Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective



1
Department of Neurosurgery, Center for Neuroregeneration, Houston Methodist Research Institute, Houston, TX 77030, USA
2
Department of Neurosurgery, Weill Medical College, New York, NY 10065, USA
*
Author to whom correspondence should be addressed.
Cells 2020, 9(4), 1065; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041065
Submission received: 28 February 2020
/
Revised: 15 April 2020
/
Accepted: 21 April 2020
/
Published: 24 April 2020
(This article belongs to the Section Mitochondria)
Abstract
:Mitochondria plays privotal role in diverse pathways that regulate cellular function and survival, and have emerged as a prime focus in aging and age-associated motor neuron diseases (MNDs), such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Accumulating evidence suggests that many amyloidogenic proteins, including MND-associated RNA/DNA-binding proteins fused in sarcoma (FUS) and TAR DNA binding protein (TDP)-43, are strongly linked to mitochondrial dysfunction. Animal model and patient studies have highlighted changes in mitochondrial structure, plasticity, replication/copy number, mitochondrial DNA instability, and altered membrane potential in several subsets of MNDs, and these observations are consistent with the evidence of increased excitotoxicity, induction of reactive oxygen species, and activation of intrinsic apoptotic pathways. Studies in MND rodent models also indicate that mitochondrial abnormalities begin prior to the clinical and pathological onset of the disease, suggesting a causal role of mitochondrial dysfunction. Our recent studies, which demonstrated the involvement of specific defects in DNA break-ligation mediated by DNA ligase 3 (LIG3) in FUS-associated ALS, raised a key question of its potential implication in mitochondrial DNA transactions because LIG3 is essential for both mitochondrial DNA replication and repair. This question, as well as how wild-type and mutant MND-associated factors affect mitochondria, remain to be elucidated. These new investigation avenues into the mechanistic role of mitochondrial dysfunction in MNDs are critical to identify therapeutic targets to alleviate mitochondrial toxicity and its consequences. In this article, we critically review recent advances in our understanding of mitochondrial dysfunction in diverse subgroups of MNDs and discuss challenges and future directions.
1. Introduction
Aging-associated neurological disorders are considered the foremost public health challenge of our time, having devastating effects on quality of life and forming a major burden on health-care systems. Motor neuron diseases (MNDs) are one such group of progressive neurodegenerative disorders characterized by dysfunction of lower motor neurons in the ventral horn and/or upper motor neurons in the precentral gyrus of the spinal cord. This typically results in loss of control over voluntary muscle activities such as walking, speaking, breathing, and swallowing. MNDs are predominantly sporadic with multifactorial etiology, but a smaller subset of ~5–10% can be familial, involving one or more mutations or genetic factors usually of autosomal dominant inheritance [1]. The incidence of motor neuron disease in the U.S. is approximately 3–5 cases for 100,000 population per year, and the average time from diagnosis to death is about 3 years [2].
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD; also known as frontotemporal lobar dementia or FTLD) are the two major MNDs, with both overlapping and unique etiologies and clinical features. Notably, the disease name defines the important features of the disease. Amyotrophy refers to atrophy of muscle fibers, which leads to weakness in affected muscles and observable fasciculations. Lateral sclerosis refers to the hardening of lateral and anterior corticospinal tracts, as motor neurons within these areas degenerate and are replaced by gliosis. Unlike ALS, FTD involves a progressive neuronal atrophy with loss of frontal and temporal cortices, which is characterized by changes in personality and behavior, and gradual impairment of language skills. In ALS, familial and sporadic cases account for ~10% and ~90%, respectively, whereas in FTD, a strong genetic contribution is reflected by the higher percentage (50%) of patients having familial history [3]. The overlapping existence of ALS and FTD has been observed in many patients; about 15% of patients with FTD develop ALS and 5–22% of patients with ALS develop FTD (refered here as ALS-FTD) [4,5,6]. Patients with ALS-FTD generally have early onset symptoms in their 50s, similar to ALS without dementia, and it is more common in men than in women [7]. ALS symptoms may precede, occur simultaneously with, or follow the signs of FTD, although more frequently, cognitive changes precede motor weakness [8,9]. The interval between cognitive symptoms and weakness can range from months up to 7 years, with an average of 2 years [10]. ALS-FTD is normally more aggressive in comparison to either disease alone. Survival post-diagnosis varies from 2 to 5 years in ALS, whereas it is ≤2 years for ALS-FTD. Notably, ALS patients with dysexecutive function have worse survival, whereas those with abnormalities that are limited to language or visuospatial skills have comparable survival to cognitively normal ALS patients. ALS patients with dementia are mainly characterized by poor memory and cognitive defects, similar to Alzheimer’s disease (AD); however, this does not seem to affect overall survival [3].
