Xanthohumol, a Prenylated Flavonoid from Hops, Induces DNA Damages in Colorectal Cancer Cells and Sensitizes SW480 Cells to the SN38 Chemotherapeutic Agent

Abstract
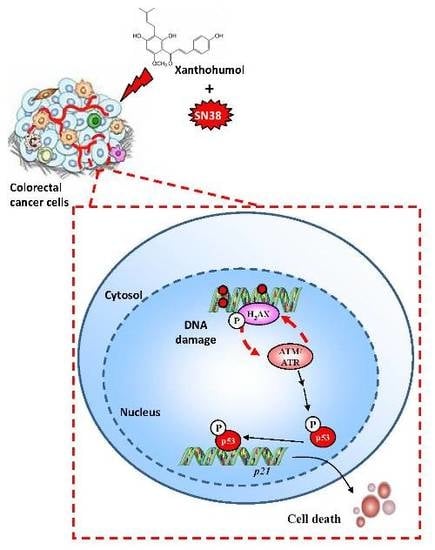
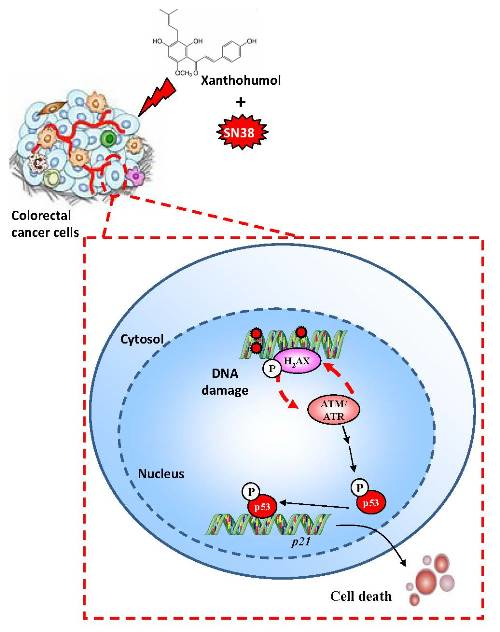
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents and Antibodies
2.3. Cell Viability Assays
2.4. Combination Index Analysis
2.5. Flow Cytometry
2.5.1. Cell Cycle Analysis
2.5.2. Apoptosis Analysis
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
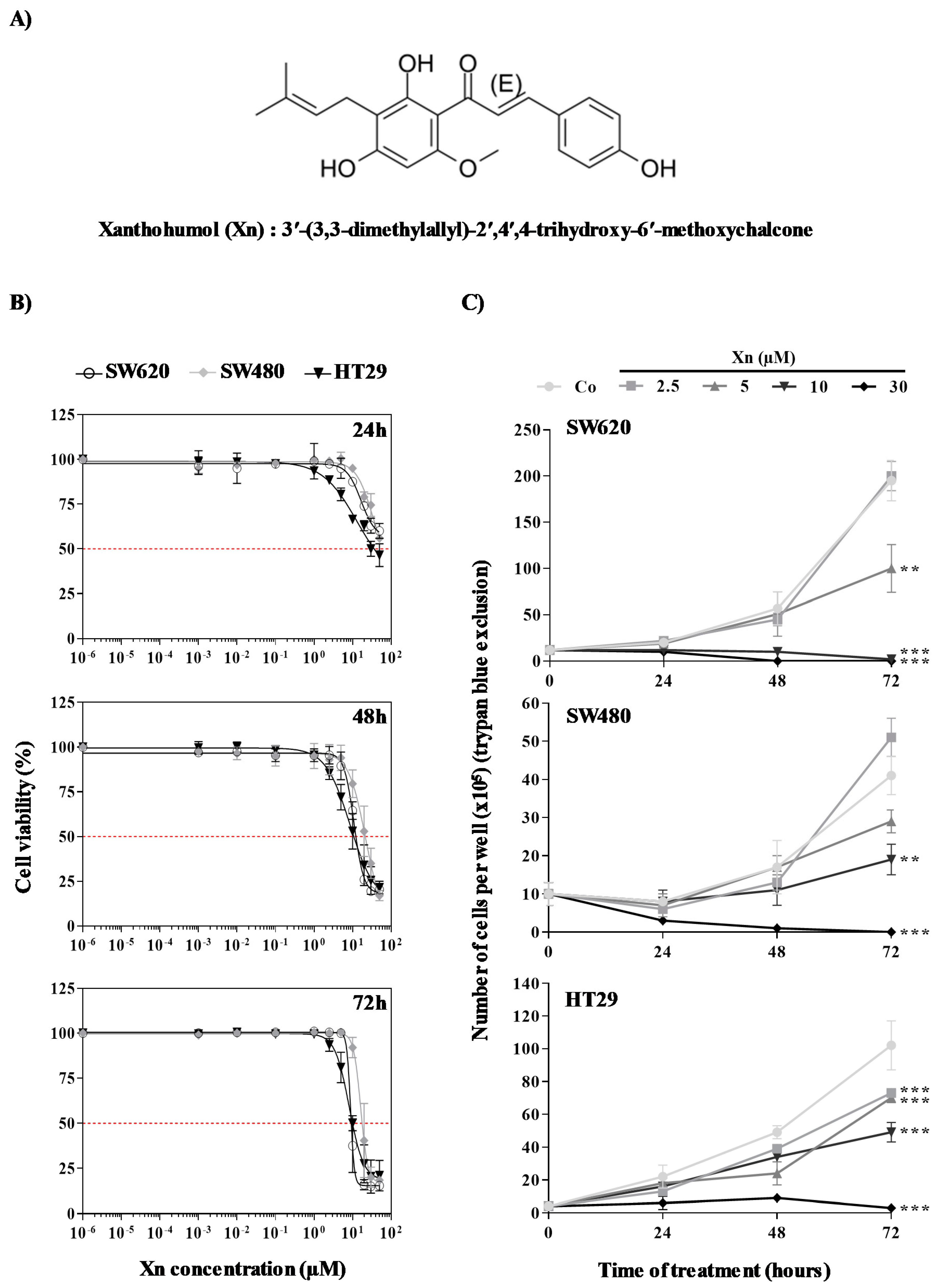
3.1. Antiproliferative Properties of Xanthohumol on Colorectal Cancer Cells
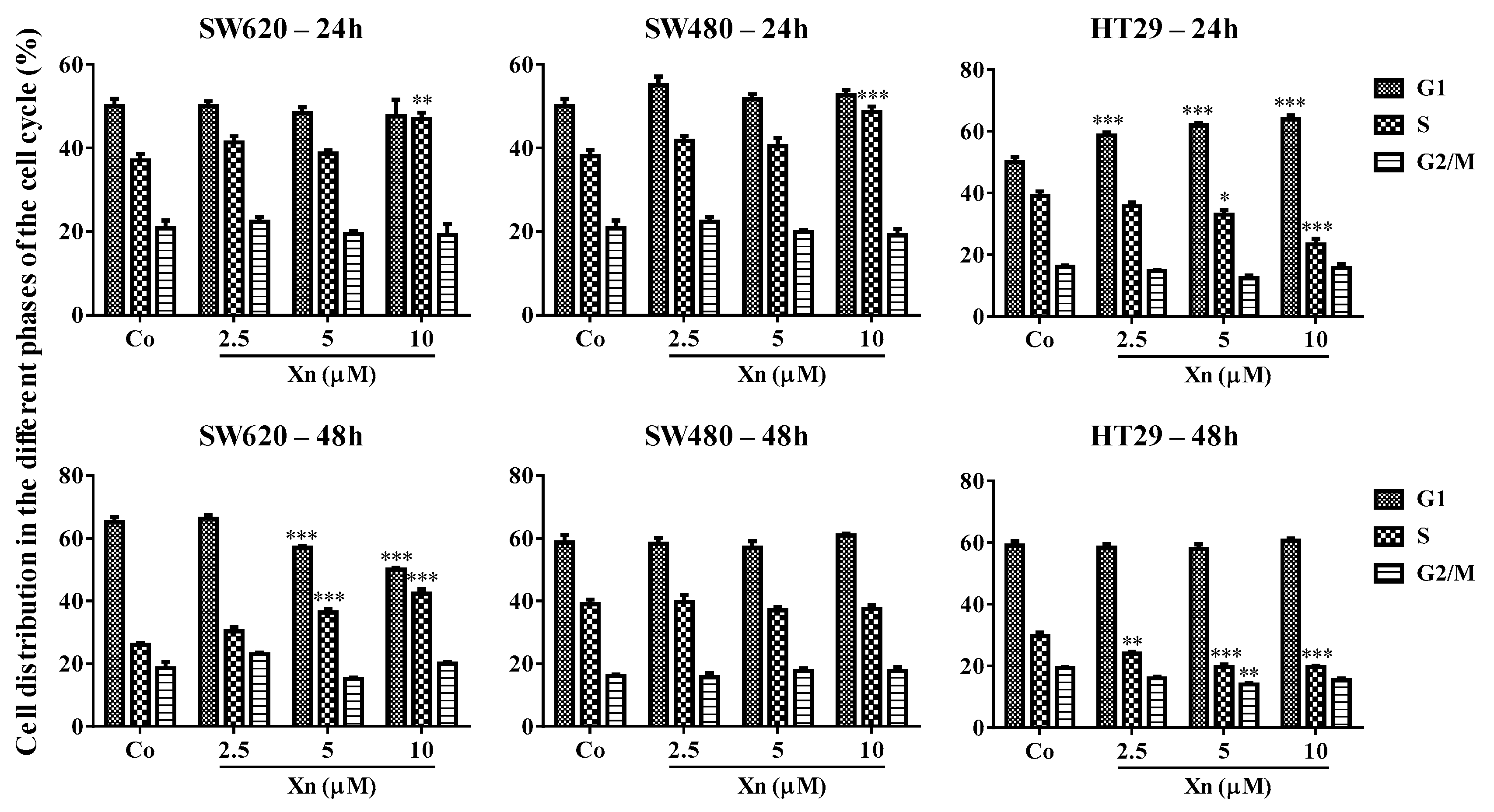
3.2. Xanthohumol Modulates Distribution of Colorectal Cancer Cells in the Cell Cycle
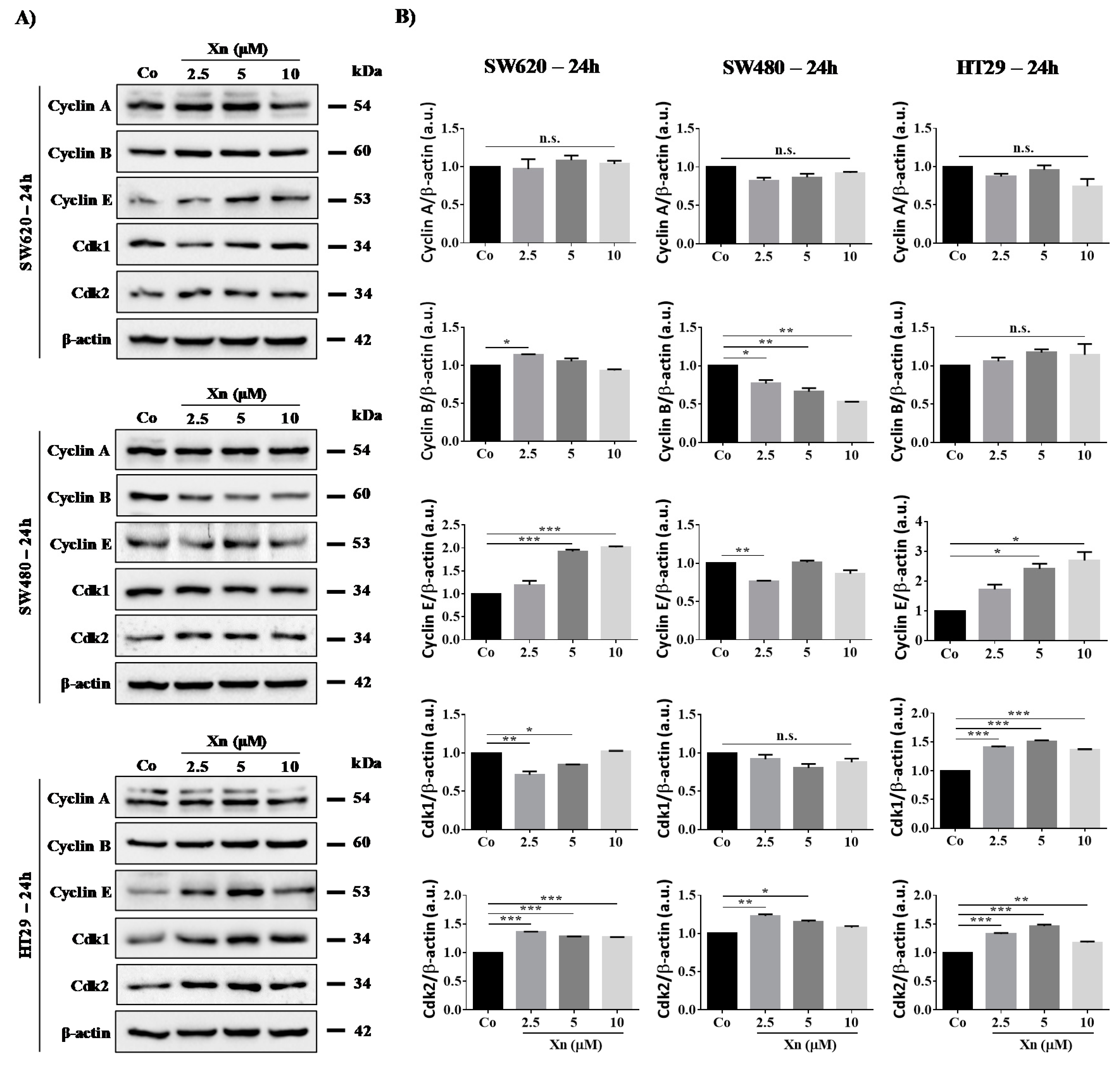
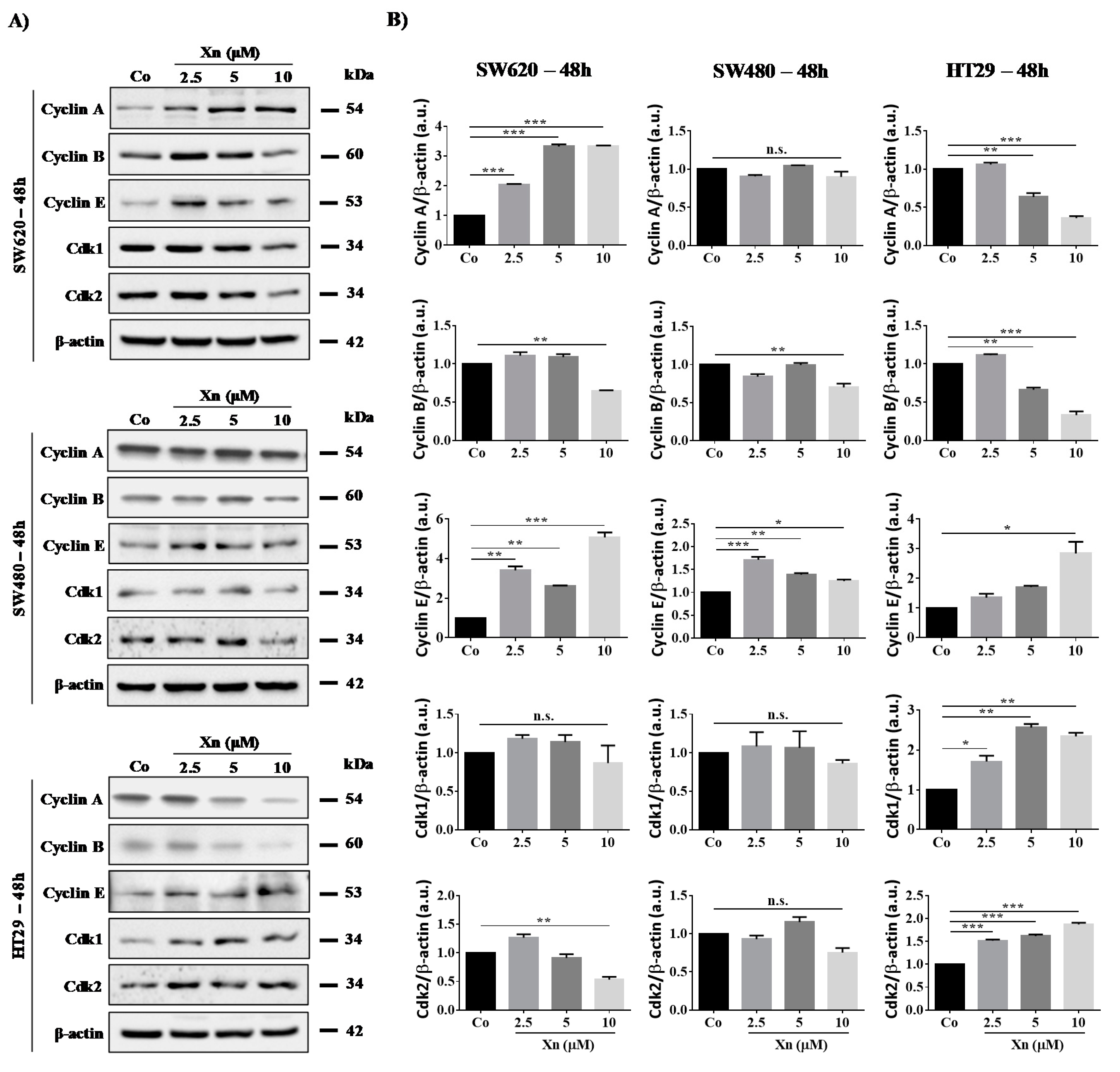
3.3. Involvement of Cyclins and their Cdks in Xanthohumol-Induced Cell Cycle Disruption
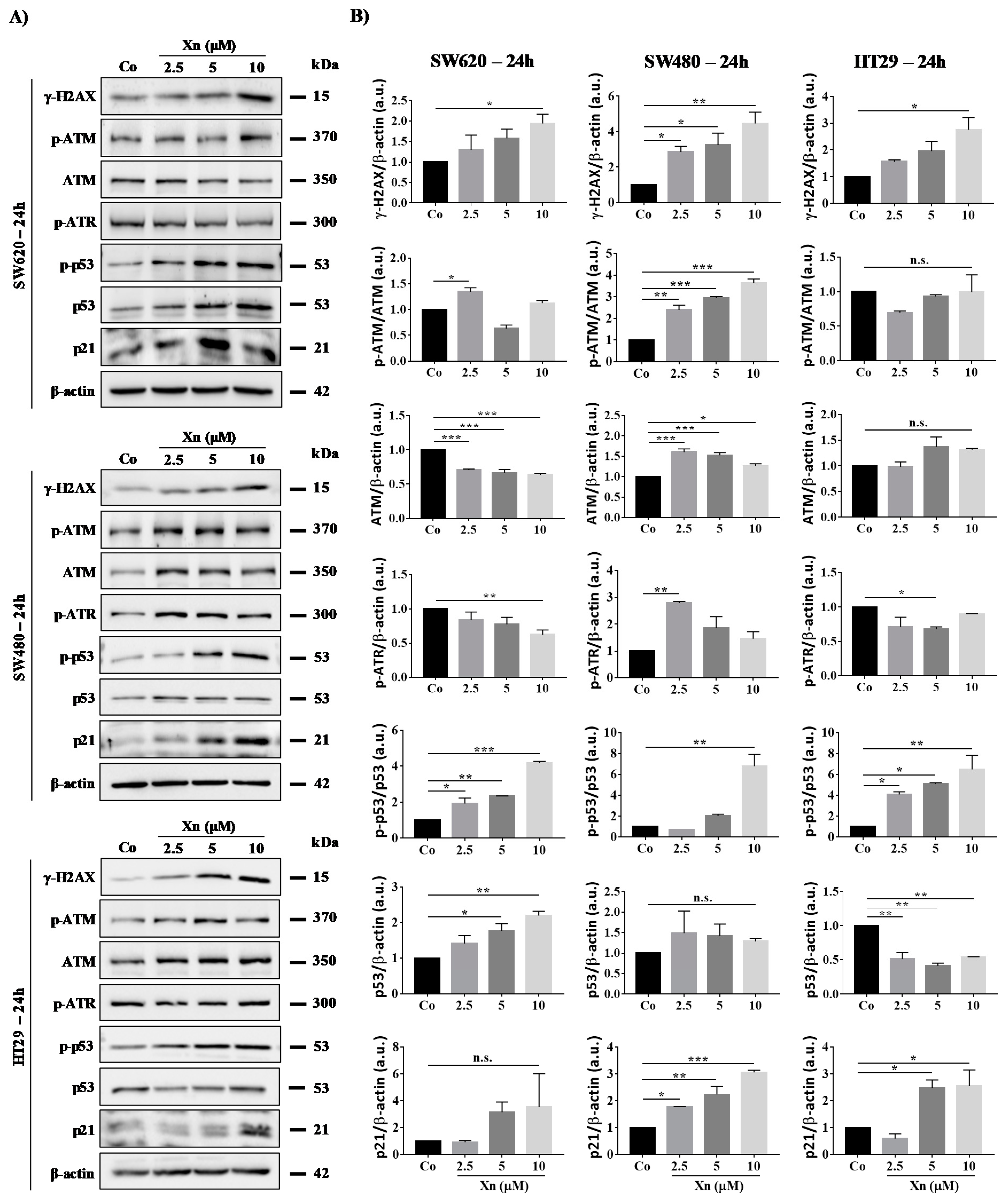
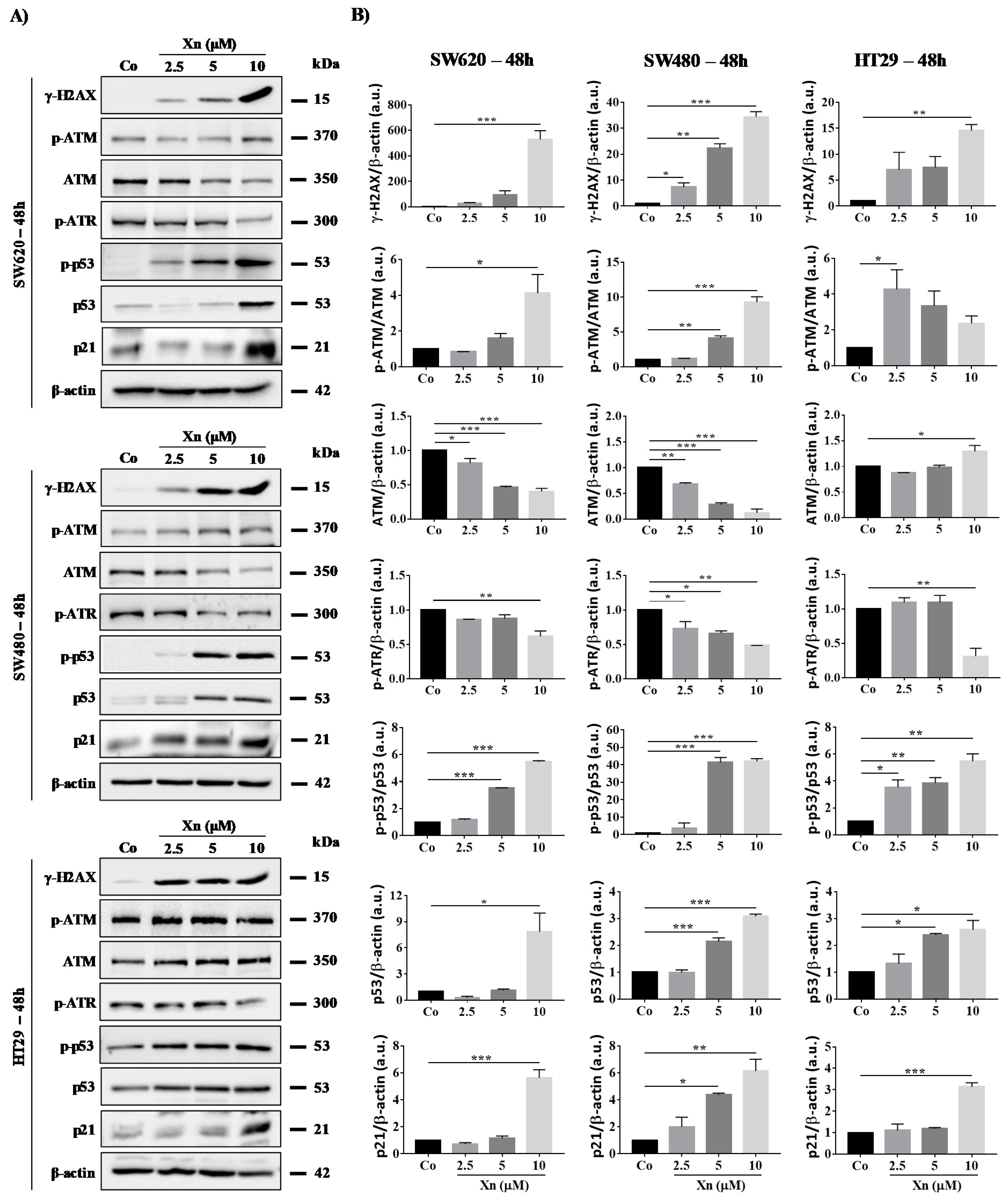
3.4. Xanthohumol Induces DNA Damage in Colon Cancer Cells
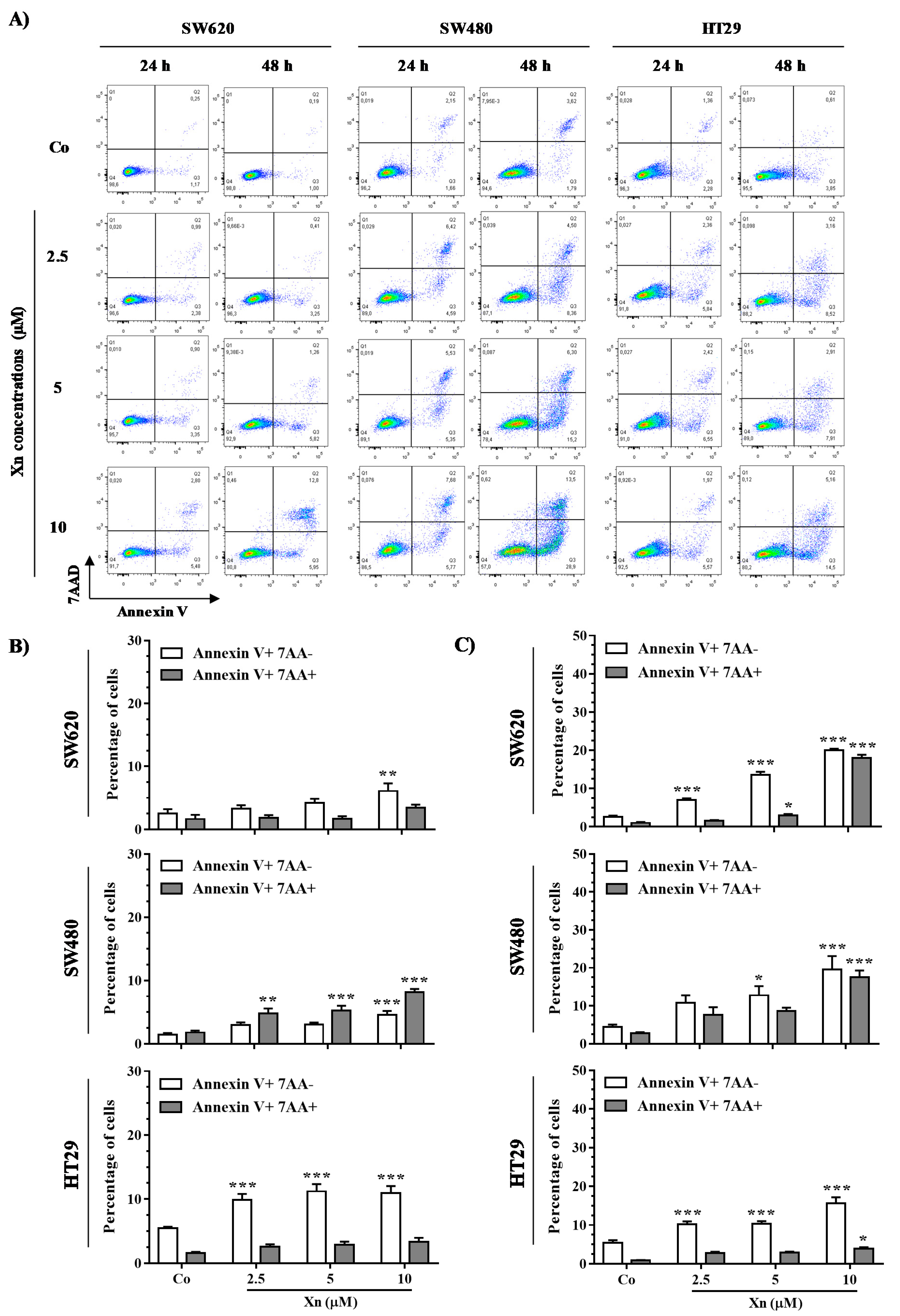
3.5. Xanthohumol Induces Apoptosis in Colon Cancer Cells
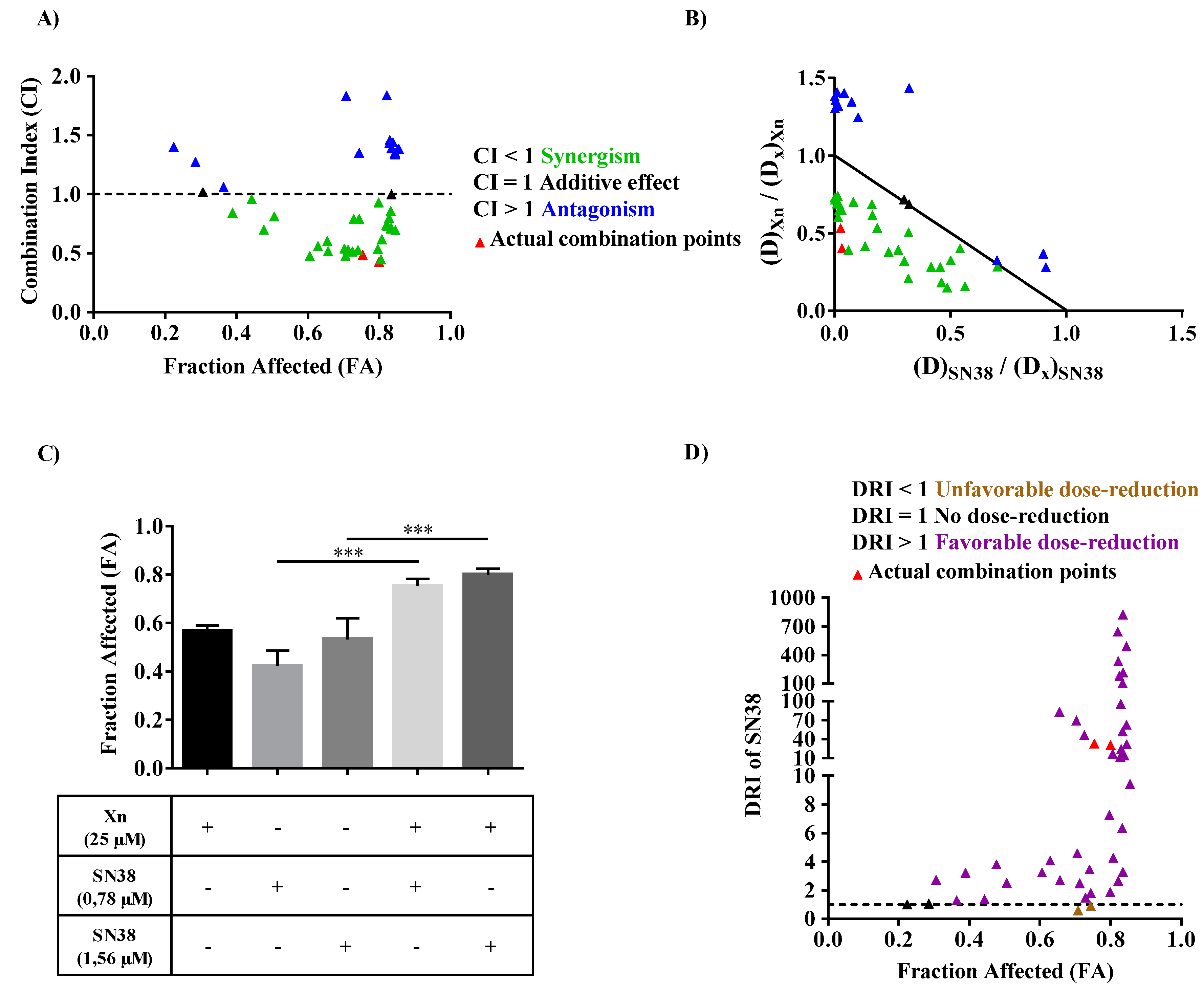
3.6. Xanthohumol Synergized with SN38 to Inhibit SW480 Colorectal Cancer Cell Proliferation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2013, 394, 1467–1480. [Google Scholar] [CrossRef]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangere, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; De Barros, J.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef]
- Cotte, A.K.; Cottet, V.; Aires, V.; Mouillot, T.; Rizk, M.; Vinault, S.; Binquet, C.; De Barros, J.P.; Hillon, P.; Delmas, D. Phospholipid profiles and hepatocellular carcinoma risk and prognosis in cirrhotic patients. Oncotarget 2019, 10, 2161–2172. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef]
