The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem
by
 , ,
, ,
Isabelle Corre
1,2,*, 
Franck Verrecchia
1,
Vincent Crenn
1,3,
Francoise Redini
1 and
Valérie Trichet
1,2,* 1
INSERM, Nantes University, UMR1238 Phy-Os “Bone Sarcomas and Remodeling of Calcified Tissues”, F-44035 Nantes, France
2
CNRS GDR3697 MicroNit, F-37044 Tours, France
3
Department of Orthopedic, Nantes Hospital, CHU Hotel-Dieu, F-44035 Nantes, France
*
Authors to whom correspondence should be addressed.
Cells 2020, 9(4), 976; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040976
Submission received: 20 March 2020
/
Revised: 6 April 2020
/
Accepted: 11 April 2020
/
Published: 15 April 2020
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Cancers: Bone Sarcomas)
Abstract
:Osteosarcomas are the most frequent primary bone sarcomas, affecting mainly children, adolescents, and young adults, and with a second peak of incidence in elderly individuals. The current therapeutic management, a combined regimen of poly-chemotherapy and surgery, still remains largely insufficient, as patient survival has not improved in recent decades. Osteosarcomas are very heterogeneous tumors, both at the intra- and inter-tumor level, with no identified driver mutation. Consequently, efforts to improve treatments using targeted therapies have faced this lack of specific osteosarcoma targets. Nevertheless, these tumors are inextricably linked to their local microenvironment, composed of bone, stromal, vascular and immune cells and the osteosarcoma microenvironment is now considered to be essential and supportive for growth and dissemination. This review describes the different actors of the osteosarcoma microenvironment and gives an overview of the past, current, and future strategies of therapy targeting this complex ecosystem, with a focus on the role of extracellular vesicles and on the emergence of multi-kinase inhibitors.
1. Introduction
Osteosarcomas (OSs) are the most common primary malignant bone sarcomas, with a bimodal age distribution. The highest incidence is in children and adolescents (median of age of 18), with a second smaller peak of incidence in elderly individuals over 60 years. Worldwide, the incidence of OS is around one to three cases annually per million individuals [1]. These tumors develop mainly in the long bones (femur, tibia, humerus), close to the growth plate in the bone metaphysis, and less frequently in the skull, jaw, and pelvis. OSs are characterized by the presence of transformed osteoblastic cells producing osteoid matrix. Nevertheless, the precise identity of the cell at the origin of the tumor remains unknown. Evidence supports the idea of an origin of OS in mesenchymal stem/stromal cells (MSCs) and/or in more committed osteoblastic precursors [2,3]. Since the introduction of chemotherapies to treat OS the late 70s, patients diagnosed with OS receive a neo-adjuvant treatment followed by a post-surgery adjuvant therapy with a cocktail of chemotherapies, i.e., high-dose methotrexate (12 g/m2), etoposide, and ifosfamide for children and young adults (<25 years) in the French OS2006/sarcome-09 study [4], or other protocols combining doxorubicin, cisplatin, and ifosfamide with or without high-dose methotrexate [5,6,7]. With these therapeutic regimens, the 5 year survival has reached 78% for children and young adults with localized disease, but still remains at only 20% in patients with metastasis at diagnosis or in relapse [1,4]. Moreover, in the last 40 years, survival has not notably improved for patients without metastases and has not improved at all for metastatic patients [8]. Therefore, improving therapy for OS remains a constant and major goal for many worldwide research and clinical groups.
A major characteristic of OSs tumors is their heterogeneity, both at the intra-tumoral level and also between individuals. Therefore, the common genomic initiating biological processes driving osteosarcomagenesis are still not identified. The complexity of the somatic genome of OS is a major cause of intra-tumoral heterogeneity, characterized by chromosomal aneuploidy, alteration of genes by mutation and/or variation of copy number, genomic instability featured by massive rearrangement through chromotripsis, and the presence of patterns of localized hypermutated regions, named kataegis [9]. A small set of genes has been found to be recurrently mutated in OS (TP53, RB, MDM2, ATRX, and DLG2) [10]. Recently, a subset of OSs was described with genomic alterations in genes of the DNA repair pathways, reminiscent of BRCA1/2-deficient tumors [11]. Several inherited syndromes such as Li–Fraumeni, Rothmund–Thomson, Werner, Bloom, and retinoblastoma familial cancers have also been associated with a predisposition to developing OS [9]. Nevertheless, in the vast majority of cases (95%), OSs appear as sporadic events. Overall, poorly defined oncogenic events associated with high cellular heterogeneity of tumor cells make the development of molecular targeted therapies devoted exclusively to tumor cells difficult.
Bone sarcomas, and in particular OSs, grow in the bone microenvironment, a very specialized, complex, and highly dynamic environment composed of bone cells (osteoclasts, osteoblasts, osteocytes), stromal cells (MSCs, fibroblasts), vascular cells (endothelial cells and pericytes), immune cells (macrophages, lymphocytes), and a mineralized extracellular matrix (ECM). In physiological conditions, a coordinated and fine-tuned orchestrated activity of bone, vascular, and stromal cells ensures bone homeostasis through intense paracrine and cellular communications. According to Paget’s theory [12], tumor cells find in this microenvironment a fertile soil to seed and manage to highjack bone physiological pathways to their advantage in order to survive and grow. Cross-talk between OS and the bone microenvironment involves numerous environmental signals, induced by multiple cytokines, chemokines, and soluble growth factors [13], but also conveyed by extracellular vesicles (EVs), considered today to be effective vectors of communication between cells [14].
In OS, the difficulty of designing and validating new therapies rests on two levels of complexity: first, a high heterogeneity in tumor cells with no evident targetable event, and second, an active and reacting microenvironment composed of active cells, interconnected and intensively communicating through paracrine secretion of soluble factors and EVs. In this review, we describe the different actors of the OS microenvironment in the context of their complex interaction with tumors cells. We also discuss the past, current, and future therapeutic strategies, regarding the complex ecosystem of OS, with a focus on the emergence of multi-kinase inhibitors (MKI) that target tumor cells and the cells of their microenvironment, and on the role of EVs as essential conveyors of information in bone sarcoma biology.
2. OS-Induced Bone Remodeling
2.1. Osteoclasts and Osteolysis
OS development is associated with para-tumor osteolysis, causing frequent painful bone fragility at the time of the detection of OS in patients. OS aggressiveness has been associated with osteolysis markers in a few clinical cases [15]. Notably, the binding of the soluble molecule Receptor Activator of Nuclear Factor kappa B Ligand (RANKL), alias TNFSF11, to its receptor (RANK), mainly regulates osteolysis through paracrine regulation. RANKL is produced by osteoblasts and osteocytes in the bone environment [16], while RANK is expressed on the cell surface of osteoclast precursors [17]. In OSs, osteoclast activity leads to a vicious cycle between OS cell proliferation and bone degradation, leading to the release of pro-tumor factors such as insulin-like growth factor 1 (IGF1) or transforming growth factor-β (TGF-β) from the bone matrix [13,18]. However, clinical trials using monoclonal antibody therapy to block the IGF receptor tyrosine kinase in patients with OS showed limited and unpredictable response rates, leading to the cessation of this therapy [19].
The link of osteolysis in the vicious cycle observed in OS has been demonstrated in preclinical studies, using either chemical inhibitors (mainly zoledronic acid, ZOL) [20,21] or RANKL receptor competitors (including osteoprotegerin (OPG) [22], RANK-Fc [23]), or RANKL silencing [24]. Thus, osteolysis inhibition became an attractive therapeutic target in combination with chemotherapeutics to treat OS. However, initiated on the basis of promising preclinical studies, OS2006, a Phase III clinical trial combining ZOL with chemotherapy and surgery gave very disappointing results, with no improvement but slightly worse therapeutic results [25]. Despite the fact that ZOL has also been described in vitro to have a direct effect on OS cells, its efficacy against OS primary growth and pulmonary metastasis remains controversial [26].
Direct implication of osteoclast activity in OS development and progression in patients is still difficult to decipher. Indeed, a loss of osteoclasts was associated with increased metastasis in a preclinical model of OS [27], while co-injection of pre-osteoclasts with human OS cells had no effect on OS local growth and lung metastases in nude mice [28]. Denosumab, an antibody directed against RANKL, efficiently inhibits osteoclast activity and is currently used to treat bone loss in bone metastasis, multiple myeloma, or giant cell tumors. However, no clinical results have been reported to date for denosumab in OS patients, except in combination with the MKI sorafenib for one patient [29,30]. Even following a more specific targeting of RANKL, denosumab does not have differentiated action towards different cell types. Indeed, the RANKL/RANK pathway is involved not only in osteoclasts, but also in many other cells of the tumor environment, including osteoblasts, stromal cells, immune cells (T and B lymphocytes, dendritic cells), and endothelial cells.
Local coupling between bone resorption and formation is essential to preserve bone density and should occur in basic multicellular units, including osteoclasts and osteoblasts, which are covered by bone lining cells forming a canopy, as originally described by Lassen et al. [31]. Under the canopy, RANKL secreted by osteoblasts induces osteoclast differentiation, as described in a well-demonstrated paradigm. Interestingly, a new paradigm model of intercellular communication of osteoclasts towards osteoblasts may be relevant (Figure 1), as it was recently reported that mature osteoclasts were able to produce EVs bearing RANK, allowing interaction with RANKL on osteoblasts [32]. RANK-bearing EVs were initially identified in mouse primary osteoclasts and precursors derived from bone marrow [33]. Recently, Ikebuchi et al. effectively demonstrated that RANK-bearing EVs issued from mouse mature osteoclasts were able to interact with RANKL-expressing osteoblasts, and therefore to induce osteoblastic differentiation coupled with bone formation involving RUNX2 signaling [32]. RANKL-reverse signaling in osteoblasts was demonstrated using RANK-masking on EVs and by creating a mutant mouse model RanklP29A, where RANKL intracellular signaling domain was suppressed. Consequently, RANK–RANKL interaction appears to be bi-directional, dual, and complementary in the coupling of bone resorption and formation: RANK transduction on osteoclasts and precursors activates osteolysis, while RANKL transduction on osteoblasts and precursors activates osteogenesis.
In the context of OS, bone remodeling is linked to a vicious cycle between osteoclasts and tumor cells [22], which is established through the release of growth factors from the degraded bone matrix. Nevertheless, this vicious cycle may be additionally enhanced by EVs secreted by osteoclasts and OS cells [34]. Indeed, EVs secreted by OS cells were able to enhance osteolysis, while RANK-EVs secreted by osteoclasts may activate RANKL expressed on OS cells [35], suggesting a possible RANK–RANKL reverse signaling in OS, as previously described in normal bone physiology [32]. In one retrospective clinical study involving 40 patients, RANKL expression was observed in 75% of OS biopsy samples and its high expression level was correlated to a poor patient outcome [36]. Branstetter et al. [37] detected RANKL in 68% of human OSs, but only 37% OS samples showed more than 10% of tumor cells expressing RANKL. The same year, it was reported that the proliferation of RANKL-expressing OS cell lines was increased through transduction signaling involving AKT and ERK activation when cells were exposed to OPG [38]. One could hypothesize that RANKL expressed on the surface of OS cells could have been activated by OPG, as this protein is the decoy and soluble form of RANK that binds RANKL (Figure 2). Nevertheless, this pro-proliferative effect of OPG was believed to be independent of RANKL because soluble RANK did not induce similar effects. Thus, it was proposed that OPG’s pro-proliferative effect was mediated by an unknown receptor. In regard to the innovative identification of the RANKL reverse signaling as described above (Figure 1 and Figure 2) [32], RANKL activation in OS cells should be revisited, as RANK-EVs released by osteoclasts may have an unexpected role in OS through a possible RANK–RANKL reverse signaling in OS cells.
2.2. Osteoblasts and Bone Formation
Primary bone tumors have potent local influences on bone and the clinical consequences of these influences can be devastating. OS is characterized by the formation of osteoid matrix surrounding anaplastic tumor cells [39,40], and it can stimulate the formation of various bone structures, such as Codman’s triangles or bone spines, designed as the sunburst periosteal reaction. The sunburst pattern of bone is due to new layers of collagen fibers stretching out perpendicularly to the bone. This process is mainly due to a deregulation of bone remodeling and in part to the activity of non-tumor osteoblasts, as observed in mouse OS models.
Osteoblastic progenitors are MSCs mainly present in the bone marrow, and more specifically multipotent skeletal stem cells (MSSCs), which are a subset of MSCs that were recently identified [41]. Under the control of different specific transcription factors, MSCs are able to differentiate into osteoblasts, chondroblasts, myoblasts, and adipocytes, while MSCCs differentiate into osteoblasts and chondroblasts, but not into myoblasts and adipocytes. However, there is not yet evidence indicating that either MSCs or MSSCs are the most important cells in the pathogenesis of OS. Current knowledge on osteoblastogenesis is based on MSC rather than MSSC differentiation. Briefly, RUNX2 and Osterix or SOX9 transcription factor expression leads to MSC differentiation, respectively towards the osteoblastic and chondroblastic lineages [42]. The differentiation of MSCs into mature osteoblasts involves a complex series of proliferation and differentiation steps (Figure 3). Briefly, RUNX2 (also known as CBFA1) is a transcriptional factor that binds a consensus site, called OSE2, present along the proximal promoters of many genes including those of the α1 chain of type I collagen (COL1A1), bone sialoprotein (BSP), osteocalcin (OCN), and osteopontin (OPN) [43,44]. RUNX2 is crucial for the early steps of MSC differentiation into pre-osteoblasts and to maintain osteoblastic function, while Osterix (also known as SP7) is involved in osteoblastic differentiation mainly downstream of RUNX2 by allowing the differentiation of pre-osteoblasts into functional mature osteoblasts [45]. Upstream of those transcriptional factors, a signal transduction cascade has to be activated by cytokines or growth factors such as TGF-βs, fibroblast growth factors (FGFs), or wingless-type MMTV integration site family members (WNTs). Most of these cytokines or growth factors are implicated in OS development.