Multiple environmental and genetic factors have been identified to contribute to ALS incidence and disease progression. Some putative environmental risk factors for ALS are history of brain trauma, radiation exposure, electrical shocks, smoking in the case of post-menopausal women, dietary factors, diabetes, physical fitness, viruses, and metal exposure [11,12,13,14,15,16,17]. The role of exogenous risk factors in ALS is reported inconsistently, indicating that these risk factors do not always cause pathology; instead, they reflect the potential interaction between various environmental factors and specific gene susceptibilities.
ALS was initially thought to be a sporadic disease [18]; however, evidence of the autosomal dominant inheritance of ALS was observed in subsequent studies, leading to associations with more than 100 genes [19]. Various bioinformatics repositories are available that list known ALS and FTD genes [20,21]. Many of these genes are, however, considered to be ‘minor genes’ because they have poor penetrance. Major genes that have been reported in multiple subsets of ALS or FTD include superoxide dismutase 1 (SOD1), TAR DNA binding protein (TARDBP coding for TDP-43 protein), fused in sarcoma (FUS), chromosome 9 open reading frame 72 (C9ORF72), microtubule-associated protein tau (MAPT), progranulin (PGRN), and valosin containing protein (VCP). C9ORF72 hexanucleotide repeat expansion is the most common cause of familial FTD and ALS, associated with ~30–50% of familial ALS, 5–7% of sporadic, ALS, 25% of familial FTD, and 6% of sporadic FTD [22,23]. Repeat expansion of the hexanucleotide repeat GGGGCC (G4C2) in the range of several hundred to thousands of repeats is seen in patients, in comparison to between two and 10 for the majority of the population [24,25]. The SOD1 gene, which encodes the Cu-Zn superoxide dismutase, is one of three proteins involved in the conversion of free superoxide radicals to molecular oxygen and hydrogen peroxide. Mutations in the SOD1 gene are found in 10–20% of familial ALS cases and 1–5% of sporadic ALS cases globally [26]; so far, more than 170 mutations of the SOD1 gene are known in ALS. However, SOD1 knockout in rodents does not cause the disease phenotype, suggesting that the pathogenicity does not involve loss of function, and rather involves gain of toxic function, which can be due to the formation of aggregates caused by protein instability [27,28]. Mutations in FUS have been identified in nearly 5% of familial ALS patients along with 1% of sporadic ALS cases and about 10–20% of familial FTD [29,30]. Most mutations in FUS are clustered in the C-terminal nuclear localization sequence (NLS), thus inducing nuclear depletion and cytosolic aggregation. FUS binds both RNA and DNA and plays roles in RNA metabolism and maintaining genome integrity by being involved in the DNA damage response [31,32,33]. Pathogenic inclusions of FUS without TDP-43 or Tau containing aggregates are present in about 10% of FTLD cases, also known as FTLD-FUS [34]. Similar to FUS, TDP-43 is an RNA/DNA-binding protein known to function in RNA metabolism and transport of subcellular RNA. In healthy neurons, TDP-43 is localized to the nucleus, whereas toxic or mutated TDP-43 is cleaved and phosphorylated abnormally, and accumulated in ubiquitinated cytoplasmic inclusions in ALS [35]. More than 30 mutations are reported in the TARDBP gene with ~4–5% familial and ~2% sporadic ALS association [36]. MAPT mutations are associated with ~10–30% of familial FTD and normally occur together with TDP-43 and other pathology [37]. The MAPT gene encodes a 758-amino acid-long Tau protein, which is important for the binding and stabilization of microtubules located in neuronal axons. The mutated Tau protein becomes hyperphosphorylated and accumulates as abnormal filaments within neuronal and glial cells [38]. The PGRN gene encodes a precursor of granulin. PGRN is a growth factor involved in various metabolic events such as wound healing, tumor growth, and inflammation. PGRN also activates several kinase-dependent signaling cascades involved in controlling the cell cycle and motility [39]. In the United States, ~10% of FTD cases carry a mutation in PGRN gene, among which ~22% are familial [40]. Immunohistochemical studies show an increase in PGRN expression with disease progression in the spinal cords of transgenic animals with MNDs, and reveal strong expression of PGRN in the microglia of ALS patients [41,42,43].