- Allison, J.D.; Tanavin, T.; Yang, Y.; Birnbaum, G.; Khalid, U. Various Manifestations of 5-Fluorouracil Cardiotoxicity: A Multicenter Case Series and Review of Literature. Cardiovasc. Toxicol. 2020. [Google Scholar] [CrossRef]
- Tsalic, M.; Bar-Sela, G.; Beny, A.; Visel, B.; Haim, N. Severe toxicity related to the 5-fluorouracil/leucovorin combination (the Mayo Clinic regimen): A prospective study in colorectal cancer patients. Am. J. Clin. Oncol. 2003, 26, 103–106. [Google Scholar] [CrossRef]
- Aires, V.; Colin, D.J.; Doreau, A.; Di Pietro, A.; Heydel, J.M.; Artur, Y.; Latruffe, N.; Delmas, D. P-Glycoprotein 1 Affects Chemoactivities of Resveratrol against Human Colorectal Cancer Cells. Nutrients 2019, 11, 2098. [Google Scholar] [CrossRef] [Green Version]
- Delmas, D.; Solary, E.; Latruffe, N. Resveratrol, a phytochemical inducer of multiple cell death pathways: Apoptosis, autophagy and mitotic catastrophe. Curr. Med. Chem. 2011, 18, 1100–1121. [Google Scholar] [CrossRef]
- Gongora, C.; Vezzio-Vie, N.; Tuduri, S.; Denis, V.; Causse, A.; Auzanneau, C.; Collod-Beroud, G.; Coquelle, A.; Pasero, P.; Pourquier, P.; et al. New Topoisomerase I mutations are associated with resistance to camptothecin. Mol. Cancer 2011, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Petitprez, A.; Poindessous, V.; Ouaret, D.; Regairaz, M.; Bastian, G.; Guerin, E.; Escargueil, A.E.; Larsen, A.K. Acquired irinotecan resistance is accompanied by stable modifications of cell cycle dynamics independent of MSI status. Int. J. Oncol. 2013, 42, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Aires, V.; Limagne, E.; Cotte, A.K.; Latruffe, N.; Ghiringhelli, F.; Delmas, D. Resveratrol metabolites inhibit human metastatic colon cancer cells progression and synergize with chemotherapeutic drugs to induce cell death. Mol. Nutr. Food Res. 2013, 57, 1170–1181. [Google Scholar] [CrossRef]
- Al-Eitan, L.N.; Alzoubi, K.H.; Al-Smadi, L.I.; Khabour, O.F. Vitamin E protects against cisplatin-induced genotoxicity in human lymphocytes. Toxicol. In Vitro 2020, 62, 104672. [Google Scholar] [CrossRef]