The TGF-β family comprises at least 30 members in humans [46]. The role of TGF-βs during bone remodeling is complex. Regarding the mesenchymal osteoblastic lineage, TGF-β1 favors bone formation by stimulating the proliferation and migration of MSCs during the early stages of osteoblastogenesis [47,48]. In contrast, during the late stages of osteoblastogenesis, TGF-β1 inhibits the differentiation of MSCs into osteoblasts and the mineralization of mature osteoblasts in culture [49]. Interestingly, TGF-β1 is mainly implicated in OS development during either primary tumor growth or metastatic progression [50]. Blocking TGF-β activity in OS cells by SMAD7 overexpression has decreased primary tumor growth by affecting the relationships between tumor cells and non-tumor cells [51].
FGFs are also key regulators of skeletal development [52]. For example, FGF2 is important for the proliferation and maturation of pre-osteoblasts, while FGF18 is essential for mature formation of osteoblasts. Therefore, FGF receptors are receptor tyrosine kinases that may represent a therapeutic target in OS patients [53]. Indeed, Weekes et al. reported an important decrease of lung metastases upon using the inhibitor AZD4547 to block FGF receptor signaling following OS induction in mice [54].
WNTs are a family of 19 secreted glycoproteins. The binding of a WNT ligand (i.e., WNT1, WNT3a) to a frizzled (FZD) receptor, and its co-receptor LRP5/6 activates the canonical WNT pathway [55]. Activation of the WNT signaling cascade leads to the promotion of bone formation and suppression of bone resorption, leading to a balance in bone remodeling [56]. Interestingly, a monoclonal antibody against the WNT signaling inhibitor dickkopf-1 inhibited OS metastasis in a preclinical model of OS [57].
Evidence is thus emerging for a role of osteoblasts in tumor growth in bone. Osteoblasts directly regulate bone matrix synthesis by their own secretome and indirectly regulate bone resorption through the release of RANKL, which binds RANK on osteoclast precursors as previously presented (Figure 1). Additionally, RANK is expressed on MSCs and is downregulated during osteoblastogenesis. Intriguingly, Branstetter et al. did not detect RANK expression on tumor cells into OS samples [37]. Nevertheless, one might address the importance of RANK signaling in OS cells, which derived from cells committed in differentiation pathway between MSCs or pre-osteoblasts towards mature osteoblasts [58]. In this context, Navet et al. investigated the role of RANK overexpression in OS cell lines and during OS development in immune-deficient mice [59]. Activation of the RANKL–RANK pathway in these OS cell lines did not change cell proliferation or migration, nor tumor growth in vivo. Such results suggest that RANK activation in OS cells is not involved in tumor growth. However, RANK-overexpressing OS cells induced a significant increase of lung metastases that was prevented with an antibody directed against RANKL. In another study [23], whole body deletion of RANKL proteins prevented OS development and lung metastases in genetically predisposed mice while, in contrast, Rank deletion in osteoblasts did not change OS burden, nor lung metastasis. RANKL–RANK pathway activation does not seem to be directly implicated in OS development, but can be indirectly involved in OS progression. Implication of a potential RANKL reverse signaling in OS cells has not been tested in these studies, but it would be interesting now to take into account the implication of RANKL transduction on osteoblasts [32] (Figure 1). Antibodies against RANKL and the whole-body deletion of RANKL could disrupt the coupling between bone resorption and formation and modify the progression of OS by inhibiting the transduction of RANKL on osteoblasts and on OS cells expressing RANKL (Figure 2).
3. MSCs in OS Microenvironment
3.1. MSCs as Sensors and Modulators of OS Microenvironment
In OS, interactions between the tumor parenchyma and the non-tumor stroma are required during tumor development and metastatic progression [60]. MSCs are sensors of their microenvironment as they express multiple growth factors and chemokine signaling receptors. They also modulate their microenvironment as they secrete components of ECM and a large variety of mitogenic growth factors, cytokines, chemokines, and metalloproteinases (MMPs) [61]. Consequently, MSCs have autocrine and paracrine trophic properties, as their secreted growth factors stimulate cell division and differentiation of MSCs, osteoblasts, and endothelial cells [62]. Furthermore, MSCs also secrete chemokines (C-C motif) ligand 5 (CCL5), stromal derived factor 1 SDF-1 or (C-X-C motif) chemokine 12 CXCL12, interleukin 6 (IL-6), and growth factor vascular endothelial growth factor (VEGF), known, among others, to promote OS growth, metastasis spread, and angiogenesis (reviewed in Reference [63]).
In a previous study, we co-injected OS-associated stromal cells, also named OS-derived cells (OSDCs), with N-methyl-N′-nitro-N-nitrosoguanidine (MNNG)-HOS OS cells, derived from a human OS, in nude mice [64]. We found several new observations at the histological level. First, the primary OS induced by MNNG-HOS cell injection near the tibia of nude mice appeared as an undifferentiated pleiomorphic sarcoma inducing bone spines (Figure 4a), while some osteoid matrix could be observed in intra-vascular tumor emboli (Figure 4b). Subsequently, following the co-injection of MNNG-HOS cells with OSDCs in nude mice, it was surprising to observe an abundant osteoid matrix (Figure 4c), usually not observed in the MNNG-HOS mouse model, but detected as a hallmark of OS tumor in patients. The second change involved a huge infiltration of peripheral blood mononuclear cells in vessel walls surrounding OS metastases in the lungs (Figure 4d). However, we do not know how MSCs influence OS cells and how MSC-educated OS cells may in turn influence their surrounding cells, leading to more osteoid matrix and higher immune infiltration. In this context, Pietrovito et al. described how MSCs in contact with OS cells gained a cancer-associated fibroblast phenotype and, in turn, how OS-activated MSCs promoted OS cell motility, invasiveness, and transendothelial migration [60]. OS-activated MSCs increased the secretion of monocyte chemoattractant protein (MCP)-1 (alias CCL2), growth-regulated oncogene (GRO)-α (also known as CXCL1), and IL-6 and -8.
MSCs and MSC-derived osteoblasts secrete components of ECM and MMPs. Tumor infiltration of immune cells is tightly dependent on ECM plasticity and proteolysis [66]. As demonstrated by Nicolas-Boluda et al. using imaging technology, T cells may cross the blood vessels but remain trapped in the stroma surrounding tumor nodules [67]. Consequently, T cells do not secrete MMPs allowing matrix lysis, and they then cannot progress alone between dense and tight fibers. Dynamic immune cell infiltration has not yet been imaged in OS and was not yet related to fiber density and MMPS in surrounding ECM; however, induced proteolysis could be a key to achieving success of any immune-cell therapy in OS.
3.2. MSCs as Donors and Acceptors of Extracellular Vesicle Cargo in OS Microenvironment
The secretome of MSCs contains bioactive EVs, which may explain some well-known but unraveled therapeutic roles of MSCs, including their role in bone regeneration [62]. MSC-secreted EVs were shown to bear tumor supportive microRNAs and proteins, but also metabolites such as lactate and glutamate [68]. A previous study showed metabolic cooperation between OSDCs and OS cells through lactate efflux by MSCs and its uptake by OS cells [69]. This metabolic cooperation can be amplified by lactate released in EVs secreted by OSDCs. Moreover, MSC-secreted EVs were shown to increase OS cell survival and migration, especially under stress induced by serum starvation [70] (Figure 5).
Gebraad et al. isolated EVs from lipopolysaccharide-activated monocytes and RANKL-activated osteoclasts, both EVs bearing RANK, and observed EV effects on gene expression and protein secretion of MSCs [71]. EVs secreted by activated monocytes, but not those secreted by osteoclasts, promoted the expression of MMP genes such as MMP3 and MMP1, and secretion of the chemokine CXCL5. The same group showed that OS-secreted EVs on MSCs promoted expression of MMP1, VEGF-A, and intercellular adhesion molecule (ICAM1) genes, which in turn may stimulate not only bone remodeling, but also tumor angiogenesis and metastasis [72].
EVs secreted by OS induced hypomethylation on long interspersed elements (LINE-1) of MSCs [72]. Hypomethylation on LINE-1 retro-transposons correlates with chromosomal instability and may drive oncogenetic effects [73]. Genomic instability in MSCs links back to the fact that MSCs have been proposed as cells of origin for high-grade OS [74], which is characterized by complex genetic rearrangements [64]. Large genomic studies have demonstrated that high-grade OS presents one of the highest levels of chromosomal instability associated with hyper-mutations. However, the molecular mechanisms initiating and underlying such huge chromosomal rearrangements are still unknown in the pathogenesis of OS [75,76].
Endo-Munoz et al. observed that some metastatic OS cell lines, including the KHOS cell line (CRL-1544 from American Type Culture Collection), produce high level of the protease urokinase plasminogen activator (uPA) and of its receptor (uPAR), leading to an autocrine loop of activation [77]. They showed that uPAR activation drives ERK phosphorylation and stimulates in vitro migration and in vivo propagation of OS cells. Moreover they demonstrated that KHOS cells produce EVs bearing uPA, which may be biologically active. Additionally, they identified a paracrine uPA axis involving stromal cells in mouse bone marrow explants cultured for 6 days in the presence of macrophage colony-stimulating factor (M-CSF or CSF-1). These mouse stromal cells were not characterized by surface marker analysis, but likely contained a large fraction of monocytes and/or hematopoietic stem cells. The implication of MSCs in this uPA paracine activation of OS cells was not shown, whereas uPA is a well-known activator of MSCs mobilization and transendothelial migration during tissue repair [78]. Above all, they identified uPA inhibition as a promising therapeutic strategy to prevent metastatic spread in OS patients. In a mouse model of OS, OS-secreted EVs bearing TGF-β induced a pro-tumorigenic and pro-metastatic phenotype of MSCs through increased IL-6 secretion and activation of STAT3 signaling pathways in OS cells [79]. This study showed how MSCs that have been conditioned or educated by OS-EVs may favor a tumor’s progression in its microenvironment.
MSC-secreted EVs may be also involved in revascularization following treatment-induced hypoxia. Indeed, EVs released by hypoxia-treated MSCs have increased angiogenesis through activation of the protein kinase A in human umbilical vein endothelial cells (HUVECs) [80]. EVs from MSCs have been described as able to act on endothelial cells to deliver angiogenic signals [81], especially in hypoxic and ischemic conditions [82,83]. In the bone context, pro-angiogenic effects of MSC-derived EVs have been shown in an osteonecrosis model [84,85]. Angiogenic properties of MSC-EVs have not been explored yet in OS. EVs are able to transfer growth factors, cytokines, and nutrients, as presented in this review, but can also transfer also nucleic acids. DNA can be expelled from cells by EV release during drug-induced senescence in order to maintain cellular homeostasis [86], whereas microRNAs contained in EVs can contribute to cell communication in bone sarcomas [87].
Finally, the well-known immunosuppressive effects of MSCs can be mediated by their EVs, as suggested by recent results of a clinical trial involving patients with chronic kidney disease [88] and in preclinical model of osteoarthritis [89]. MSC-secreted EVs bearing TGF-β-induced gene product-h3 (TGFβI/BIGH3) could ameliorate osteoarthritis, however they can worsen tumor development by suppressing the activity of immune cells.
4. Vascular Microenvironment in OS Biology
4.1. Physiologic Angiogenesis during Bone Development
During both normal development and regenerative processes, blood vessels are major actors in bone biology, providing all necessary nutrients and oxygen to the highly dynamic bone tissue [90]. Beside their role as an efficient transport network for molecules and hematopoietic cells, blood vessels also support bone formation and homeostasis [91]. In long bones, the vasculature has a unique complex architecture, composed of a set of arteries penetrating the cortical zone and forming a heterogeneous network of capillaries, drained by a central vein [92]. New insights into the anatomical architecture and physiology of the vascular system in long bones have emerged with the description of trans-cortical microvessels originating in the bone marrow and forming connections between the endosteal and periosteal circulation [93]. Recently, a new bone-specific capillary subtype was identified in mouse juvenile tibia [94]. It is characterized by type-H endothelial cells that are rich in CD31 high Endomucinhigh and are specifically localized close to the growth plate in the metaphysis and in the endosteal regions. During bone growth and modeling, these type-H cells have high proliferative capacities and are responsible for promoting the bone angiogenesis required for long bone growth. Furthermore, these type-H capillaries provide a favorable niche for Osterix+ osteoprogenitors, as they couple angiogenesis to osteogenesis through hypoxia-induced secreted Notch-ligand Noggin [94,95]. These populations of endothelial cells, the amount of which decreased with age in mice, have also been described in humans and have been proposed as an indicator of bone loss [96]. The roles of these bone-specific endothelial populations still needs to be elucidated in the context of bone tumor angiogenesis.
4.2. Neo-Vascularization in OS
Vascularization is an essential factor for tumor growth and dissemination, providing oxygen and nutrients, and supporting intravasation and extravasation of cancer cells. Elaboration of a tumor-dedicated vascular environment is one of the main hallmarks of cancer [97], and the formation of a tumor vascular network relies mainly on tumor piracy of physiological sprouting neo-angiogenesis mechanisms from existing vessels. Nevertheless, additional processes like vasculogenesis and vascular mimicry may also contribute to the expansion of a tumor vascular network [98].