Despite the association of multiple genes with ALS and FTD in the past decade, the precise etiology and mechanism of disease progression remain elusive. Defects in oxidative phosphorylation, calcium (Ca2+) buffering, and mitochondrial transport, normally seen at disease onset in the majority of patients, suggests the involvement of mitochondrial dysfunction in the etiology of ALS. In addition to their role as energy producers, mitochondria play a central role in Ca2+ homeostasis, phospholipid biogenesis, and apoptosis. Mitochondrial function is particularly crucial in the brain; despite constituting only 2% of body mass, the brain consumes 20% of the body’s resting ATP production. In neurotransmission, mitochondria act as essential Ca2+ buffering organelles. Due to their long life span, neurons may be more susceptible to damage caused by mitochondrial dysfunction. Furthermore, many proteins linked to ALS and FTD, including SOD1, TDP-43, FUS, and C9ORF72, are shown to interact with mitochondria. The association of these proteins with mitochondria is emerging as a critical factor in triggering disease onset and progression.
This review focuses on the central role of mitochondria in MNDs, the implications of recent findings regarding the role of TDP-43 and FUS in DNA repair and how they may play a critical role in the maintenance of genome stability in mitochondria.
2. Role of Mitochondrial Function and Plasticity in Central Nervous System (CNS)
The mitochondria are sites of aerobic respiration and supply energy for cellular needs in the form of ATP. Apart from ATP production, mitochondria are involved in a myriad of other cellular processes including amino acid and nucleotide metabolism, protein synthesis, fatty acid metabolism, ion homeostasis, and apoptosis [44,45]. Maintenance of mitochondrial quality control is important for cell survival. A mitochondrion has its own proteolytic system, which degrades misfolded/toxic proteins [46]. Damaged proteins on the outer mitochondrial membrane (OMM) are degraded by proteosome [47]. These multiple quality control mechanisms cumulatively help in maintaining healthy mitochondrial homeostasis. In addition, the fission and fusion processes within and among mitochondria help in clearing/redistribution of damaged components [48]. The mitochondrial respiratory chain, comprised of four multienzyme complexes designated as NADH coenzyme Q reductase (Complex I), succinate coenzyme Q reductase (Complex II), ubiquinol cytochrome c reductase (Complex III), and cytochrome c oxidase (Complex IV), is localized in the inner mitochondrial membrane (IMM) along with two electron carriers, cytochrome C and ubiquinone [49]. The mammalian mitochondrial proteome is comprised of ~1200 proteins, the majority of which are expressed from the nuclear genome, whereas a small subset of these proteins are expressed by the heritably and spatially separate mitochondrial genome [50]. Human mitochondrial DNA encodes 11 mRNAs, 2 rRNAs, and 22 tRNAs [51]. In higher metazoans, the mitochondrial genome is attached firmly to the IMM, packaged into DNA-protein complexes with mitochondrial transcription factor A (TFAM) [52].