- Shruthi, S.; Bhasker Shenoy, K. Genoprotective effects of gallic acid against cisplatin induced genotoxicity in bone marrow cells of mice. Toxicol. Res. 2018, 7, 951–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hu, P.C.; Gao, F.F.; Lv, J.W.; Xu, S.; Kuang, C.C.; Wei, L.; Zhang, J.W. The protective effect of curcumin on hepatotoxicity and ultrastructural damage induced by cisplatin. Ultrastruct. Pathol. 2014, 38, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Colin, D.; Gimazane, A.; Lizard, G.; Izard, J.C.; Solary, E.; Latruffe, N.; Delmas, D. Effects of resveratrol analogs on cell cycle progression, cell cycle associated proteins and 5fluoro-uracil sensitivity in human derived colon cancer cells. Int. J. Cancer 2009, 124, 2780–2788. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.; van Breemen, R.B. Analytical methods for quantitation of prenylated flavonoids from hops. Curr. Anal. Chem. 2013, 9, 71–85. [Google Scholar] [CrossRef]
- Benelli, R.; Vene, R.; Ciarlo, M.; Carlone, S.; Barbieri, O.; Ferrari, N. The AKT/NF-kappaB inhibitor xanthohumol is a potent anti-lymphocytic leukemia drug overcoming chemoresistance and cell infiltration. Biochem. Pharmacol. 2012, 83, 1634–1642. [Google Scholar] [CrossRef]
- Ho, Y.C.; Liu, C.H.; Chen, C.N.; Duan, K.J.; Lin, M.T. Inhibitory effects of xanthohumol from hops (Humulus lupulus L.) on human hepatocellular carcinoma cell lines. Phytother. Res. 2008, 22, 1465–1468. [Google Scholar] [CrossRef]
- Yoo, Y.B.; Park, K.S.; Kim, J.B.; Kang, H.J.; Yang, J.H.; Lee, E.K.; Kim, H.Y. Xanthohumol inhibits cellular proliferation in a breast cancer cell line (MDA-MB231) through an intrinsic mitochondrial-dependent pathway. Indian J. Cancer 2014, 51, 518–523. [Google Scholar]