Tumor angiogenesis is initiated by environmental stresses (hypoxia, acidosis) leading to disequilibrium of the pro-/anti-angiogenic balance, and consequently to the elevated expression of pro-angiogenic factors such as hypoxia-induced factor (HIF) and VEGF. OSs are highly vascularized bone tumors, lying in a hypoxic and acidic bone microenvironment. How neovascularization occurs in OS remains poorly understood. Nevertheless, OSs appear preferentially in the region of bone growth close to the metaphysis, where type-H endothelial cells promoting angiogenesis are located [94,95], suggesting a possible implication of these cells in OS neo-angiogenesis. In tumors, it is commonly admitted that neo-angiogenesis sprouting from pre-existing vessels is the most relevant process of angiogenesis, but the implication of endothelial progenitors cells (EPCs), able to differentiate into mature endothelial cells, has to be mentioned in tumor vascular growth, even if there is less of a consensus on these cells’ involvement [99]. EPCs may be recruited from their initial location in the bone marrow to the tumor site, or EPCs may reside in the tumor microenvironment, where tumors and stromal cells can provide plasticity and differentiation paracrine signals to EPCs. In Ewing sarcoma bone tumor, bone marrow EPC-induced vasculogenesis has been identified as an essential step of neovascularization and is crucial for tumor growth, with 10% of neo-vessels containing bone-marrow-derived cells [100]. In OS, a role for EPCs in neovascularization was also recently highlighted in an in vitro rat model, where co-injection of encapsulated OS cells with EPCs enhanced tumor vascularization [101]. In that case, OS rat tumor cells promoted migration and angiogenic properties of EPCs, through their specific secretome of angiogenesis-related factors (VEGF, TGF-β1, MCP-1, Activin A, and OPN).
Initially described in uveal melanoma, vascular mimicry, an alternative process to angiogenesis and vasculogenesis, is characterized by the de novo formation of perfusable, matrix-rich, vasculogenic-like micro-channels generated by transformation of aggressive tumor cells [102,103]. OS tumors appear to develop such an endothelial-free tumor-derived vasculogenic network, as vascular mimicry is found in 22.7% of osteoblastic-type OS samples and associated with unfavorable prognostis [104,105]. Human OS cell lines (U2OS [106], MG-63 [105]) and aggressive canine cell lines [107] are able to form vessel-like structures in three-dimensional cultures. Vascular mimicry mechanisms remain largely unknown, often being assessed via inappropriate and biased in vitro assays [108]. Nevertheless, a subpopulation of OS tumor cells co-expressing high levels of both VEGF and its receptor VEGFR-1 has been described as an important factor of aggressiveness [109]. This autocrine VEGF/VEGFR-1 signaling, associated with increased tumor growth and tumor vascularity, may possibly confer to OS cells the capacity to develop vasculogenic properties, leading to vascular mimicry. In fact, silencing VEGF suppresses vasculogenic mimicry in OS in vitro [110].
For many years, pro-angiogenic factors like VEGFs and angiopoietins have been considered paracrine soluble factors secreted by tumor cells and measurable in patient serum. However, EVs now appear to be essential players of intercellular communication, especially in tumors and in particular in the dialogue promoting angiogenesis. Indeed, stimulation of angiogenesis by tumor-derived EV cargo has been highlighted in numerous tumors [111]. In the context of OS, two recent studies established the pro-angiogenic role of OS-EVs through their cargo containing angiocrines and angiogenesis-related miRNAs [112,113].
4.3. Vascular and Angiogenic Factors in OS Patients
Several analyses of cohorts of OS patients have revealed the importance of neo-vascularization markers in patient samples. Amplification of genes in the VEGF pathway, in particular VEGF-A, has been described in OS patients, and was confirmed at the protein level [114]. Expression of high VEGF is positively associated with tumor stages and with metastasis [115,116]. Accordingly, a significant increase in vascularity density appears to be a hallmark of primary OS tumor in metastatic vs. non-metastatic patients [117]. Indeed, several clinical studies correlated high expression of VEGF in biopsies with worse disease-free survival and lower overall survival either in untreated [115] or in pre-operative treated patients [118]. Along these lines, a systematic review issued from a meta-analysis including 559 patients from 12 retrospective studies suggested that VEGF expression could be considered an effective biomarker of prognosis on OS patients [119]. On the other hand, conclusions drawn from another meta-analysis [120] underlined the importance of considering heterogeneity and geographic origin of patients. Beside VEGF, the expression of its receptor VEGFR-2 is increased in OS as compared to normal bone tissues, and high VEGFR-2 expression is associated with poor prognosis [121]. Investigation of angiogenic circulating factors also revealed that serum concentration of VEGF in bone sarcoma (Ewing sarcoma, OS, chondrosarcoma) was higher than in healthy samples or benign tumors [122]. The prognostic value of circulating VEGF was addressed in one study [123], as patients with metastasis at diagnosis or within a year of diagnosis had significantly higher serum levels of VEGF than non-metastatic patients without early metastatic disease. Level of circulating VEGF was independent of the tumor size and serum concentrations of two angiogenic factors, FGF2 and placental growth factor (PGF) were not different in metastatic vs. non-metastatic stage patients. Therefore, considering tissue or circulating angiogenic factors as objective prognostic factors still needs to be evaluated in larger prospective studies.
4.4. Targeting the Vascular Microenvironment with Anti-Angiogenic Agents
Targeting neo-vascularization has been considered in OS, with several Phase I/II clinical trials conducted during the past decade, mainly in advanced-stage OS [124]. The most promising clinical trials have been those including anti-angiogenic agents targeting VEGFRs, such as sorafenib alone [125] or associated with everolimus, an inhibitor of mTOR2 [126]. These trials published by the Italian Sarcoma Group in recurrent OS showed clinical benefits with an increase in 4–6 months progression-free survival for 43–46% of patients. On the other side, associating the well-described anti-VEGF antibody bevacizumab with pre- and post-operative chemotherapies in OS patients with resectable and localized OS did not improve the percentage of good responders to neo-adjuvant therapy, nor did it improve the outcomes of patients. Nonetheless, the treatment could induce post-surgical wound healing troubles [127]. In one case of spinal OS, association of sorafenib with the antibody denosumab, targeting RANKL present in the osteoid matrix, led to an efficient metabolic tumor regression [29]. A recent anti-angiogenic therapeutic strategy explored in preclinical studies is the targeting of VEGFR-2, a VEGF receptor expressed mainly on angiogenic vessels but also on OS tumor cells [121]. Results showed that the highly specific VEGFR-2 inhibitor, apatinib, was associated with direct anti-tumoral activity through VEGFR2/STAT3/BCL2 signaling [121,128]. Targeting VEGFR-2 with the monoclonal antibody ramucirumab was also shown to exert anti-angiogenic activity in vitro [129]. Surprisingly, injection of anti-mouse Vegfr-2 antibody in preclinical OS pediatric cancer models did not affect tumor growth, even when combined with cytotoxic chemotherapy using doxorubucin, suggesting that targeting both tumor and vascular microenvironment is mandatory in OS, even if it is difficult to achieve efficiently.
5. Cells of the Immune System in OS Microenvironment
The immune context of the OS microenvironment is mainly composed of tumor-associated macrophages (TAMs), with a significant number of dendritic, lymphoid, and myeloid cells [130]. Indeed, both myeloid and lymphoid cells have been detected in OS [130,131]. In contrast to the majority of epithelial tumors [132,133], studies have shown that TAMs were present in the immune infiltrate in a high proportion of biopsies and that an increased infiltration was associated with reduced metastasis and improved survival in high-grade OS [117,134,135]. The mechanism by which TAMs inhibit metastasis in OS remains unclear, but the observation by different teams that OSs were infiltrated with a heterogeneous population of pro-inflammatory M1 and anti-inflammatory M2 TAMs may suggest that in this precise case, the constitutive presence of M2 macrophages may have an anti-metastatic rather than a pro-metastatic effect [134]. Based on these data, OS patients were enrolled in a randomized clinical trial using the macrophage-activating agent mifamurtide in addition to a standard chemotherapy regimen, resulting in a significant improvement in 6 year overall survival [136,137]. Similar results were obtained in earlier studies in human and canine OS [138,139,140]. Mifamurtide has thus become the first new therapeutic drug used for the treatment of OS in the last 20 years [137]. More recently, the SARCOMA13 clinical trial (NCT03643133) proposed the use of the combination of mifamurtide with conventional chemotherapy in the treatment of French OS patients after surgery.
The observation that cytotoxic CD8 T lymphocytes are less abundant than myeloid cells in OS biopsies suggests that OSs are poorly immunogenic tumors, with a lack of tumor neo-antigens and scarce infiltrate of immune cytotoxic lymphocytes. This would define OSs as cold tumors, characteristic of pediatric tumors [141]. In recent studies, the presence of CD8 T lymphocytes was described in half of OS patient samples at diagnosis, and their presence was significantly associated with a lower rate of metastasis [135,142]. In addition, the ratio of intra-tumor CD8 T cells to regulatory immunosuppressive FOXP3 CD4 T cells in initial biopsies allowed discrimination of OS patients with prolonged survival from non-survivors [142]. As for most other tumors, OS infiltration by antigen presenting cells (APCs), including CD1a dendritic cells (DCs) and CD68 macrophages, has been correlated with poorer prognosis. Moreover, tumor programmed death-ligand 1 (PD-L1) expression has been associated with a poorer 5 year event-free survival [143]. Programmed death 1 (PD-1) is expressed at the surface of activated CD8 T lymphocytes, B lymphocytes, and natural killer (NK) cells. The involvement of the PD-1/PD-L1 checkpoint in OS varies from one study to another. In a recent systematic meta-analysis of 14 studies with a total of 868 patients, OS patients had 14–75% higher PD-L1 expression in tumor tissues, which was significantly correlated with metastasis, mortality risk, and poorer overall survival [144,145]. However, PD-L1 staining was detected in few OS specimens in two independent studies [135,146], which suggested that the role of the PD1/PD-L1 checkpoint is not predominant in the pathogenesis of OS. Nevertheless, several clinical trials using targeting PD-1/PD-L1 have been proposed. Among them, only one using anti-PD-L1 avelumab (NCT03006848) is recruiting, while three trials using anti-PD-1, penbrolizumab (NCT02301039 and NCT03013127), or nivolumab (NCT02304458) are either not recruiting or suspended, likely due to risk of immune-related side effects such as skin reactions, pneumonitis, colitis, and hepatitis [147,148].
With the extraordinary clinical success of adoptive therapies based on the use of chimeric antigen receptor (CAR) engineered T cells in B-cell malignancies [149], attempts to develop such therapeutic strategies are emerging in solid tumors. In the context of OS, CAR-T-cell therapy still remains largely unexplored. The main challenge of this immune approach is the identification of an optimal target expressed restrictively on tumor cells, in order to avoid toxicity and to selectively eradicate cancer cells. Up to now, few potential targets have been identified in OS [150]—the Human epidermal growth factor 2 (HER2), the Insulin-like Growth factor receptor-1 (IGFR1), and the tyrosine orphan-like receptor 1 (ROR1). HER-2 CAR-T cells have been shown to drive tumor regression in an animal model [151]. Tested in a phase I/II clinical trial in HER-2 positive sarcoma patients, infusion of HER-2 CAR-T was shown to induce no toxicity, and the 6 week stability of these cells [152] paved the way for further trials. Preclinical studies highlighted that IGFR1 and ROR1 that are overexpressed in OS cells lines are also relevant targets. An adoptive transfer of IGFR1-CAR and ROR1-CAR T cells derived from a sarcoma patient significantly suppressed tumor growth in both localized and disseminated sarcoma xenograft models [153]. In OS, the main issues with CAR-T cells would be accessing their target through a stiff osteoid bone tumor matrix, allowing their survival in a local immunosuppressive microenvironment. Despite promising preclinical results, launching the use of CAR-T cell therapies in OS is still a matter of debate.
6. The Multi-Kinase Inhibitors (MKIs) as Promising Therapies in OS Treatment
Targeted therapies using MKIs proved their efficacy initially in renal cell cancer, hepatocellular carcinoma, and thyroid cancer [154]. These molecules exert an anti-cancer activity by simultaneously targeting several kinases. Initially developed as potent VEGFR signaling inhibitors, it is now admitted that their anti-tumor action depends on both the inhibition of angiogenic but also non-angiogenic pathways.
In the context of OS, several in vitro and preclinical results have highlighted the relevance of MKIs as potential therapeutic tools. Anlotinib, a VEGFR2 and MET kinase inhibitor, suppressed tumor growth and metastasis and increased chemosensitivity of OS cells [155]. Moreover, cabozantinib, an inhibitor of c-MET (also known as HGFR), VEGFR-2, c-KIT, and TIE-2, appears to block proliferation and migration of OS cells via autophagy. Additionally, it acts on the bone microenvironment by decreasing RANKL and increasing OPG production by osteoblasts [156], which results in a strong inhibition of RANK-positive OS cell proliferation [157]. Exploration of MKIs in clinical trials (Table 1) started initially with sorafenib [126], as described above. Subsequently, a therapeutic strategy using regorafenib, an MKI that targets angiogenic (VEGFR1-3, TIE-2), stromal (PDGFR-β, FGFR), and oncogenic kinases (KIT, RET, and RAF) has been tested. Notably, two clinical trials [158,159] were conducted in patients with recurrent, progressive, metastatic OS after failure of conventional chemotherapy and demonstrated a significant extension of progression-free survival. These studies highlighted the potential therapeutic value of MKIs in very aggressive and resistant OS. These very promising results paved the way for further exploration of this type of targeted therapy. In this context, pazopanib, a MKI inhibiting VEGR1-3, PDGFRα/β, and c-KIT, showed objective responses in metastatic or relapsed patients [160,161]. Preclinical and in vitro data reinforced the relevance of MKI in OS. One phase II multi-centric clinical trial has just been published, exploring cabozantinib in advanced relapsed OS and Ewing sarcoma [162]. Encouraging responses were reported for both pathologies. In OS, 12% patients presented a 6 months objective response and 33% patients had no progression in 6 months. Additionally, a phase II clinical study in relapsed refractory OS (NCT04154189) starting in February 2020 involves lenvatinib, a MKI inhibiting VEGFR1-3, FGFR1-4, PDGFRα, KIT, and RET and used in thyroid cancer [163].
7. Discussion
In the past decade, the supportive role of the bone microenvironment in the tumor progression and metastatic dissemination of OS has been intensively documented. This culminates in the characterization of a vicious cycle established between OS cells and osteoclasts, where tumors, by promoting osteoclastic activity, indirectly lead to the degradation of bone matrix and to the release of pro-tumor factors initially trapped in the bone matrix.