Energy production and Ca2+ homeostasis maintained by mitochondria are particularly important in the CNS due to the high metabolic activity and energy needs of neurons. Local Ca2+ concentrations are critical for inter-neuronal communications [53]. Notably, bioenergetics, Ca2+ transport, and redox homeostasis in mitochondria form a tight network and these processes influence each other both under normal and disease conditions. For example, decrease in ATP generation due to oxidative stress causes defective Ca2+ pump function on plasma membrane leading to Ca2+ overload. High Ca2+ levels may cause cell death by forming permeability transition pore and collapse of transmembrane potential, which releases cytochrome c [54]. Excessive Ca2+ can lead to degradation of structural and enzymatic proteins by activating Ca2+ binding calpain proteases [55]. Apart from rapid cell death, subtle changes in Ca2+ homeostasis coupled with aging can lead to significant consequences such as cognitive decline [56]. Although some of this ATP is provided by glycolysis, the majority is produced in mitochondria [57]. Every part of the neuron requires ATP, and mitochondria, being the major source of ATP, are abundant in neurons. Some mitochondria are retained in the soma, whereas others are trafficked along axons and dendrites to supply energy-hungry sites, including the presynaptic terminal and near the nodes of ranvier [58].
Mitochondria are highly dynamic organelles and undergo both transient and rapid morphological adaptations that are crucial for many cellular processes including mitochondrial functions as well as quality control. The classical definition of mitochondrial dynamics refers to its coordinated cycles of fission and fusion [59]. These processes not only maintain mitochondrial shape, distribution, and size within cells, but also involve mitochondrial transport, fission, fusion, and selective degradation. Mitochondrial dynamics play a crucial role in neuronal development and function [59,60]. Emerging studies suggest multifunctionality of proteins linked to mitochondrial dynamics and cross-talk between mitochondrial dynamics, biogenesis, quality control and trafficking pathways [61]. Mitochondrial fission is important for homeostasis, fusion events enable mitochondria to complement proteins, repair DNA, and for equal distribution of metabolites [48,62]. In neurons, defective mitochondrial fusion leads to swelling of these organelles and prevents them from entering into distal, smaller branches of neurons, causing their degeneration in axons and dendrites [63]. In the case of defective fission, it leads to failure in isolating damaged parts of mitochondria leading to their autophagic degradation, promoting potential neuronal apoptosis [64]. Ablating genes involved in mitochondrial dynamics leads to defects in brain development. For example, MFN2 (important for mitochondrial fusion) conditional knock out in cerebellum leads to smaller cerebella and motor defects [63]. Other studies reported importance of MFN2 as playing role in synapse formation in human induced pluripotent stem cells (iPSCs) [65]. Homozygous mutations in Opa1 (function in mitochondrial fusion) are associated with infantile-onset encephalopathy [66]. Association of DRP1 (role in mitochondrial fission) in neuronal development become apparent in the case of newborns with abnormalities in brain development and optic atrophy with a lethal dominant negative allele of DRP1 [67]. In developing brains, mitochondria assume an elongated morphology in neuronal stem cells but are fragmented in neuronal progenitor cells. In the adult hippocampus, mitochondria display a mixture of globular and tubular structures [68,69]. In developing neurons, mitochondria are concentrated at the growth cones, where they are required to satisfy the high metabolic requirements [70]. In addition to ATP synthesis, mitochondria also play an important role in regulating intracellular Ca2+ dynamics, which in turn influence growth cone extension and collapse [71]. Interestingly, mitochondrial fission and fusion are also important for mitochondrial genome stability, via their role in maintaining mitochondrial DNA copy number, distribution, and integrity. Furthermore, mitochondrial dynamics mitigate the deleterious effects of mutations in mitochondrial DNA by complementation with intact DNA from healthy mitochondria via fusion [72,73,74,75].