- Monteiro, R.; Calhau, C.; Silva, A.O.; Pinheiro-Silva, S.; Guerreiro, S.; Gartner, F.; Azevedo, I.; Soares, R. Xanthohumol inhibits inflammatory factor production and angiogenesis in breast cancer xenografts. J. Cell. Biochem. 2008, 104, 1699–1707. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Jiang, H.; Arbab, A.S.; Dulchavsky, S.A.; Gautam, S.C. Growth inhibitory and apoptosis-inducing effects of xanthohumol, a prenylated chalone present in hops, in human prostate cancer cells. Anticancer Res. 2010, 30, 3333–3339. [Google Scholar]
- Miranda, C.L.; Stevens, J.F.; Helmrich, A.; Henderson, M.C.; Rodriguez, R.J.; Yang, Y.H.; Deinzer, M.L.; Barnes, D.W.; Buhler, D.R. Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines. Food Chem. Toxicol. 1999, 37, 271–285. [Google Scholar] [CrossRef]
- Drenzek, J.G.; Seiler, N.L.; Jaskula-Sztul, R.; Rausch, M.M.; Rose, S.L. Xanthohumol decreases Notch1 expression and cell growth by cell cycle arrest and induction of apoptosis in epithelial ovarian cancer cell lines. Gynecol. Oncol. 2011, 122, 396–401. [Google Scholar] [CrossRef]
- Festa, M.; Capasso, A.; D’Acunto, C.W.; Masullo, M.; Rossi, A.G.; Pizza, C.; Piacente, S. Xanthohumol induces apoptosis in human malignant glioblastoma cells by increasing reactive oxygen species and activating MAPK pathways. J. Nat. Prod. 2011, 74, 2505–2513. [Google Scholar] [CrossRef]
- Pan, L.; Becker, H.; Gerhauser, C. Xanthohumol induces apoptosis in cultured 40-16 human colon cancer cells by activation of the death receptor- and mitochondrial pathway. Mol. Nutr. Food Res. 2005, 49, 837–843. [Google Scholar] [CrossRef]
- Saito, K.; Matsuo, Y.; Imafuji, H.; Okubo, T.; Maeda, Y.; Sato, T.; Shamoto, T.; Tsuboi, K.; Morimoto, M.; Takahashi, H.; et al. Xanthohumol inhibits angiogenesis by suppressing nuclear factor-kappaB activation in pancreatic cancer. Cancer Sci. 2018, 109, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, W.; Liu, H.; Yu, X. Xanthohumol inhibits colorectal cancer cells via downregulation of Hexokinases II-mediated glycolysis. Int. J. Biol. Sci. 2019, 15, 2497–2508. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Song, M.; Wang, P.; Zhao, R.; Chen, H.; Zhang, M.; Shi, Y.; Liu, K.; Liu, F.; Yang, R.; et al. Targeted therapy of the AKT kinase inhibits esophageal squamous cell carcinoma growth in vitro and in vivo. Int. J. Cancer 2019, 145, 1007–1019. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, D.O.; Freitas, J.; Nogueira, P.; Henriques, S.N.; Carmo, A.M.; Castro, M.A.; Guido, L.F. Xanthohumol inhibits cell proliferation and induces apoptosis in human thyroid cells. Food Chem. Toxicol. 2018, 121, 450–457. [Google Scholar] [CrossRef]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjornslett, M.; Meza-Zepeda, L.A.; Eknaes, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—A resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Traganos, F.; Ardelt, B.; Halko, N.; Bruno, S.; Darzynkiewicz, Z. Effects of genistein on the growth and cell cycle progression of normal human lymphocytes and human leukemic MOLT-4 and HL-60 cells. Cancer Res. 1992, 52, 6200–6208. [Google Scholar]
- Hosokawa, N.; Hosokawa, Y.; Sakai, T.; Yoshida, M.; Marui, N.; Nishino, H.; Kawai, K.; Aoike, A. Inhibitory effect of quercetin on the synthesis of a possibly cell-cycle-related 17-kDa protein, in human colon cancer cells. Int. J. Cancer 1990, 45, 1119–1124. [Google Scholar] [CrossRef]
- Zi, X.; Grasso, A.W.; Kung, H.J.; Agarwal, R. A flavonoid antioxidant, silymarin, inhibits activation of erbB1 signaling and induces cyclin-dependent kinase inhibitors, G1 arrest, and anticarcinogenic effects in human prostate carcinoma DU145 cells. Cancer Res. 1998, 58, 1920–1929. [Google Scholar]
- Pagano, M.; Pepperkok, R.; Lukas, J.; Baldin, V.; Ansorge, W.; Bartek, J.; Draetta, G. Regulation of the cell cycle by the cdk2 protein kinase in cultured human fibroblasts. J. Cell Biol. 1993, 121, 101–111. [Google Scholar] [CrossRef]