Intuitively, targeting the bone resorption in order to limit tumor growth thus appeared to be a promising therapeutic option. Therefore, the bisphosphonate ZOL was the first anti-resorptive agent to be explored as a combined treatment for OS. Unfortunately, its translation into the clinical setting was abandoned due to disappointing results obtained in the clinical trial OS2006, since it showed no benefit for OS patients [25]. Along the same line, blocking osteolysis by using denosumab, a humanized antibody directed against RANKL, is no longer considered a potential combined treatment for OS patients. Indeed, if RANK signaling transduces signal in different cell types, including osteoclasts, stromal cells, endothelial cells, and dendritic cells, it is not directly implicated in OS cell division and survival. Importantly, the description of the reverse RANK-RANKL signaling, involving membrane RANKL activation by RANK-bearing EVs, is a new and unexpected regulatory pathway of bone remodeling that has to be considered. Such bidirectional regulation occurring simultaneously in osteoblasts and osteoclasts within a bone multicellular unit has been previously described for Ephrin membrane ligands and their receptors [164]. Therefore, the reverse RANK-RANKL signaling should be explored in other cell types and pathological conditions including OS. Moreover, new anti-RANKL antibodies should be produced to inhibit osteoclasts without effect on osteoblasts.
EVs have now to be considered as key actors of cellular communication in the OS tumor microenvironment ecosystem. EVs originate from the tumor itself, but also from MSCs and osteoclasts. EVs appear to be essential for mediating signals for bone remodeling, angiogenesis, and also for immune responses. EVs modulate gene expression by transferring their cargo to cells and drive communication between cells through many combinations. As reported in this review, EVs are able to transfer growth factors, cytokines, and nutrients. In addition, they can also transfer nucleic acids. Notably, DNA can be expelled from cells by EV release during drug-induced senescence in order to maintain cellular homeostasis [86], whereas microRNAs contained in EVs can contribute to modulating cell communication in bone sarcomas [87]. Blocking EV-mediated signaling would be an appealing strategy to counteract deleterious and pro-tumor communications in the OS environment. Additionally, EVs are currently being explored as potential vectors of therapies but not yet as targetable entities [165].
Targeting the immune system by the macrophage-activating agent mifamurtide resulted in the most recent improvement of OS therapy since polychemotherapy. However, stimulating the immune system by using antibodies directed against PD-1/PD-L1 in OS is still debated. This debate includes toxic effects of antibodies, poor CD8 T lymphocytes infiltrating OS, no histological evidence of the PD-1/PD-L1 in most OS samples, and no correlation with tumor outcome to date [135].
With the same logic of hitting the microenvironment in order to block tumor progression, targeting the vascular environment, which is particularly developed and supportive in OS, has also been evaluated. If the first clinical trial including the anti-VEGF antibody bevacizumab was disappointing [127], encouraging results emerged from initial clinical trials using the MKI sorafenib, which targets intracellular kinase activity of VEGFRs [125,126]. Since then, several MKIs like regorafenib [158,159], pazopanib [160,161], and more recently cabozantinib, have been reported to have beneficial effects in advanced OS and Ewing sarcoma bone tumors [162]. Accordingly, these agents had promising therapeutic results in selected patients, probably through the combined action on both angiogenic vascular compartment and OS cells in which they inhibit multiple growth factor pathways with potential oncogenic activity (c-MET, c-KIT).
In conclusion, the MKI class of small molecules represents a significant major advance in OS therapy in recent decades, and reinforces the notion that considering OS in its whole ecosystem and thereby targeting several of its components, including EVs, is the way to successful therapies.
Funding
This research was funded by INSERM, CNRS and by the association La Ligue contre le Cancer (Comities 22, 29, 37, 44, 56).
Acknowledgments
The authors are deeply grateful to Prof. Jacques Huot (Laval University, QC, Canada) for his critical reading and comments of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, M.; Silvestri, A.; Haybaeck, J.; Reichardt, P.; Lowery, C.D.; Stancato, L.F.; Zybarth, G.; Regenbrecht, C.R.A. Three-Dimensional Patient-Derived In Vitro Sarcoma Models: Promising Tools for Improving Clinical Tumor Management. Front. Oncol. 2017, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, A.J.; Walkley, C.R. Cells of origin in osteosarcoma: Mesenchymal stem cells or osteoblast committed cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Occean, B.V.; Pacquement, H.; Bompas, E.; Bouvier, C.; Brisse, H.J.; Castex, M.P.; Cheurfa, N.; Corradini, N.; Delaye, J.; et al. Results of methotrexate-etoposide-ifosfamide based regimen (M-EI) in osteosarcoma patients included in the French OS2006/sarcome-09 study. Eur. J. Cancer 2018, 88, 57–66. [Google Scholar] [CrossRef]
- Bacci, G.; Lari, S. Current treatment of high grade osteosarcoma of the extremity: Review. J. Chemother. 2001, 13, 235–243. [Google Scholar] [CrossRef]
- Bishop, M.W.; Janeway, K.A.; Gorlick, R. Future directions in the treatment of osteosarcoma. Curr. Opin. Pediatrics 2016, 28, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.; Kleinerman, E.S.; Betcher, D.; Bernstein, M.L.; Conrad, E.; Ferguson, W.; Gebhardt, M.; Goorin, A.M.; et al. Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2004–2011. [Google Scholar] [CrossRef]
- Allison, D.C.; Carney, S.C.; Ahlmann, E.R.; Hendifar, A.; Chawla, S.; Fedenko, A.; Angeles, C.; Menendez, L.R. A meta-analysis of osteosarcoma outcomes in the modern medical era. Sarcoma 2012, 2012, 704872. [Google Scholar] [CrossRef] [Green Version]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Alfranca, A.; Martinez-Cruzado, L.; Tornin, J.; Abarrategi, A.; Amaral, T.; de Alava, E.; Menendez, P.; Garcia-Castro, J.; Rodriguez, R. Bone microenvironment signals in osteosarcoma development. Cell. Mol. Life Sci. 2015, 72, 3097–3113. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Avnet, S.; Longhi, A.; Salerno, M.; Halleen, J.M.; Perut, F.; Granchi, D.; Ferrari, S.; Bertoni, F.; Giunti, A.; Baldini, N. Increased osteoclast activity is associated with aggressiveness of osteosarcoma. Int. J. Oncol. 2008, 33, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef]
- Lamora, A.; Talbot, J.; Mullard, M.; Brounais-Le Royer, B.; Redini, F.; Verrecchia, F. TGF-beta Signaling in Bone Remodeling and Osteosarcoma Progression. J. Clin. Med. 2016, 5, 96. [Google Scholar] [CrossRef]
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.V.; Ranchere, D.; Kurtz, J.E.; et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035. [Google Scholar] [CrossRef]
- Heymann, D.; Ory, B.; Blanchard, F.; Heymann, M.F.; Coipeau, P.; Charrier, C.; Couillaud, S.; Thiery, J.P.; Gouin, F.; Redini, F. Enhanced tumor regression and tissue repair when zoledronic acid is combined with ifosfamide in rat osteosarcoma. Bone 2005, 37, 74–86. [Google Scholar] [CrossRef]
- Ohba, T.; Cole, H.A.; Cates, J.M.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Bisphosphonates inhibit osteosarcoma-mediated osteolysis via attenuation of tumor expression of MCP-1 and RANKL. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, F.; Richard, P.; Wittrant, Y.; Battaglia, S.; Pilet, P.; Trichet, V.; Blanchard, F.; Gouin, F.; Pitard, B.; Heymann, D.; et al. Therapeutic relevance of osteoprotegerin gene therapy in osteosarcoma: Blockade of the vicious cycle between tumor cell proliferation and bone resorption. Cancer Res. 2007, 67, 7308–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Di Grappa, M.A.; Molyneux, S.D.; McKee, T.D.; Waterhouse, P.; Penninger, J.M.; Khokha, R. RANKL blockade prevents and treats aggressive osteosarcomas. Sci. Transl. Med. 2015, 7, 317ra197. [Google Scholar] [CrossRef]
- Rousseau, J.; Escriou, V.; Lamoureux, F.; Brion, R.; Chesneau, J.; Battaglia, S.; Amiaud, J.; Scherman, D.; Heymann, D.; Redini, F.; et al. Formulated siRNAs targeting Rankl prevent osteolysis and enhance chemotherapeutic response in osteosarcoma models. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 2452–2462. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Le Deley, M.C.; Redini, F.; Pacquement, H.; Marec-Berard, P.; Petit, P.; Brisse, H.; Lervat, C.; Gentet, J.C.; Entz-Werle, N.; et al. Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 1070–1080. [Google Scholar] [CrossRef]
- Ouyang, Z.; Li, H.; Zhai, Z.; Xu, J.; Dass, C.R.; Qin, A.; Dai, K. Zoledronic Acid: Pleiotropic Anti-Tumor Mechanism and Therapeutic Outlook for Osteosarcoma. Curr. Drug Targets 2018, 19, 409–421. [Google Scholar] [CrossRef]
- Endo-Munoz, L.; Cumming, A.; Rickwood, D.; Wilson, D.; Cueva, C.; Ng, C.; Strutton, G.; Cassady, A.I.; Evdokiou, A.; Sommerville, S.; et al. Loss of osteoclasts contributes to development of osteosarcoma pulmonary metastases. Cancer Res. 2010, 70, 7063–7072. [Google Scholar] [CrossRef] [Green Version]
- Avril, P.; Le Nail, L.R.; Brennan, M.A.; Rosset, P.; De Pinieux, G.; Layrolle, P.; Heymann, D.; Perrot, P.; Trichet, V. Mesenchymal stem cells increase proliferation but do not change quiescent state of osteosarcoma cells: Potential implications according to the tumor resection status. J. Bone Oncol. 2016, 5, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Cathomas, R.; Rothermundt, C.; Bode, B.; Fuchs, B.; von Moos, R.; Schwitter, M. RANK ligand blockade with denosumab in combination with sorafenib in chemorefractory osteosarcoma: A possible step forward? Oncology 2015, 88, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Savvidou, O.D.; Bolia, I.K.; Chloros, G.D.; Papanastasiou, J.; Koutsouradis, P.; Papagelopoulos, P.J. Denosumab: Current Use in the Treatment of Primary Bone Tumors. Orthopedics 2017, 40, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Lassen, N.E.; Andersen, T.L.; Ploen, G.G.; Soe, K.; Hauge, E.M.; Harving, S.; Eschen, G.E.T.; Delaisse, J.M. Coupling of Bone Resorption and Formation in Real Time: New Knowledge Gained From Human Haversian BMUs. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 1395–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.; VonMoss, L.; Smith, D.; Rahman, I.; Felemban, M.F.; Zuo, J.; Rody, W.J., Jr.; McHugh, K.P.; Holliday, L.S. Characterization of Regulatory Extracellular Vesicles from Osteoclasts. J. Dent. Res. 2016, 95, 673–679. [Google Scholar] [CrossRef]
- Cappariello, A.; Rucci, N. Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone. Int. J. Mol. Sci. 2019, 20, 4805. [Google Scholar] [CrossRef] [Green Version]
- Garimella, R.; Washington, L.; Isaacson, J.; Vallejo, J.; Spence, M.; Tawfik, O.; Rowe, P.; Brotto, M.; Perez, R. Extracellular Membrane Vesicles Derived from 143B Osteosarcoma Cells Contain Pro-Osteoclastogenic Cargo: A Novel Communication Mechanism in Osteosarcoma Bone Microenvironment. Transl. Oncol. 2014, 7, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.A.; Jung, J.S.; Kim, D.H.; Lim, J.S.; Kim, M.S.; Kong, C.B.; Song, W.S.; Cho, W.H.; Jeon, D.G.; Lee, S.Y.; et al. RANKL expression is related to treatment outcome of patients with localized, high-grade osteosarcoma. Pediatric Blood Cancer 2011, 56, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Branstetter, D.; Rohrbach, K.; Huang, L.Y.; Soriano, R.; Tometsko, M.; Blake, M.; Jacob, A.P.; Dougall, W.C. RANK and RANK ligand expression in primary human osteosarcoma. J. Bone Oncol. 2015, 4, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Marley, K.; Bracha, S.; Seguin, B. Osteoprotegerin activates osteosarcoma cells that co-express RANK and RANKL. Exp. Cell Res. 2015, 338, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Toledo, S.R.; Oliveira, I.D.; Okamoto, O.K.; Zago, M.A.; de Seixas Alves, M.T.; Filho, R.J.; Macedo, C.R.; Petrilli, A.S. Bone deposition, bone resorption, and osteosarcoma. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2010, 28, 1142–1148. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Shimizu, K.; Nakase, T.; Takaoka, K. Periosteal sunburst spiculation in osteosarcoma. A possible role for bone morphogenetic protein. Clin. Orthop. Relat. Res. 1994, 308, 213–219. [Google Scholar]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the Human Skeletal Stem Cell. Cell 2018, 175, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marie, P.J. Transcription factors controlling osteoblastogenesis. Arch. Biochem. Biophys. 2008, 473, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, R.; Wakabayashi, M.; Hata, K.; Matsubara, T.; Honma, S.; Wakisaka, S.; Kiyonari, H.; Shioi, G.; Yamaguchi, A.; Tsumaki, N.; et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J. Biol. Chem. 2012, 287, 33179–33190. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [Green Version]