Neurons are highly polarized cells with a cell body, numerous short and thick dendrites, and a long thin axon. Distinct structural and functional domains of neurons have different metabolic demands and display very non-uniform mitochondrial distribution. In comparison to any other region of the neuron, the synaptic terminal contains more mitochondria, where they are required for powering neurotransmission through the production of ATP, and for maintaining Ca2+ buffering. Neuronal mitochondria are dynamic, undergoing bidirectional movement in sync with neuronal processes; about one-third of mature mitochondria in axons are motile. Long-distance axonal transport is supported by motor proteins and microtubules. Both anterograde and retrograde transportation of mitochondria occur more often during development and become less frequent with age [76].
For proper synaptic function, stationary mitochondrial presence is required at regions with high energy requirements. Mitochondrial distribution and transport are highly correlated with local energy changes and metabolic demands, which is regulated by synaptic activation [77]. Sustained synaptic activity causes elevation of intracellular Ca2+ levels, and increased Ca2+ levels further inhibit mitochondrial movement either by activating voltage dependent Ca2+ channels or N-methyl-D-aspartate (NMDA) receptors. Docked mitochondria serve as stationary power plants, and for sequestering intracellular Ca2+ to sustain Ca2+ homeostasis [78,79].
Human brain circuitry is comprised of trillions of neurons and quadrillion synapses, and this connectivity underlies all human perception, emotion, thought and behavior. Neural circuits in response to a variety of stimuli undergo extensive sculpting and rewiring; this process of changes in synaptic connections in response to experience is called synaptic plasticity [80]. During synaptic activation and long-term potentiation, many studies reported changes in mitochondria, including increased Ca2+ pump activity, altered mitochondrial energy production, and increased mitochondrial gene expression [81,82,83,84]. Pharmacological studies revealed that inhibition of mitochondrial activity results in impairment of synaptic potentiation and neurotransmission [85,86].
An early and essential step for the formation of neuronal circuits is the establishment of neuronal polarity. Depletion of mitochondria from cells prevents axon formation, and even maintaining cellular ATP levels does not rescue this effect, suggesting a mechanism other than the ATP-generating function of mitochondria [87].
Maintenance of an appropriate pool of healthy mitochondria throughout the life of a neuron is extremely important. The challenge for neurons is the maintenance of a large number of neuronal mitochondria that require constant rejuvenation, even though their residence is far removed from the soma. One way neurons overcome this is by axonal protein synthesis mediated by soma-to-axon delivery of mRNAs. Intra axonal translation is necessary for regulation of growth cones, persynaptic plasticity, and injury response [88]. The majority of proteins generated by axonal translation including CoxIV and Atp5g1 are normally targeted to mitochondria [89,90]. In the human substantia nigra, a single neuron has been estimated to generate 1 to 2 million striatal synapses; this indicates that each cell has ~2 million mitochondria, throughout the several meters of axons [91,92,93]. The functional properties and behavior of mitochondria vary in axons and dendrites. In cultured hippocampal neurons, axons have twice as many motile mitochondria in comparison to dendrites, whereas dendrites have a higher proportion of highly charged mitochondria that are metabolically more active [94,95].
3. Mitochondrial Dysfunction in Common Neurodegenerative Diseases
As a dynamic organelle, the mitochondrion constantly undergoes fusion, fission, and transport in a regulated fashion. Increasing evidence of alteration in trafficking and fusion–fission dynamics in AD, Parkinson’s disease (PD), Huntington’s disease (HD), and ALS have been shown. In AD brains and peripheral cells derived from patients, energy deficiency is the fundamental characteristic feature. A reduction in the activity of OXPHOS complexes I, III, and IV are reported in the platelets and lymphocytes of AD patients and postmortem brain samples [96,97]. DRP1, a protein involved in mitochondrial fission, is reported to be abnormally expressed in postmortem brain samples of AD patients [98]. In PD patients, a mild deficiency in the mitochondrial electron transport chain (Complex I) is reported in the substantia nigra, and the inhibition of complex 1 in a mouse model causes similar behavioral and neuropathological symptoms as PD [99]. Many PD-linked proteins including Parkin, α-synuclein, PINK1, DJ-1, and leucine-rich repeat kinase 2, are associated with either mitochondria or mitochondrial proteins [100]. HD is caused by an abnormal expansion of CAG repeats in the HTT gene. There is extensive evidence for defects in bioenergetics and mitochondrial dysfunction in HD; for example, weight loss despite required caloric intake, increased lactate in the cerebral cortex and basal ganglia when observed using NMR spectroscopy, and reduced activity of the OXPHOS complexes II and III observed in cortical biopsies obtained from patients with both adult and juvenile onset HD. Besides, the number of mitochondria in HD postmortem brain tissue is reduced in mutant Htt knock-in mice [101].