- Delmas, D.; Passilly-Degrace, P.; Jannin, B.; Malki, M.C.; Latruffe, N. Resveratrol, a chemopreventive agent, disrupts the cell cycle control of human SW480 colorectal tumor cells. Int. J. Mol. Med. 2002, 10, 193–199. [Google Scholar] [CrossRef]
- Kuwajerwala, N.; Cifuentes, E.; Gautam, S.; Menon, M.; Barrack, E.R.; Reddy, G.P. Resveratrol induces prostate cancer cell entry into s phase and inhibits DNA synthesis. Cancer Res. 2002, 62, 2488–2492. [Google Scholar]
- Heiss, E.H.; Schilder, Y.D.; Dirsch, V.M. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J. Biol. Chem. 2007, 282, 26759–26766. [Google Scholar] [CrossRef] [Green Version]
- Young, L.F.; Martin, K.R. Time-dependent resveratrol-mediated mRNA and protein expression associated with cell cycle in WR-21 cells containing mutated human c-Ha-Ras. Mol. Nutr. Food Res. 2006, 50, 70–77. [Google Scholar] [CrossRef]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes (Basel) 2016, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Teng, L.; Legerski, R.J. DNA damage checkpoint recovery and cancer development. Exp. Cell Res. 2015, 334, 350–358. [Google Scholar] [CrossRef]
- Liu, X.; An, L.J.; Li, Y.; Wang, Y.; Zhao, L.; Lv, X.; Guo, J.; Song, A.L. Xanthohumol chalcone acts as a powerful inhibitor of carcinogenesis in drug-resistant human colon carcinoma and these effects are mediated via G2/M phase cell cycle arrest, activation of apoptotic pathways, caspase activation and targeting Ras /MEK/ERK pathway. J. BUON Off. J. Balk. Union Oncol. 2019, 24, 2442–2447. [Google Scholar]
- Logan, I.E.; Miranda, C.L.; Lowry, M.B.; Maier, C.S.; Stevens, J.F.; Gombart, A.F. Antiproliferative and Cytotoxic Activity of Xanthohumol and Its Non-Estrogenic Derivatives in Colon and Hepatocellular Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 1203. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, M.; Ahlawat, P.; Srinivas, N.R. Irinotecan and its active metabolite, SN-38: Review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed. Chromatogr. 2010, 24, 104–123. [Google Scholar] [CrossRef]