- Janssens, K.; ten Dijke, P.; Janssens, S.; Van Hul, W. Transforming growth factor-beta1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef] [Green Version]
- Juarez, P.; Guise, T.A. TGF-beta in cancer and bone: Implications for treatment of bone metastases. Bone 2011, 48, 23–29. [Google Scholar] [CrossRef]
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 2004, 23, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Verrecchia, F.; Redini, F. Transforming Growth Factor-beta Signaling Plays a Pivotal Role in the Interplay Between Osteosarcoma Cells and Their Microenvironment. Front. Oncol. 2018, 8, 133. [Google Scholar] [CrossRef]
- Lamora, A.; Talbot, J.; Bougras, G.; Amiaud, J.; Leduc, M.; Chesneau, J.; Taurelle, J.; Stresing, V.; Le Deley, M.C.; Heymann, M.F.; et al. Overexpression of smad7 blocks primary tumor growth and lung metastasis development in osteosarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5097–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factors in skeletal development. Curr. Top. Dev. Biol. 2019, 133, 195–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Y.; Zheng, H.; Du, X.L.; Yang, J.L. Characterization of FGFR signaling pathway as therapeutic targets for sarcoma patients. Cancer Biol. Med. 2016, 13, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weekes, D.; Kashima, T.G.; Zandueta, C.; Perurena, N.; Thomas, D.P.; Sunters, A.; Vuillier, C.; Bozec, A.; El-Emir, E.; Miletich, I.; et al. Regulation of osteosarcoma cell lung metastasis by the c-Fos/AP-1 target FGFR1. Oncogene 2016, 35, 2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danieau, G.; Morice, S.; Redini, F.; Verrecchia, F.; Royer, B.B. New Insights about the Wnt/beta-Catenin Signaling Pathway in Primary Bone Tumors and Their Microenvironment: A Promising Target to Develop Therapeutic Strategies? Int. J. Mol. Sci. 2019, 20, 3751. [Google Scholar] [CrossRef] [Green Version]
- Amjadi-Moheb, F.; Akhavan-Niaki, H. Wnt signaling pathway in osteoporosis: Epigenetic regulation, interaction with other signaling pathways, and therapeutic promises. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Goldstein, S.D.; Trucco, M.; Bautista Guzman, W.; Hayashi, M.; Loeb, D.M. A monoclonal antibody against the Wnt signaling inhibitor dickkopf-1 inhibits osteosarcoma metastasis in a preclinical model. Oncotarget 2016, 7, 21114–21123. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, R.; Roth, M.; Piperdi, S.; Zhang, W.; Dorfman, H.; Rao, P.; Park, A.; Tripathi, S.; Freeman, C.; et al. Genetically transforming human osteoblasts to sarcoma: Development of an osteosarcoma model. Genes Cancer 2017, 8, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Navet, B.; Ando, K.; Vargas-Franco, J.W.; Brion, R.; Amiaud, J.; Mori, K.; Yagita, H.; Mueller, C.G.; Verrecchia, F.; Dumars, C.; et al. The Intrinsic and Extrinsic Implications of RANKL/RANK Signaling in Osteosarcoma: From Tumor Initiation to Lung Metastases. Cancers 2018, 10, 398. [Google Scholar] [CrossRef] [Green Version]
- Pietrovito, L.; Leo, A.; Gori, V.; Lulli, M.; Parri, M.; Becherucci, V.; Piccini, L.; Bambi, F.; Taddei, M.L.; Chiarugi, P. Bone marrow-derived mesenchymal stem cells promote invasiveness and transendothelial migration of osteosarcoma cells via a mesenchymal to amoeboid transition. Mol. Oncol. 2018, 12, 659–676. [Google Scholar] [CrossRef]
- Ponte, A.L.; Marais, E.; Gallay, N.; Langonne, A.; Delorme, B.; Herault, O.; Charbord, P.; Domenech, J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: Comparison of chemokine and growth factor chemotactic activities. Stem Cells 2007, 25, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Humbert, P.; Brennan, M.A.; Davison, N.; Rosset, P.; Trichet, V.; Blanchard, F.; Layrolle, P. Immune Modulation by Transplanted Calcium Phosphate Biomaterials and Human Mesenchymal Stromal Cells in Bone Regeneration. Front. Immunol. 2019, 10, 663. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, G.; Chen, R.; Hua, Y.; Cai, Z. Mesenchymal stem cells in the osteosarcoma microenvironment: Their biological properties, influence on tumor growth, and therapeutic implications. Stem Cell Res. Ther. 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Nail, L.R.; Brennan, M.; Rosset, P.; Deschaseaux, F.; Piloquet, P.; Pichon, O.; Le Caignec, C.; Crenn, V.; Layrolle, P.; Herault, O.; et al. Comparison of Tumor- and Bone Marrow-Derived Mesenchymal Stromal/Stem Cells from Patients with High-Grade Osteosarcoma. Int. J. Mol. Sci. 2018, 19, 707. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.A.; Renaud, A.; Amiaud, J.; Rojewski, M.T.; Schrezenmeier, H.; Heymann, D.; Trichet, V.; Layrolle, P. Pre-clinical studies of bone regeneration with human bone marrow stromal cells and biphasic calcium phosphate. Stem Cell Res. Ther. 2014, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Nicolas-Boluda, A.; Donnadieu, E. Obstacles to T cell migration in the tumor microenvironment. Comp. Immunol. Microbiol. Infect. Dis. 2019, 63, 22–30. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Penfornis, P.; Dhule, S.; Guillonneau, F.; Adams, K.V.; Mo, Y.Y.; Xu, R.; Liu, Y.; Watabe, K.; Vemuri, M.C.; et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget 2015, 6, 4953–4967. [Google Scholar] [CrossRef] [Green Version]
- Bonuccelli, G.; Avnet, S.; Grisendi, G.; Salerno, M.; Granchi, D.; Dominici, M.; Kusuzaki, K.; Baldini, N. Role of mesenchymal stem cells in osteosarcoma and metabolic reprogramming of tumor cells. Oncotarget 2014, 5, 7575–7588. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Hassler, M.Y.; Abraham, A.; Whitt, J.; Mo, Y.Y.; Atfi, A.; Pochampally, R. Mesenchymal Stem/Stromal Cells under Stress Increase Osteosarcoma Migration and Apoptosis Resistance via Extracellular Vesicle Mediated Communication. PLoS ONE 2016, 11, e0166027. [Google Scholar] [CrossRef]
- Gebraad, A.; Kornilov, R.; Kaur, S.; Miettinen, S.; Haimi, S.; Peltoniemi, H.; Mannerstrom, B.; Seppanen-Kaijansinkko, R. Monocyte-derived extracellular vesicles stimulate cytokine secretion and gene expression of matrix metalloproteinases by mesenchymal stem/stromal cells. FEBS J. 2018, 285, 2337–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannerstrom, B.; Kornilov, R.; Abu-Shahba, A.G.; Chowdhury, I.M.; Sinha, S.; Seppanen-Kaijansinkko, R.; Kaur, S. Epigenetic alterations in mesenchymal stem cells by osteosarcoma-derived extracellular vesicles. Epigenetics 2019, 14, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, B.; Kimhi, S.; Howard, G.; Eden, A.; Lyko, F. Demethylation of a LINE-1 antisense promoter in the cMet locus impairs Met signalling through induction of illegitimate transcription. Oncogene 2010, 29, 5775–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleton-Jansen, A.M.; Anninga, J.K.; Briaire-de Bruijn, I.H.; Romeo, S.; Oosting, J.; Egeler, R.M.; Gelderblom, H.; Taminiau, A.H.; Hogendoorn, P.C. Profiling of high-grade central osteosarcoma and its putative progenitor cells identifies tumourigenic pathways. Br. J. Cancer 2009, 101, 2064. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Pandya, P.H.; Liu, E.; Chandra, P.; Wang, L.; Murray, M.E.; Carter, J.; Ferguson, M.; Saadatzadeh, M.R.; Bijangi-Visheshsaraei, K.; et al. Integration of genomic copy number variations and chemotherapy-response biomarkers in pediatric sarcoma. BMC Med. Genom. 2019, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Smida, J.; Xu, H.; Zhang, Y.; Baumhoer, D.; Ribi, S.; Kovac, M.; von Luettichau, I.; Bielack, S.; O’Leary, V.B.; Leib-Mosch, C.; et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int. J. Cancer 2017, 141, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Endo-Munoz, L.; Cai, N.; Cumming, A.; Macklin, R.; Merida de Long, L.; Topkas, E.; Mukhopadhyay, P.; Hill, M.; Saunders, N.A. Progression of Osteosarcoma from a Non-Metastatic to a Metastatic Phenotype Is Causally Associated with Activation of an Autocrine and Paracrine uPA Axis. PLoS ONE 2015, 10, e0133592. [Google Scholar] [CrossRef] [Green Version]
- Vallabhaneni, K.C.; Tkachuk, S.; Kiyan, Y.; Shushakova, N.; Haller, H.; Dumler, I.; Eden, G. Urokinase receptor mediates mobilization, migration, and differentiation of mesenchymal stem cells. Cardiovasc. Res. 2011, 90, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Baglio, S.R.; Lagerweij, T.; Perez-Lanzon, M.; Ho, X.D.; Leveille, N.; Melo, S.A.; Cleton-Jansen, A.M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Shen, Y.; Li, X.; Li, B.; Zhao, S.; Gu, J.; Chen, Y.; Ma, B.; Wei, J.; Han, Q.; et al. Exosomes Derived from Hypoxia-Treated Human Adipose Mesenchymal Stem Cells Enhance Angiogenesis Through the PKA Signaling Pathway. Stem Cells Dev. 2018, 27, 456–465. [Google Scholar] [CrossRef]
- Gong, M.; Yu, B.; Wang, J.; Wang, Y.; Liu, M.; Paul, C.; Millard, R.W.; Xiao, D.S.; Ashraf, M.; Xu, M. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget 2017, 8, 45200–45212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadaran, P.; Rajendran, R.L.; Lee, H.W.; Kalimuthu, S.; Hong, C.M.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Extracellular vesicles from mesenchymal stem cells activates VEGF receptors and accelerates recovery of hindlimb ischemia. J. Control. Release Off. J. Control. Release Soc. 2017, 264, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-King, H.; Garcia, N.A.; Ontoria-Oviedo, I.; Ciria, M.; Montero, J.A.; Sepulveda, P. Hypoxia Inducible Factor-1alpha Potentiates Jagged 1-Mediated Angiogenesis by Mesenchymal Stem Cell-Derived Exosomes. Stem Cells 2017, 35, 1747–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Q.; Niu, X.; Hu, B.; Chen, S.; Song, W.; Ding, J.; Zhang, C.; Wang, Y. Exosomes Secreted from Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Prevent Osteonecrosis of the Femoral Head by Promoting Angiogenesis. Int. J. Biol. Sci. 2017, 13, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Sakai, K.; Urata, Y.; Toyama, N.; Nakamichi, E.; Hibi, H. Extracellular Vesicles of Stem Cells to Prevent BRONJ. J. Dent. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Chicon-Bosch, M.; Tirado, O.M. Exosomes in Bone Sarcomas: Key Players in Metastasis. Cells 2020, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Nassar, W.; El-Ansary, M.; Sabry, D.; Mostafa, M.A.; Fayad, T.; Kotb, E.; Temraz, M.; Saad, A.N.; Essa, W.; Adel, H. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomater. Res. 2016, 20, 21. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, M.; Toupet, K.; Maumus, M.; Rozier, P.; Jorgensen, C.; Noel, D. TGFBI secreted by mesenchymal stromal cells ameliorates osteoarthritis and is detected in extracellular vesicles. Biomaterials 2020, 226, 119544. [Google Scholar] [CrossRef]
- Ramasamy, S.K. Structure and Functions of Blood Vessels and Vascular Niches in Bone. Stem Cells Int. 2017, 2017, 5046953. [Google Scholar] [CrossRef] [Green Version]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, E.C.; Adams, R.H. Biology of Bone: The Vasculature of the Skeletal System. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Gruneboom, A.; Hawwari, I.; Weidner, D.; Culemann, S.; Muller, S.; Henneberg, S.; Brenzel, A.; Merz, S.; Bornemann, L.; Zec, K.; et al. A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat. Metab. 2019, 1, 236–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.K.; Kusumbe, A.P.; Wang, L.; Adams, R.H. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014, 507, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhou, F.; Zhang, P.; Wang, H.; Qu, Z.; Jia, P.; Yao, Z.; Shen, G.; Li, G.; Zhao, G.; et al. Human type H vessels are a sensitive biomarker of bone mass. Cell Death Dis. 2017, 8, e2760. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Marcola, M.; Rodrigues, C.E. Endothelial progenitor cells in tumor angiogenesis: Another brick in the wall. Stem Cells Int. 2015, 2015, 832649. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Su, B.; Hollomon, M.; Deng, Y.; Facchinetti, V.; Kleinerman, E.S. Vasculogenesis driven by bone marrow-derived cells is essential for growth of Ewing’s sarcomas. Cancer Res. 2010, 70, 1334–1343. [Google Scholar] [CrossRef] [Green Version]
- An, R.; Schmid, R.; Klausing, A.; Robering, J.W.; Weber, M.; Bauerle, T.; Detsch, R.; Boccaccini, A.R.; Horch, R.E.; Boos, A.M.; et al. Proangiogenic effects of tumor cells on endothelial progenitor cells vary with tumor type in an in vitro and in vivo rat model. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 5587–5601. [Google Scholar] [CrossRef] [PubMed]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef] [Green Version]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Ren, K.; Yao, N.; Wang, G.; Tian, L.; Ma, J.; Shi, X.; Zhang, L.; Zhang, J.; Zhou, X.; Zhou, G.; et al. Vasculogenic mimicry: A new prognostic sign of human osteosarcoma. Hum. Pathol. 2014, 45, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Zhang, J.; Gu, X.; Wu, S.; Shi, X.; Ni, Y.; Chen, Y.; Lu, J.; Gao, Z.; Wang, C.; et al. Migration-inducing gene-7 independently predicts poor prognosis of human osteosarcoma and is associated with vasculogenic mimicry. Exp. Cell Res. 2018, 369, 80–89. [Google Scholar] [CrossRef]
