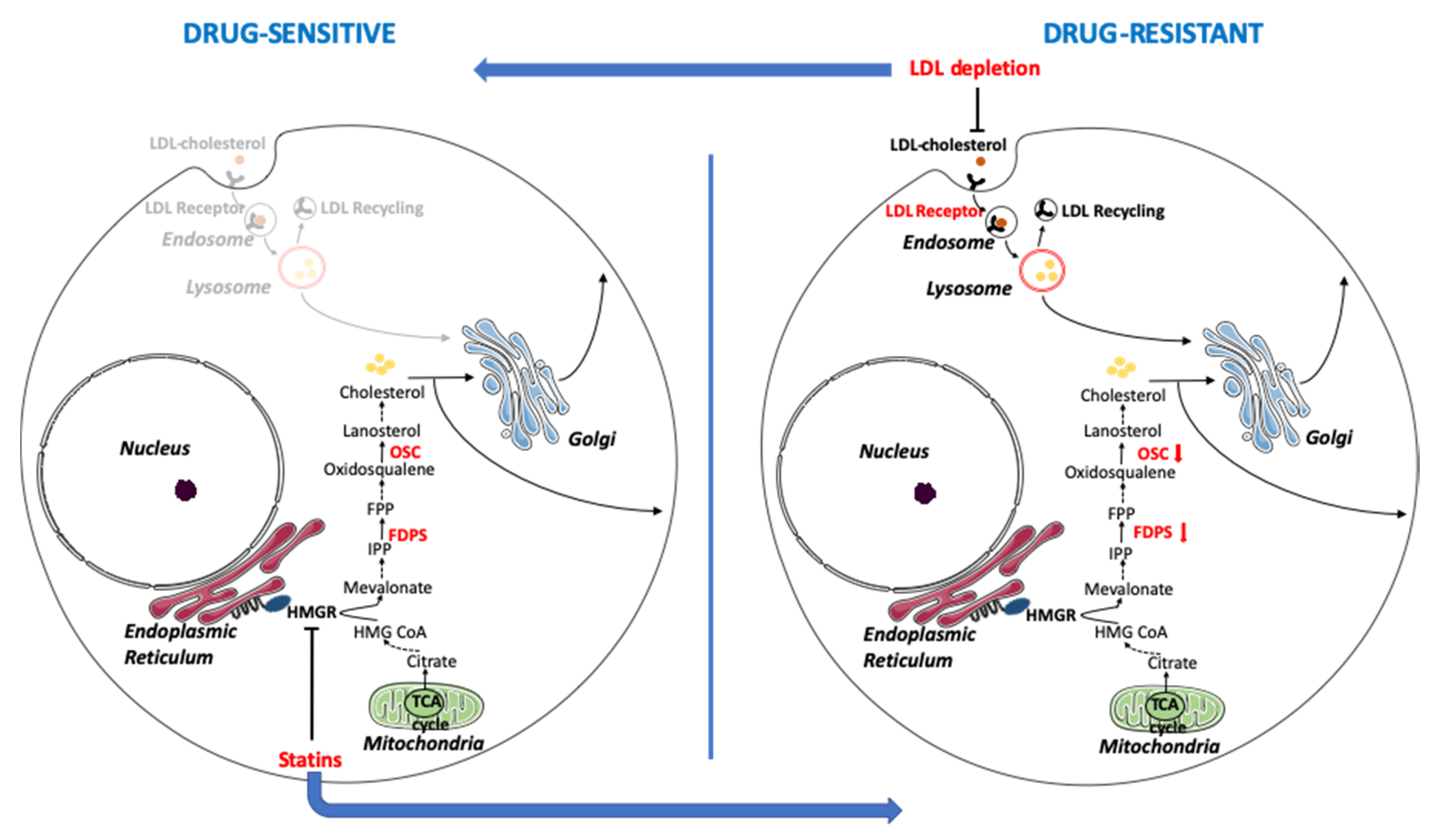
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The Dualistic Model of Ovarian Carcinogenesis. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossaï, M.; Leary, A.; Scoazec, J.Y.; Genestie, C. Ovarian Cancer: A Heterogeneous Disease. Pathobiology 2018, 85, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A. PARP inhibitors in ovarian cancer. Ann. Oncol. 2016, 27, 40–44. [Google Scholar] [CrossRef]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef] [Green Version]
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014, 24, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Lu, H.; Sisinni, L.; Lettini, G.; Gabra, H.; Landriscina, M.; Esposito1, F. TRAP1 downregulation in human ovarian cancer enhances invasion and epithelial–mesenchymal transition. Cell Death Dis. 2016, 7, 2522. [Google Scholar] [CrossRef] [Green Version]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; de Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of Endosomal Trafficking Is Associated with Early-Onset Parkinsonism Caused by SYNJ1 Mutations. Cell Death Dis. 2018. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, 661–667. [Google Scholar] [CrossRef]
- Yu, J.E.; Han, S.Y.; Wolfson, B.; Zhou, Q. The role of endothelial lipase in lipid metabolism, inflammation, and cancer. Histol. Histopathol. 2018, 33, 1–10. [Google Scholar]
- He, M.; Smith, L.D.; Chang, R.; Li, X.; Vockley, J. The role of sterol-C4-methyl oxidase in epidermal biology. Biochim. Biophys. Acta 2014, 1841, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, J.; Yang, E.J.; Shim, J.S. Cholesterol Trafficking: An Emerging Therapeutic Target for Angiogenesis and Cancer. Cells 2019, 8, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stronach, E.A.; Alfraidi, A.; Rama, N.; Datler, C.; Studd, J.B.; Agarwal, R.; Guney, T.G.; Gourley, C.; Hennessy, B.T.; Mills, G.B.; et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011, 71, 4412–4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloch, K. The biological synthesis of cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kandutsch, A.A.; Russell, A.E. Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J. Biol. Chem. 1960, 235, 2256–2261. [Google Scholar] [PubMed]