- Ambroz, M.; Lnenickova, K.; Matouskova, P.; Skalova, L.; Bousova, I. Antiproliferative Effects of Hop-derived Prenylflavonoids and Their Influence on the Efficacy of Oxaliplatine, 5-fluorouracil and Irinotecan in Human ColorectalC Cells. Nutrients 2019, 11, 879. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.L.; Stevens, J.F.; Ivanov, V.; McCall, M.; Frei, B.; Deinzer, M.L.; Buhler, D.R. Antioxidant and prooxidant actions of prenylated and nonprenylated chalcones and flavanones in vitro. J. Agric. Food Chem. 2000, 48, 3876–3884. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C. Broad spectrum anti-infective potential of xanthohumol from hop (Humulus lupulus L.) in comparison with activities of other hop constituents and xanthohumol metabolites. Mol. Nutr. Food Res. 2005, 49, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Legette, L.L.; Luna, A.Y.; Reed, R.L.; Miranda, C.L.; Bobe, G.; Proteau, R.R.; Stevens, J.F. Xanthohumol lowers body weight and fasting plasma glucose in obese male Zucker fa/fa rats. Phytochemistry 2013, 91, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.; Massinger, S.; Wuzik, A.; Heilmann, J.; Hellerbrand, C. Xanthohumol suppresses inflammatory response to warm ischemia-reperfusion induced liver injury. Exp. Mol. Pathol. 2013, 94, 10–16. [Google Scholar] [CrossRef]
- Jiang, C.H.; Sun, T.L.; Xiang, D.X.; Wei, S.S.; Li, W.Q. Anticancer Activity and Mechanism of Xanthohumol: A Prenylated Flavonoid from Hops (Humulus lupulus L.). Front. Pharmacol. 2018, 9, 530. [Google Scholar] [CrossRef]
- Pichler, C.; Ferk, F.; Al-Serori, H.; Huber, W.; Jager, W.; Waldherr, M.; Misik, M.; Kundi, M.; Nersesyan, A.; Herbacek, I.; et al. Xanthohumol Prevents DNA Damage by Dietary Carcinogens: Results of a Human Intervention Trial. Cancer Prev. Res. 2017, 10, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Dietz, B.M.; Kang, Y.H.; Liu, G.; Eggler, A.L.; Yao, P.; Chadwick, L.R.; Pauli, G.F.; Farnsworth, N.R.; Mesecar, A.D.; van Breemen, R.B.; et al. Xanthohumol isolated from Humulus lupulus Inhibits menadione-induced DNA damage through induction of quinone reductase. Chem. Res. Toxicol. 