- Xia, Y.; Cai, X.; Fan, J.; Zhang, L.; Li, Z.; Ren, J.; Wu, G.; Zhu, F. RhoA/ROCK pathway inhibition by fasudil suppresses the vasculogenic mimicry of U2OS osteosarcoma cells in vitro. Anti-Cancer Drugs 2017, 28, 514–521. [Google Scholar] [CrossRef]
- Massimini, M.; De Maria, R.; Malatesta, D.; Romanucci, M.; D’Anselmo, A.; Della Salda, L. Establishment of three-dimensional canine osteosarcoma cell lines showing vasculogenic mimicry and evaluation of biological properties after treatment with 17-AAG. Vet. Comp. Oncol. 2019, 17, 376–384. [Google Scholar] [CrossRef]
- Valdivia, A.; Mingo, G.; Aldana, V.; Pinto, M.P.; Ramirez, M.; Retamal, C.; Gonzalez, A.; Nualart, F.; Corvalan, A.H.; Owen, G.I. Fact or Fiction, It Is Time for a Verdict on Vasculogenic Mimicry? Front. Oncol. 2019, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Ohba, T.; Cates, J.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Autocrine VEGF/VEGFR1 signaling in a subpopulation of cells associates with aggressive osteosarcoma. Mol. Cancer Res. 2014, 12, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Gao, Y.; Zhang, L.; Cai, X.; Qian, Z.; Huang, H.; Huang, W. VEGF-siRNA silencing induces apoptosis, inhibits proliferation and suppresses vasculogenic mimicry in osteosarcoma in vitro. Exp. Oncol. 2008, 30, 29–34. [Google Scholar]
- Aslan, C.; Maralbashi, S.; Salari, F.; Kahroba, H.; Sigaroodi, F.; Kazemi, T.; Kharaziha, P. Tumor-derived exosomes: Implication in angiogenesis and antiangiogenesis cancer therapy. J. Cell. Physiol. 2019, 234, 16885–16903. [Google Scholar] [CrossRef] [PubMed]
- Perut, F.; Roncuzzi, L.; Zini, N.; Massa, A.; Baldini, N. Extracellular Nanovesicles Secreted by Human Osteosarcoma Cells Promote Angiogenesis. Cancers 2019, 11, 779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, L.; De Luca, A.; Gallo, A.; Costa, V.; Russelli, G.; Cuscino, N.; Manno, M.; Raccosta, S.; Carina, V.; Bellavia, D.; et al. Osteosarcoma cell-derived exosomes affect tumor microenvironment by specific packaging of microRNAs. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Yang, J.; Yang, D.; Sun, Y.; Sun, B.; Wang, G.; Trent, J.C.; Araujo, D.M.; Chen, K.; Zhang, W. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer 2011, 117, 4925–4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, M.; Wada, T.; Akatsuka, T.; Kawaguchi, S.; Nagoya, S.; Shindoh, M.; Higashino, F.; Mezawa, F.; Okada, F.; Ishii, S. Vascular endothelial growth factor expression in untreated osteosarcoma is predictive of pulmonary metastasis and poor prognosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 572–577. [Google Scholar]
- Lammli, J.; Fan, M.; Rosenthal, H.G.; Patni, M.; Rinehart, E.; Vergara, G.; Ablah, E.; Wooley, P.H.; Lucas, G.; Yang, S.Y. Expression of Vascular Endothelial Growth Factor correlates with the advance of clinical osteosarcoma. Int. Orthop. 2012, 36, 2307–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumars, C.; Ngyuen, J.M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef] [Green Version]
- Charity, R.M.; Foukas, A.F.; Deshmukh, N.S.; Grimer, R.J. Vascular endothelial growth factor expression in osteosarcoma. Clin. Orthop. Relat. Res. 2006, 448, 193–198. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Y.J.; Zhu, K.W.; Wang, W.C. A systematic review of vascular endothelial growth factor expression as a biomarker of prognosis in patients with osteosarcoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 1895–1899. [Google Scholar] [CrossRef]
- Yu, X.W.; Wu, T.Y.; Yi, X.; Ren, W.P.; Zhou, Z.B.; Sun, Y.Q.; Zhang, C.Q. Prognostic significance of VEGF expression in osteosarcoma: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 155–160. [Google Scholar] [CrossRef]
- Liu, K.; Ren, T.; Huang, Y.; Sun, K.; Bao, X.; Wang, S.; Zheng, B.; Guo, W. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 2017, 8, e3015. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Kaminska, J.; Kowalska, M.; Ruka, W.; Steffen, J. Cytokine and cytokine receptor serum levels in adult bone sarcoma patients: Correlations with local tumor extent and prognosis. J. Surg. Oncol. 2003, 84, 151–159. [Google Scholar] [CrossRef]
- Kaya, M.; Wada, T.; Kawaguchi, S.; Nagoya, S.; Yamashita, T.; Abe, Y.; Hiraga, H.; Isu, K.; Shindoh, M.; Higashino, F.; et al. Increased pre-therapeutic serum vascular endothelial growth factor in patients with early clinical relapse of osteosarcoma. Br. J. Cancer 2002, 86, 864–869. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Ji, T.; Guo, W. Anti-angiogenesis target therapy for advanced osteosarcoma (Review). Oncol. Rep. 2017, 38, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.G.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Navid, F.; Santana, V.M.; Neel, M.; McCarville, M.B.; Shulkin, B.L.; Wu, J.; Billups, C.A.; Mao, S.; Daryani, V.M.; Stewart, C.F.; et al. A phase II trial evaluating the feasibility of adding bevacizumab to standard osteosarcoma therapy. Int. J. Cancer 2017, 141, 1469–1477. [Google Scholar] [CrossRef]
- Zheng, B.; Ren, T.; Huang, Y.; Guo, W. Apatinib inhibits migration and invasion as well as PD-L1 expression in osteosarcoma by targeting STAT3. Biochem. Biophys. Res. Commun. 2018, 495, 1695–1701. [Google Scholar] [CrossRef]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Renschler, M.; Perez, L.V.; Stephens, J.; Pytowski, B.; Wasserstrom, H.; Stancato, L.F.; Falcon, B. Anti-VEGFR2 therapy delays growth of preclinical pediatric tumor models and enhances anti-tumor activity of chemotherapy. Oncotarget 2019, 10, 5523–5533. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, Y.; Hookway, E.; Williams, K.A.; Hassan, A.B.; Oppermann, U.; Tanaka, Y.; Soilleux, E.; Athanasou, N.A. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin. Sarcoma Res. 2016, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Fang, X.C.; Ding, Z.; Sun, Z.G.; Sun, L.M.; Wang, Y.L. Pre-operative lymphocyte-to-monocyte ratio as a predictor of overall survival in patients suffering from osteosarcoma. FEBS Open Bio 2015, 5, 682–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonomura, N.; Takayama, H.; Nakayama, M.; Nakai, Y.; Kawashima, A.; Mukai, M.; Nagahara, A.; Aozasa, K.; Tsujimura, A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011, 107, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008, 84, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Burger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; Larousserie, F.; Entz-Werle, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Kleinerman, E.S.; Meyers, P.A.; Raymond, A.K.; Gano, J.B.; Jia, S.F.; Jaffe, N. Combination therapy with ifosfamide and liposome-encapsulated muramyl tripeptide: Tolerability, toxicity, and immune stimulation. J. Immunother. Emphas. Tumor Immunol. Off. J. Soc. Biol. Ther. 1995, 17, 181–193. [Google Scholar] [CrossRef]
- Meyers, P.A. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev. Anticancer Ther. 2009, 9, 1035–1049. [Google Scholar] [CrossRef]
- Fidler, I.J.; Sone, S.; Fogler, W.E.; Barnes, Z.L. Eradication of spontaneous metastases and activation of alveolar macrophages by intravenous injection of liposomes containing muramyl dipeptide. Proc. Natl. Acad. Sci. USA 1981, 78, 1680–1684. [Google Scholar] [CrossRef] [Green Version]
- Kleinerman, E.S.; Jia, S.F.; Griffin, J.; Seibel, N.L.; Benjamin, R.S.; Jaffe, N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: The cytokine cascade and monocyte activation following administration. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1992, 10, 1310–1316. [Google Scholar] [CrossRef]
- MacEwen, E.G.; Kurzman, I.D.; Rosenthal, R.C.; Smith, B.W.; Manley, P.A.; Roush, J.K.; Howard, P.E. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J. Natl. Cancer Inst. 1989, 81, 935–938. [Google Scholar] [CrossRef]
- Casey, D.L.; Cheung, N.V. Immunotherapy of Pediatric Solid Tumors: Treatments at a Crossroads, with an Emphasis on Antibodies. Cancer Immunol. Res. 2020, 8, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sapi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendroi, M.; Bosch, A.L.; et al. CD8(+)/FOXP3(+)-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Lussier, D.M.; O’Neill, L.; Nieves, L.M.; McAfee, M.S.; Holechek, S.A.; Collins, A.W.; Dickman, P.; Jacobsen, J.; Hingorani, P.; Blattman, J.N. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 2015, 38, 96–106. [Google Scholar] [CrossRef]
- Zhu, Z.; Jin, Z.; Zhang, M.; Tang, Y.; Yang, G.; Yuan, X.; Yao, J.; Sun, D. Prognostic value of programmed death-ligand 1 in sarcoma: A meta-analysis. Oncotarget 2017, 8, 59570–59580. [Google Scholar] [CrossRef] [Green Version]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Hasan Ali, O.; Diem, S.; Markert, E.; Jochum, W.; Kerl, K.; French, L.E.; Speiser, D.E.; Fruh, M.; Flatz, L. Characterization of nivolumab-associated skin reactions in patients with metastatic non-small cell lung cancer. Oncoimmunology 2016, 5, e1231292. [Google Scholar] [CrossRef] [Green Version]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Koksal, H.; Muller, E.; Inderberg, E.M.; Bruland, O.; Walchli, S. Treating osteosarcoma with CAR T cells. Scand. J. Immunol. 2019, 89, e12741. [Google Scholar] [CrossRef] [Green Version]
- Rainusso, N.; Brawley, V.S.; Ghazi, A.; Hicks, M.J.; Gottschalk, S.; Rosen, J.M.; Ahmed, N. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012, 19, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Wang, G.; Sun, M.; Jiang, Y.; Zhang, T.; Sun, W.; Wang, H.; Yin, F.; Wang, Z.; Sang, W.; Xu, J.; et al. Anlotinib, a novel small molecular tyrosine kinase inhibitor, suppresses growth and metastasis via dual blockade of VEGFR2 and MET in osteosarcoma. Int. J. Cancer 2019, 145, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Fioramonti, M.; Santini, D.; Iuliani, M.; Ribelli, G.; Manca, P.; Papapietro, N.; Spiezia, F.; Vincenzi, B.; Denaro, V.; Russo, A.; et al. Cabozantinib targets bone microenvironment modulating human osteoclast and osteoblast functions. Oncotarget 2017, 8, 20113–20121. [Google Scholar] [CrossRef] [Green Version]
- Fioramonti, M.; Fausti, V.; Pantano, F.; Iuliani, M.; Ribelli, G.; Lotti, F.; Pignochino, Y.; Grignani, G.; Santini, D.; Tonini, G.; et al. Cabozantinib Affects Osteosarcoma Growth Through A Direct Effect On Tumor Cells and Modifications In Bone Microenvironment. Sci. Rep. 2018, 8, 4177. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.E.; Bolejack, V.; Ryan, C.W.; Ganjoo, K.N.; Loggers, E.T.; Chawla, S.; Agulnik, M.; Livingston, M.B.; Reed, D.; Keedy, V.; et al. Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1424–1431. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Aggerholm-Pedersen, N.; Rossen, P.; Rose, H.; Safwat, A. Pazopanib in the Treatment of Bone Sarcomas: Clinical Experience. Transl. Oncol. 2019, 13, 295–299. [Google Scholar] [CrossRef]
- Longhi, A.; Paioli, A.; Palmerini, E.; Cesari, M.; Abate, M.E.; Setola, E.; Spinnato, P.; Donati, D.; Hompland, I.; Boye, K. Pazopanib in relapsed osteosarcoma patients: Report on 15 cases. Acta Oncol. 2019, 58, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werle, N.; Saada, E.; et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Zhu, C.; Ma, X.; Hu, Y.; Guo, L.; Chen, B.; Shen, K.; Xiao, Y. Safety and efficacy profile of lenvatinib in cancer therapy: A systematic review and meta-analysis. Oncotarget 2016, 7, 44545–44557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, K.; Otaki, N. Bone cell interactions through Eph/ephrin: Bone modeling, remodeling and associated diseases. Cell Adhes. Migr. 2012, 6, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklander, O.P.B.; Brennan, M.A.; Lotvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
RANK–RANKL interactions under the canopy of bone remodeling compartments: old and new paradigms. The canopy is generated by bone lining cells and insulates a basic multicellular unit with osteoclasts (OC) and osteoblasts (OB). OC and OB precursors are recruited under the canopy, from respectively bone marrow- and blood stream-supplied hematopoietic stem cells and bone marrow-issued mesenchymal stromal cells. In the old and well-demonstrated paradigm, RANKL secreted by OB induces OC differentiation through RANK intracellular signaling (RANKL-RANK), while a new paradigm proposes a reverse signaling through RANKL intracellular signaling (RANK-RANKL) mediated by RANK-bearing extracellular vesicles EVs from OC [32]. RANK: receptor activator of nuclear factor kappa-B; RANKL: RANK ligand.
Figure 1.
RANK–RANKL interactions under the canopy of bone remodeling compartments: old and new paradigms. The canopy is generated by bone lining cells and insulates a basic multicellular unit with osteoclasts (OC) and osteoblasts (OB). OC and OB precursors are recruited under the canopy, from respectively bone marrow- and blood stream-supplied hematopoietic stem cells and bone marrow-issued mesenchymal stromal cells. In the old and well-demonstrated paradigm, RANKL secreted by OB induces OC differentiation through RANK intracellular signaling (RANKL-RANK), while a new paradigm proposes a reverse signaling through RANKL intracellular signaling (RANK-RANKL) mediated by RANK-bearing extracellular vesicles EVs from OC [32]. RANK: receptor activator of nuclear factor kappa-B; RANKL: RANK ligand.