4. Impaired Mitochondrial Dynamics and Plasticity in MND Pathogenesis
Many proteins linked to both familial and sporadic ALS and FTD are shown to be associated with mitochondria, including FUS, TDP-43, SOD1, and C9ORF72. Furthermore, the interactions of these proteins with mitochondria appear to be a key step in mitochondrial damage associated with ALS and FTD.
The pathology of TDP-43 is comprised of ubiquitination, hyperphosphorylation, and proteolytic cleavage of TDP-43, which leads to 35-kDa and 25-kDa insoluble aggregates, which are major components of inclusion bodies [102]. In cultured neural stem cells (NSC34 line), overexpression of full-length TDP-43 or its C-terminal fragment (25 kDa) induces mitophagy. In some mouse models of TDP-43, distinct changes in mitochondrial dynamics and aggregation are reported [103,104]. Mitochondrial dynamics are coordinated by regulators of fusion, which include mitofusin 1 and 2 (MFN1 and MFN2), along with dynamin-related protein 1 (DRP1), which is a regulator of fission [105]. Overexpression of either wild-type TDP-43 or the mutant form causes impairment of mitochondrial morphology and movement; overexpression of MFN2, which forms a complex with TDP-43, is shown to rescue TDP-43-induced mitochondrial dysfunction [106]. Selective expression of TDP-43 in the cortex and hippocampus of 4-month-old mice resulted in an increase in phosphorylation on the serine at position 637 of the DRP1 protein. This causes a decrease in fission and underscores a role of TDP-43 in fission dynamics (Figure 1) [107].
Full-length TDP-43 is shown to be localized to the mitochondrial fraction upon stress in autosomal dominant ALS. In mitochondria, both wild-type and mutant TDP-43 bind preferentially to the mitocondrial-transcribed mRNAs ND3 and ND6, which encode subunits of complex I and inhibit oxidative phosphorylation (Figure 2). Surprisingly, suppression of TDP-43 localization to the mitochondria abolishes the toxicity of mutant and wild-type TDP-43 in mitochondria and neurons in ALS transgenic mice, and resulted in improved motor function [108]. In contrast, studies in TDP-43 A315T mutant mice and TDP-43 mutant fibroblasts revealed no impairment in mitochondrial bioenergetics [109]. Increased TDP-43 expression, due to either mutation or cellular stresses, causes the activation of the mitochondrial unfolded protein response (UPRmt) LonP1 which is a key mitochondrial protease involved in UPRmt and plays an important role in the degradation of mitochondrial TDP-43. Downregulation of LonP1 expression in TDP-43 expressing flies results in more severe mitochondrial damage and advances disease onset [110]. Overexpression of wild-type and mutant TDP-43 affects endoplasmic reticulum (ER)-mitochondrial associations. TDP-43 activates glycogen synthase kinase 3β (GSK-3β), a kinase implicated in both ALS and FTD. Modulation of GSK-3β activity affects the Vesicle-associated membrane protein-associated protein B/C-Protein tyrosine phosphatase interacting protein 51 (VAPB-PTPIP51) interaction, inhibition of GSK-3β causes an increase whereas overexpression of GSK-3β causes a decrease in the VAPB-PTPIP51 interaction. Ca2+ homeostasis is affected by TDP-43 inhibition of the VAPB-PTIP51 interaction, leading to a decrease in mitochondrial Ca2+, which is essential for many pathways ranging from ATP synthesis to mitochondrial biogenesis (Figure 3) [111,112,113]. Ca2+ dysregulation caused by miscommunication between the ER and mitochondria may be considered to be a primary cause of motor neuron death in ALS [114].