2005, 18, 1296–1305. [Google Scholar] [CrossRef]
- Ferk, F.; Huber, W.W.; Filipic, M.; Bichler, J.; Haslinger, E.; Misik, M.; Nersesyan, A.; Grasl-Kraupp, B.; Zegura, B.; Knasmuller, S. Xanthohumol, a prenylated flavonoid contained in beer, prevents the induction of preneoplastic lesions and DNA damage in liver and colon induced by the heterocyclic aromatic amine amino-3-methyl-imidazo[4,5-f]quinoline (IQ). Mutat. Res. 2010, 691, 17–22. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 | 24 h | 48 h | 72 h | |
|---|---|---|---|---|
| Cell lines | SW620 | - | 12 ± 3.57 µM | 7 ± 1.38 µM |
| SW480 | - | 22 ± 6.49 µM $ | 20 ± 3.30 µM ££ | |
| HT29 | 39 ± 6.48 µM *** | 12 ± 2.80 µM | 10 ± 1.75 µM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scagliarini, A.; Mathey, A.; Aires, V.; Delmas, D. Xanthohumol, a Prenylated Flavonoid from Hops, Induces DNA Damages in Colorectal Cancer Cells and Sensitizes SW480 Cells to the SN38 Chemotherapeutic Agent. Cells 2020, 9, 932. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040932
Scagliarini A, Mathey A, Aires V, Delmas D. Xanthohumol, a Prenylated Flavonoid from Hops, Induces DNA Damages in Colorectal Cancer Cells and Sensitizes SW480 Cells to the SN38 Chemotherapeutic Agent. Cells. 2020; 9(4):932. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040932
Chicago/Turabian StyleScagliarini, Alessandra, Aline Mathey, Virginie Aires, and Dominique Delmas. 2020. "Xanthohumol, a Prenylated Flavonoid from Hops, Induces DNA Damages in Colorectal Cancer Cells and Sensitizes SW480 Cells to the SN38 Chemotherapeutic Agent" Cells 9, no. 4: 932. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040932