Figure 2.
Proposed model of OPG/RANK–RANKL interactions in osteosarcoma (OS). RANK transduction induces the differentiation of osteoclasts (OC), leading to osteolysis, which in turn activates tumor cell proliferation, described as the vicious cycle. OPG is a decoy form of RANK, binding and neutralizing RANKL. Additionally, OPG increases proliferation of RANKL-expressing OS cells following its binding to an unknown receptor [38], possibly RANKL. The reverse signaling of RANKL, described recently in osteoblasts [32], could be also induced in OS cells. To the same extend, OS cells expressing RANKL could be activated by RANK-extracellular vesicles (EVs) produced by OC. RANK: receptor activator of nuclear factor kappa-B; RANKL: RANK ligand; OPG: osteoprotegerin.
Figure 2.
Proposed model of OPG/RANK–RANKL interactions in osteosarcoma (OS). RANK transduction induces the differentiation of osteoclasts (OC), leading to osteolysis, which in turn activates tumor cell proliferation, described as the vicious cycle. OPG is a decoy form of RANK, binding and neutralizing RANKL. Additionally, OPG increases proliferation of RANKL-expressing OS cells following its binding to an unknown receptor [38], possibly RANKL. The reverse signaling of RANKL, described recently in osteoblasts [32], could be also induced in OS cells. To the same extend, OS cells expressing RANKL could be activated by RANK-extracellular vesicles (EVs) produced by OC. RANK: receptor activator of nuclear factor kappa-B; RANKL: RANK ligand; OPG: osteoprotegerin.

Figure 3.
Osteoblastic differentiation from mesenchymal stem/stromal cells (MSCs) to osteocytes. Transcription factor RUNX2 promotes MSC commitment toward the osteoblastic lineage at the early stages while repressing maturation in osteocytes. SP7 allows the differentiation of pre-osteoblasts into functional mature osteoblasts. RUNX2 induces expression of genes coding for expression of collagen type 1 (COL1), osteopontin (OPN), bone sialoprotein (BSP) and osteocalcin (OCN) proteins. Transforming growth factor-β (TGF-β1) and WNT stimulate early stages of osteoblastic differentiation.
Figure 3.
Osteoblastic differentiation from mesenchymal stem/stromal cells (MSCs) to osteocytes. Transcription factor RUNX2 promotes MSC commitment toward the osteoblastic lineage at the early stages while repressing maturation in osteocytes. SP7 allows the differentiation of pre-osteoblasts into functional mature osteoblasts. RUNX2 induces expression of genes coding for expression of collagen type 1 (COL1), osteopontin (OPN), bone sialoprotein (BSP) and osteocalcin (OCN) proteins. Transforming growth factor-β (TGF-β1) and WNT stimulate early stages of osteoblastic differentiation.

Figure 4.
Histological analysis of experimentally induced osteosarcoma (OS) in athymic mice. Human OS-inducing cells (MNNG-HOS cells, CRL-1547, from American Type Culture Collection) were injected either alone (a,b) or co-injected with OS-derived stromal cells (OSDCs) (c,d) at tibial sites of athymic mice. Tumor samples were fixed in 10% buffered formaldehyde, embedded in paraffin wax, sectioned, and stained. Magnifications are indicated. (a) Human OS cells (brown nuclei) were distinguished from mouse cells (blue nuclei) by in situ hybridization using the human-specific repetitive Alu sequence [65]. Bone (B) spine lined by mouse cells was observed. (b) MNNG-HOS-induced tumor section was stained with hematoxylin–eosin–safran solution (HES). Tumor developed in muscle and appeared as an undifferentiated pleomorphic sarcoma (magnification in left panel), while osteoid matrix was observed only in intra-vascular tumor emboli (magnification in right panel). (c) Following co-injection with OSDC [64], MNNG-HOS-induced tumor was visible with abundant osteoid matrix surrounding anaplastic tumor cells. Tumor section was stained with HES. (d) Image of lung metastasis developed from co-injection of MNNG-HOS and OSDCs at paratibial site. Remarkable and non-usual immune cell infiltration (I) was observed in the vessel wall.
Figure 4.
Histological analysis of experimentally induced osteosarcoma (OS) in athymic mice. Human OS-inducing cells (MNNG-HOS cells, CRL-1547, from American Type Culture Collection) were injected either alone (a,b) or co-injected with OS-derived stromal cells (OSDCs) (c,d) at tibial sites of athymic mice. Tumor samples were fixed in 10% buffered formaldehyde, embedded in paraffin wax, sectioned, and stained. Magnifications are indicated. (a) Human OS cells (brown nuclei) were distinguished from mouse cells (blue nuclei) by in situ hybridization using the human-specific repetitive Alu sequence [65]. Bone (B) spine lined by mouse cells was observed. (b) MNNG-HOS-induced tumor section was stained with hematoxylin–eosin–safran solution (HES). Tumor developed in muscle and appeared as an undifferentiated pleomorphic sarcoma (magnification in left panel), while osteoid matrix was observed only in intra-vascular tumor emboli (magnification in right panel). (c) Following co-injection with OSDC [64], MNNG-HOS-induced tumor was visible with abundant osteoid matrix surrounding anaplastic tumor cells. Tumor section was stained with HES. (d) Image of lung metastasis developed from co-injection of MNNG-HOS and OSDCs at paratibial site. Remarkable and non-usual immune cell infiltration (I) was observed in the vessel wall.