- Laezza, C.; D’Alessandro, A.; Croce, L.D.; Picardi, P.; Ciaglia, E.; Pisanti, S.; Malfitano, A.M.; Comegna, M.; Faraonio, R.; Gazzerro, P.; et al. p53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonen, E. Cellular Cholesterol Trafficking and Compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-κB, IκB, and IKK: Integral Components of Immune System Signaling. In Structural Immunology; Jin, T., Yin, Q., Eds.; Springer: Singapore, 2019; Volume 1172, pp. 207–226. [Google Scholar]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Simons, K. How Cells Handle Cholesterol. Science 2000, 290, 1721–1726. [Google Scholar] [CrossRef] [Green Version]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; Ghamrasni, S.E.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [Green Version]
- Thurnher, M.; Nussbaumer, O.; Gruenbacher, G. Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3524–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siperstein, M.D.; Guest, M.J. Studies on the Site of the Feedback Control of Cholesterol Synthesis. J. Clin. Investig. 1960, 39, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivel, T.; Ramseyer, C.; Yesylevskyy, S. The asymmetry of plasma membranes and their cholesterol content influence the uptake of cisplatin. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; de Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ravnskov, U.; McCully, K.S.; Rosch, P.J. The statin-low cholesterol-cancer conundrum. QJM 2012, 105, 383–388. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, M.; Jiang, H.; Hao, J.; Zhang, Q.; Zhu, Q.; Saren, G.; Zhang, Y.; Meng, X.; Yue, X. Angiotensin II Upregulates Endothelial Lipase Expression via the NF-Kappa B and MAPK Signaling Pathways. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Maddalena, F.; Condelli, V.; Landriscina, M.; Esposito, F. Stress-Adaptive Response in Ovarian Cancer Drug Resistance: Role of TRAP1 in Oxidative Metabolism-Driven Inflammation. Adv. Protein Chem. Struct. Biol. 2017, 108, 163–198. [Google Scholar]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-ΚB Signaling Pathway during Inflammation. Front. Immunol. 2016. [Google Scholar] [CrossRef]
- Scott, C.C.; Vossio, S.; Vacca, F.; Snijder, B.; Larios, J.; Schaad, O.; Guex, N.; Kuznetsov, D.; Martin, O.; Chambon, M.; et al. Wnt Directs the Endosomal Flux of LDL -derived Cholesterol and Lipid Droplet Homeostasis. EMBO Rep. 2015. [Google Scholar] [CrossRef] [Green Version]
- Liscovitch, M.; Lavie, Y. Multidrug Resistance: A Role for Cholesterol Efflux Pathways? Trends Biochem. Sci. 2000, 25, 530–534. [Google Scholar] [CrossRef]