FUS is reported to alter mitochondrial dynamics in various studies; an electron microscope study of two spinal cord samples of ALS-FUS reported that the disorganization of mitochondria and the ER in one sample corresponded to the P525L mutation [115]. Expression of ALS-associated FUS mutants R521G or R521H in the cytoplasm are associated with shortened mitochondria in cultured motor neurons [116]. Motor neurons derived from iPSCs of ALS patients harboring FUS R521H and P525L exhibited axonal transport defects, progressive ER-mitochondrial vesicle transport defects, and reduction in mitochondria-associated membranes. These defective phenotypes are shown to be rescued by the use of HDAC6 inhibitors or by genetically knocking down HDAC6 [117]. Although it was shown previously that the TDP-43-FUS complex regulates the mRNA expression of HDAC6, no difference in HDAC6 expression is reported in motor neurons derived from FUS mutant iPSC line. Both wild-type and mutant (P525L) FUS interact with HSP60. Elevated HSP60 expression was seen in brain tissue specimens from two out of three FTD-FUS patients. Knocking down HSP60 led to a reduction in mitochondrial FUS levels in cultured cells. Increased localization of FUS to mitochondria, due to either cellular stresses or pathogenic FUS mutations, leads to moderate increase in Fis1 levels leading to mitochondrial fragmentation, loss of membrane potential, increase in reactive oxygen species (ROS) production and defective mitochondrial axonal transport [118]. FUS, when accumulated inside mitochondria, interacts with mitochondrial ATP synthase catalytic subunit ATP5B and reduces mitochondrial ATP synthesis. Accumulation of both wild-type FUS and the ALS-associated P525L mutant disrupts the formation of ATP synthase complex and suppresses the activity of ATP synthase, leading to loss of mitochondrial cristae, and thereby causing mitochondrial fragmentation (Figure 2) [119]. Expression of both wild-type and ALS-associated mutant FUS disrupts ER-mitochondrial associations. FUS activates GSK-3β which is a known regulator of the ER-mitochondria association, which causes reduction of the interaction between VAPB and PTPIP51 (Figure 3) [120].
In primary cortical and motor neuron cultures, the ALS SOD1 G93A mutation selectively reduces mitochondrial anterograde transport [121]. Transgenic mice and rats with the SOD1 G93A mutation display defective anterograde and retrograde axonal transport of mitochondria, which is correlated with reduced levels of the outer mitochondrial membrane protein Miro1, a master regulator of mitochondrial axonal transport in response to cytosolic Ca2+ levels. The ALS SOD1 mutant inhibits axonal transport of mitochondria by Miro1 degradation, by inducing the Parkin/PINK1 dependent pathway [122]. SOD1 G93A forms high molecular weight aggregates and causes elevated oxidative lipid and protein damage in mitochondria; impairment of Ca2+ handling is also a common feature. These types of mitochondrial damage activate the PINK1/Parkin pathway, which halts mitochondrial transport [123]. The spinal cords of SOD1 G93A transgenic mice show progressively low levels of Mfn1 and Opa1, which are important for fusion; however, the levels of activated Drp1 and Fis1, which are important for fission, remain stable, thus disrupting the balance between fission and fusion (Figure 1) [123]. Mutant SOD1 forms a complex with BCL-2, binds to the cytoplasmic-facing domain of VDAC1, a mitochondrial porin located on the outer membrane of mitochondria that is important for metabolic crosstalk between mitochondria and rest of the cell. Binding of misfolded SOD1 to VDAC1 leads to inhibition of conductance (Figure 3) [124]. Reduced activity of respiratory chain complexes II and IV are observed in NSC34 cells expressing the SOD1 mutants G93A and G37R [125]. In G93A spinal cord mitochondria, impaired Ca2+ handling occurs very early on in the course of disease, and precedes respiratory chain defects, whereas in G85R hSOD1 expressing transgenic mice, Ca2+ capacity impairment occurs in the absence of oxidative phosphorylation dysfunction (Figure 2). This suggests that impairment of Ca2+ capacity may occur upstream of the cascade of events that leads to mitochondrial dysfunction in familial ALS [126].