Figure 5.
Sensing and modulating roles of mesenchymal stem/stromal cells (MSCs). MSCs constitutively express many mitogenic growth factors, chemokines, and matrix metalloproteinases at various levels. High/intermediate and low levels are represented by large or small font size, respectively. MSCs respond to tumor necrosis factor alpha (TNFα) by increasing expression (indicated in red) of some growth factor receptors (GFR), growth factors (GF), chemokine receptors (CR), chemokines, interleukins (IL), and matrix metalloproteinases (MMP) [61]. MSCs have autocrine and paracrine trophic properties, as all of these growth factors are able to act on several effectors of the tumor ecosystem (tumor, bone cells, endothelial cells, and immune cells). MSCs can also secrete extracellular vesicles (EVs) that convey diverse contents (see text for details), and they can receive information through EVs from bone pre-osteoclasts (pre-OC) and osteoclasts (OC) and tumor cells. EGFR: epithelial GFR; VEGFR: vascular endothelial GFR; TIE-1: angiopoietin receptor; TGFBR: transforming growth factor beta receptor; TIMP: tissue inhibitors of MMP; CXCL: chemokine (C-X-C motif) ligand; CCL: chemokine (C-C motif) ligand; GROα/β: growth-regulated oncogene alpha/beta (also known as CXCL1/2); MCP1/3: monocyte chemoattractant protein (also known as CCL2/7); SDF1: stromal cell-derived factor (also known as CXCL12); RANTES: Regulated on Activation Normal T Cell Expressed and Secreted (alias CCL5).
Figure 5.
Sensing and modulating roles of mesenchymal stem/stromal cells (MSCs). MSCs constitutively express many mitogenic growth factors, chemokines, and matrix metalloproteinases at various levels. High/intermediate and low levels are represented by large or small font size, respectively. MSCs respond to tumor necrosis factor alpha (TNFα) by increasing expression (indicated in red) of some growth factor receptors (GFR), growth factors (GF), chemokine receptors (CR), chemokines, interleukins (IL), and matrix metalloproteinases (MMP) [61]. MSCs have autocrine and paracrine trophic properties, as all of these growth factors are able to act on several effectors of the tumor ecosystem (tumor, bone cells, endothelial cells, and immune cells). MSCs can also secrete extracellular vesicles (EVs) that convey diverse contents (see text for details), and they can receive information through EVs from bone pre-osteoclasts (pre-OC) and osteoclasts (OC) and tumor cells. EGFR: epithelial GFR; VEGFR: vascular endothelial GFR; TIE-1: angiopoietin receptor; TGFBR: transforming growth factor beta receptor; TIMP: tissue inhibitors of MMP; CXCL: chemokine (C-X-C motif) ligand; CCL: chemokine (C-C motif) ligand; GROα/β: growth-regulated oncogene alpha/beta (also known as CXCL1/2); MCP1/3: monocyte chemoattractant protein (also known as CCL2/7); SDF1: stromal cell-derived factor (also known as CXCL12); RANTES: Regulated on Activation Normal T Cell Expressed and Secreted (alias CCL5).


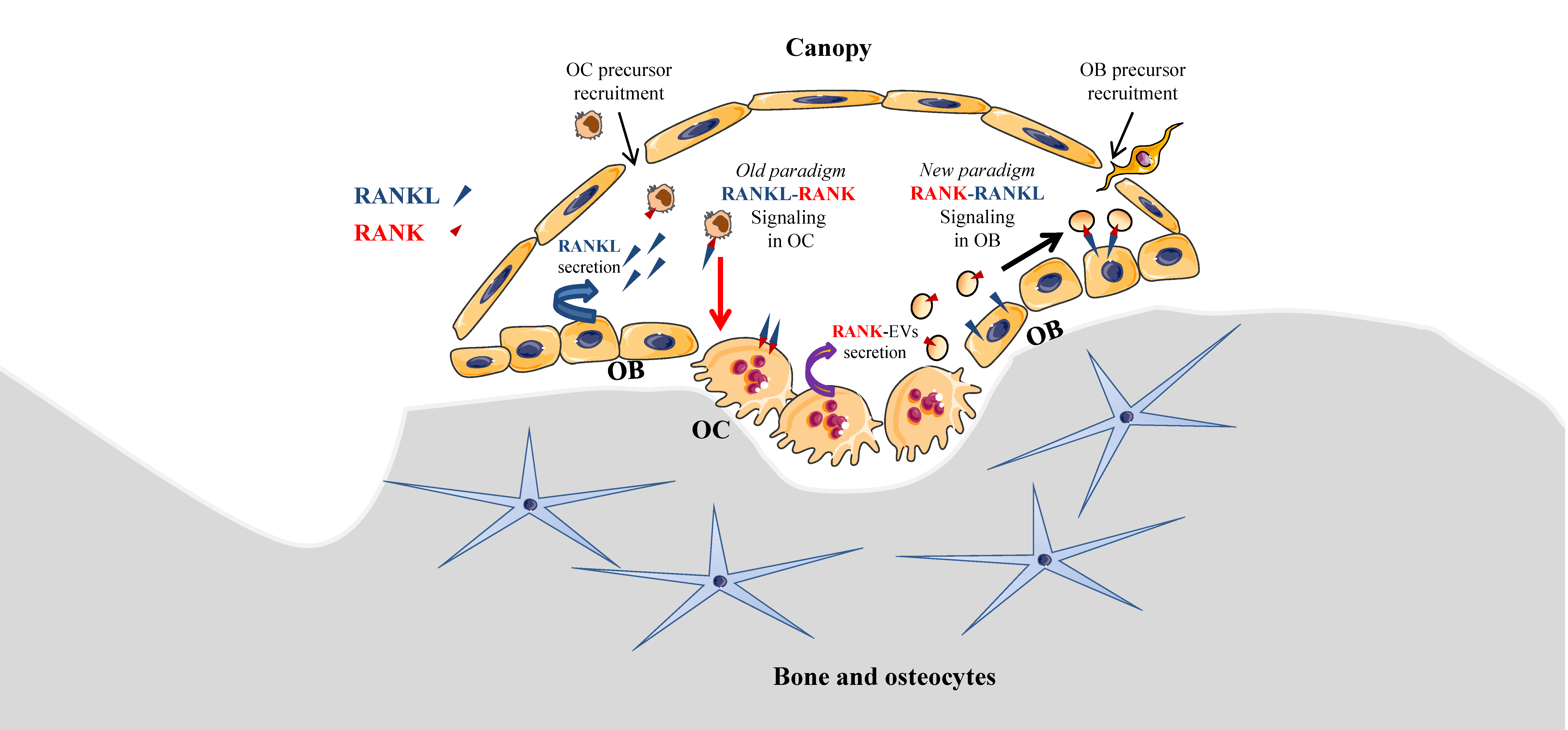{kind=link}
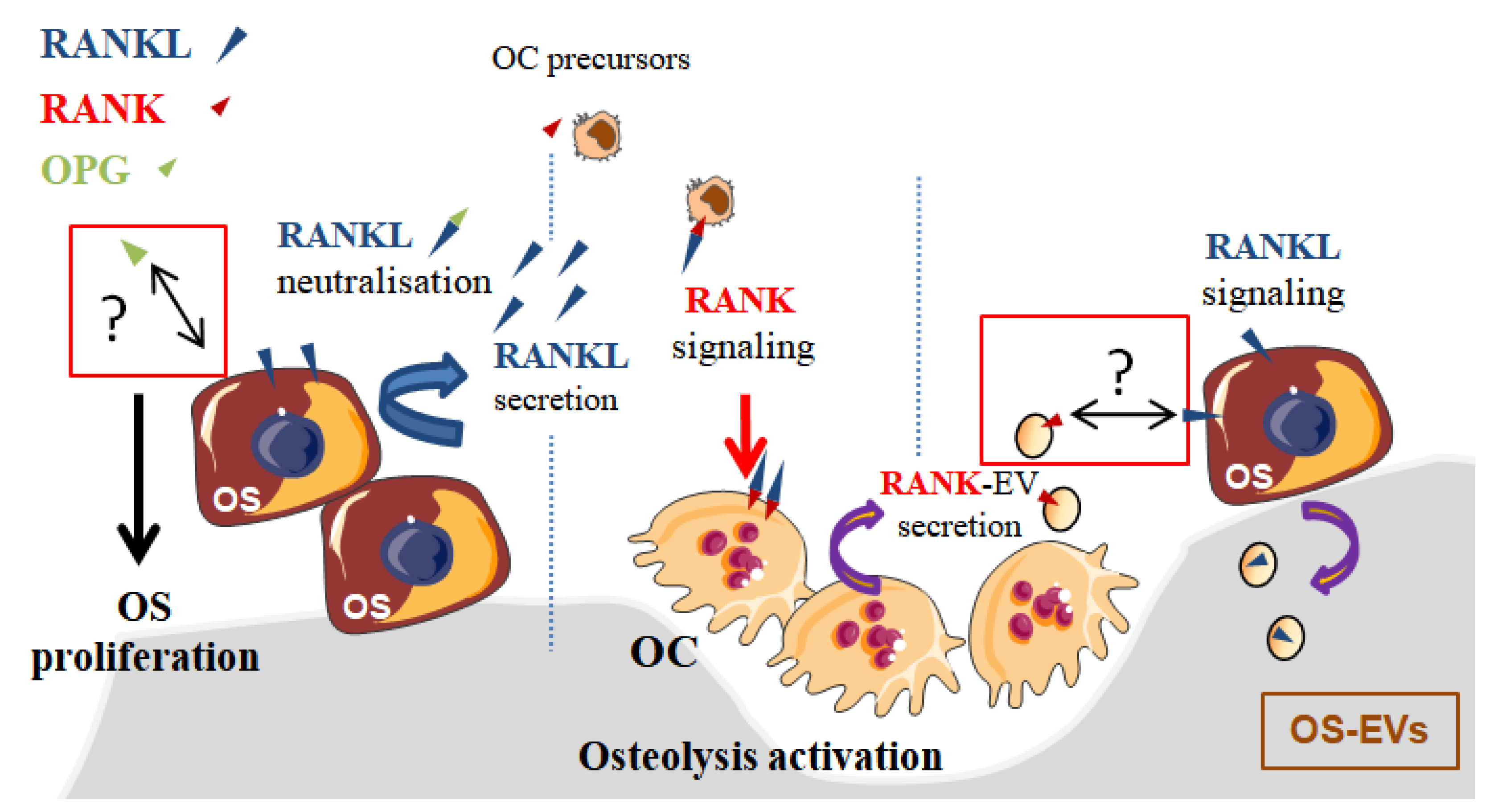{kind=link}
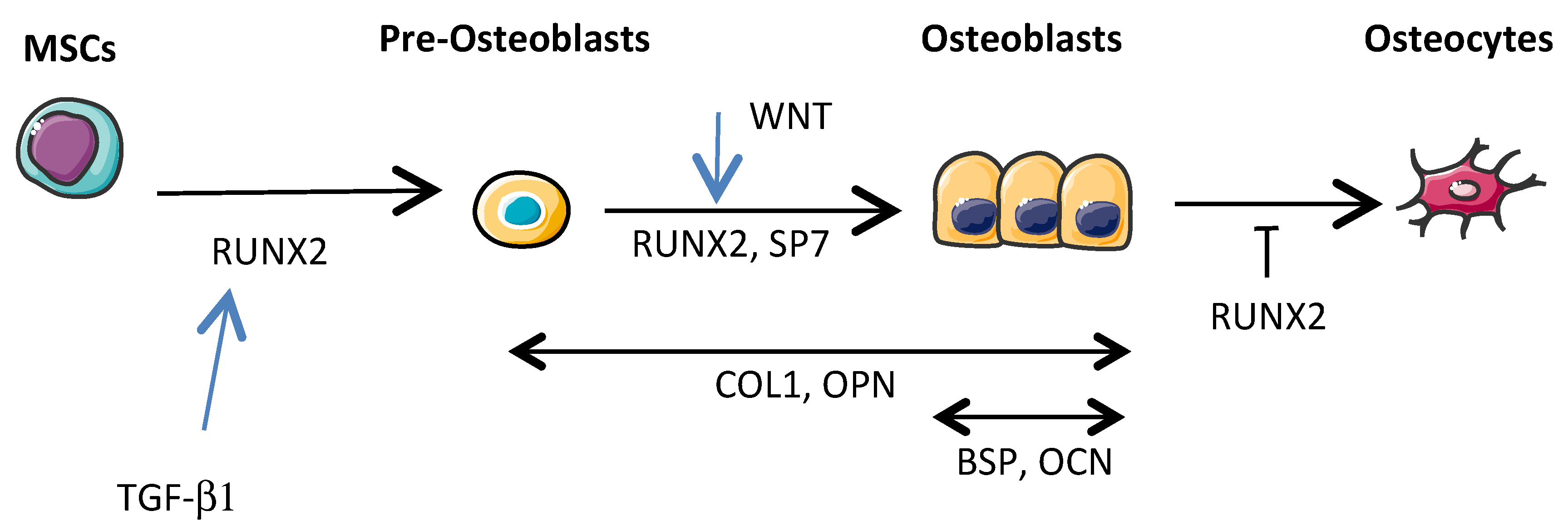{kind=link}
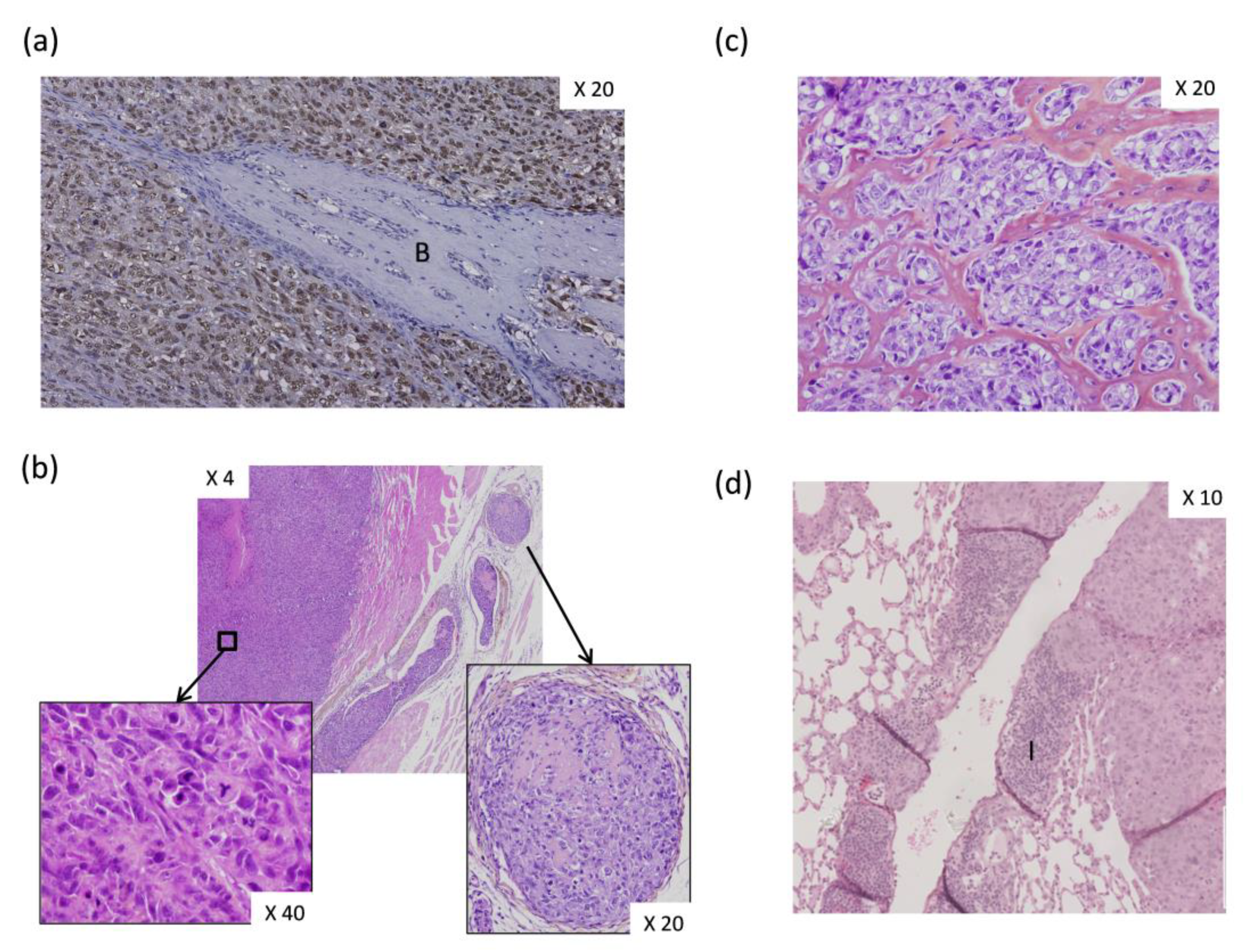{kind=link}
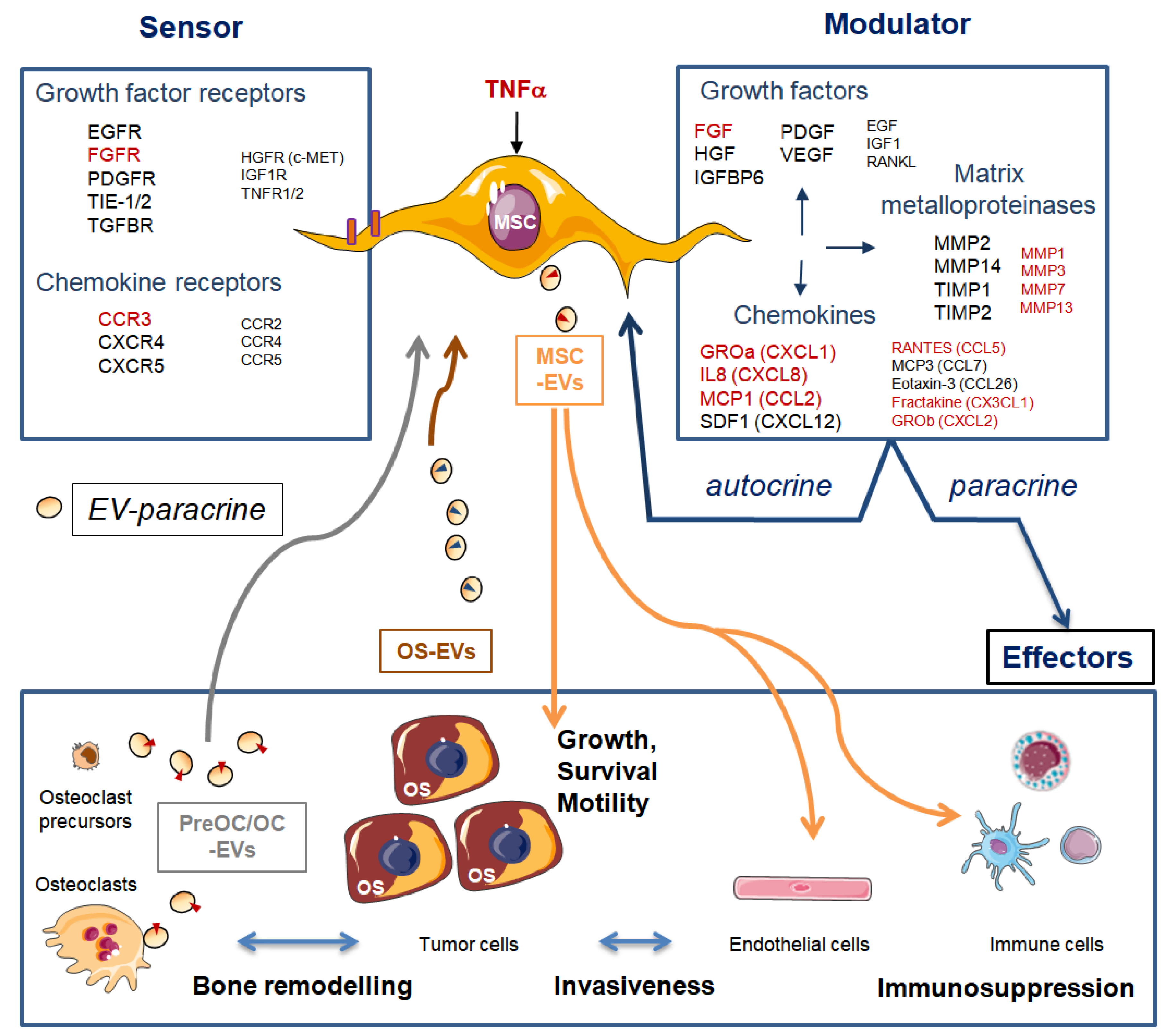{kind=link}
Table 1.
Multi-kinases inhibitors (MKI) in Osteosarcoma (OS): Molecular and Cellular Targets and Clinical Trials.
Table 1.
Multi-kinases inhibitors (MKI) in Osteosarcoma (OS): Molecular and Cellular Targets and Clinical Trials.
| MKI | Targets | Clinical Trials | |
|---|---|---|---|
| Molecular | Cellular | ClinicalTrials.gov | |
| Sorafenib | RAF, KIT, FLT3, RET VEGFR1-3 PDGFRβ | Tumor Endothelial Stromal | NCT 00889057 [125] NCT 01804374 [126] |
| Regorafenib | KIT, RET, RAF VEGFR1-3, Tie-2 PDGFRβ FGFR | Tumor Endothelial Stromal | NCT 0238244 [159] NCT 02048371 [158] |
| Pazopanib | KIT, FMS VEGFR1-3 PDGFRαβ FGFR | Tumor Endothelial Stromal | [160] |
| Cabozantinib | MET, KIT, RET VEGFR-2, Tie-2 | Tumor Osteoblasts Endothelial | NCT02243605 [162] |
| Lenvatinib | KIT, RET VEGFR-1, 2, 3 PDGFR-α FGFR1-4 | Tumor Endothelial Stromal | NCT04154189 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem. Cells 2020, 9, 976. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040976
AMA Style
Corre I, Verrecchia F, Crenn V, Redini F, Trichet V. The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem. Cells. 2020; 9(4):976. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040976
Chicago/Turabian StyleCorre, Isabelle, Franck Verrecchia, Vincent Crenn, Francoise Redini, and Valérie Trichet. 2020. "The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem" Cells 9, no. 4: 976. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040976
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.