- Bermudez, J.G.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 2019, 567, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Curtarello, M.; Tognon, M.; Venturoli, C.; Silic-Benussi, M.; Grassi, A.; Verza, M.; Minuzzo, S.; Pinazza, M.; Brillo, V.; Tosi, G.; et al. Rewiring of Lipid Metabolism and Storage in Ovarian Cancer Cells after Anti-VEGF Therapy. Cells 2019, 8, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer ID | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| ACTIN | CCTCACCCTGAAGTACCCCA | TCGTCCCAGTTGGTGACGAT |
| CSF2 | GTCATCTTGGAGGGACCAAGG | CCATGCCTGTATCAGGGTCAG |
| DHCR24 | TGAAGACAAACCGAGAGGGC | CAGCCAAAGAGGTAGCGGAA |
| FDPS | AGCAGGATTTCGTTCAGCAC | TCCCGGAATGCTACTACCAC |
| HMGCR | TACCATGTCAGGGGTACGTC | CCAGTCCTAATGAAACCTTAGAAG |
| HMGCS1 | TGTCCTTTCGTGGCTCACTC | GCCAGCAAGCTTCTGCATTC |
| IL6 | TGCAATAACCACCCCTGACC | CAATCTGAGGTGCCCATGCT |
| IL8 | CCAGTCTTGTCATTGCCAGC | TGACTGTGGAGTTTTGGCTGT |
| LDLR | GAATTTGGCCAGACACGGT | CACCGTACCCAGCTGATTTT |
| LIPG | CAGGCTGTGGACTCAACGAT | TCGGCTTGTCCTGATTCACC |
| MVK | GCTCAAGTTCCCAGAGATCG | ATGGTGCTGGTTCATGTCAA |
| MSMO1 | GGTTCCGAGGTTGGAACACCT | TTCAAATCTCTGCAGACAGCCT |
| NPC1 | TTGTGGTGTTGGCTTTTGCC | GTTCGCGCTCTGTTCCTTTG |
| OSC | ATGGTGGCCCACTTTTCCTC | CTGCACTGACCGCAGGTA |
| SC5D | TTTGCAGAGCAGTGGCGT | CTGGCCATGTGGCTGGATAC |
| TAP1 | CCTCCTTTCCAAGCTCCTCG | GGCCAGCATATGCCTTCAGT |
| TRAP1 | GACGCACCGCTCAACAT | CACATCAAACATGGACGGTTT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criscuolo, D.; Avolio, R.; Calice, G.; Laezza, C.; Paladino, S.; Navarra, G.; Maddalena, F.; Crispo, F.; Pagano, C.; Bifulco, M.; et al. Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer. Cells 2020, 9, 828. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040828
Criscuolo D, Avolio R, Calice G, Laezza C, Paladino S, Navarra G, Maddalena F, Crispo F, Pagano C, Bifulco M, et al. Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer. Cells. 2020; 9(4):828. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040828
Chicago/Turabian StyleCriscuolo, Daniela, Rosario Avolio, Giovanni Calice, Chiara Laezza, Simona Paladino, Giovanna Navarra, Francesca Maddalena, Fabiana Crispo, Cristina Pagano, Maurizio Bifulco, and et al. 2020. "Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer" Cells 9, no. 4: 828. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040828