Poly (GR), which results from repeat expansion in C9ORF72, favorably binds to mitochondrial ribosomal protein and compromises mitochondria function, which further leads to increased oxidative stress in neurons [127]. An altered balance between fission and fusion processes is evident in ALS and FTD patient fibroblasts carrying C9ORF72 mutation (mtC9ORF72) showed increased MFN1 levels and alterations in mitochondrial morphology (Figure 1) [128]. In mtC9ORF72 fibroblasts, the median mitochondrial membrane potential value increased significantly, suggesting hyperpolarized mitochondria. An increase in mitochondrial DNA content and mass is seen in mtC9ORF72 fibroblasts. Fragmentation of the mitochondrial network, deformation, and loss of mitochondrial cristae is observed in fibroblasts of ALS patients with repeat expansions [129]. In motor neurons derived from C9ORF72-associated ALS patient iPSC lines, swollen mitochondria were reported. Altered mitochondrial morphology and structural abnormalities in cristae are seen in 88% of C9ORF72 neurons in comparison to 2% of healthy neurons [128,130].
5. Mitochondrial Genome Instability in MND: Potential Role of TDP-43, FUS, and C9ORF72
Mitochondrial biogenesis and function require coordinated action of both nuclear and mitochondrial genomes. Mammalian mitochondria DNA is a circular molecule with 16,569 base pairs and can represent approximately 1% of total DNA in some cells. Mitochondrial DNA replication is not limited to the S phase but can occur throughout the cell cycle. Similar to nuclear DNA, mitochondrial DNA is constantly exposed to both external and internal DNA damaging agents, but mitochondrial DNA is generally more susceptible to DNA damaging agents as it is not protected by chromatinization and also lack some DNA repair factors [131]. Mitochondrial DNA is modified by oxidation and alkylation ~10 fold more than nuclear DNA [132,133,134,135]. In addition, hydrolytic damage, adduct formation, mismatched bases, and formation of single- and double-stranded breaks are reported in mitochondria [131,136,137,138]. Various DNA repair pathways have been reported in mitochondria, including base excision repair (BER), translesion DNA synthesis (TLS), homologous recombination (HR), non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and the novel mismatch repair (MMR) pathway distinctive from nuclear MMR [139,140,141,142,143,144,145,146], although the level of proficiency of each of these pathways is not known. The nucleotide excision repair (NER) and Fanconi anemia (FA) pathways are not reported in mitochondria, but evidence of mitochondrial localization of multiple proteins involved in these pathways has been reported (Table 1) [147,148].
The ALS proteins TDP-43, FUS, and C9ORF72 were recently shown to be important for maintaining nuclear genomic stability due to their involvement in DNA damage repair. For example, FUS is recruited to the DNA damage site in a PARP1-dependent manner, where it recruits XRCC1/LIG3 and is required for efficient function of LIG3 [31]. Our laboratory identified TDP-43 as a key component of NHEJ-mediated DNA double-strand break repair machinery, where it acts as a scaffold for the recruitment of XRCC4-LIG4 [149]. Furthermore, C9ORF72 iPSC differentiated motor neurons show increased oxidative stress and DNA damage in an age-dependent manner [129]. Although the roles of these proteins in mitochondrial dynamics and plasticity have been explored, their emerging DNA damage response role in the nuclear genome raises the question of whether these proteins are also important for the maintenance of genomic stability in mitochondria. It is important to note that mitochondria have a limited number of back up DNA repair pathways, unlike the nucleus. For example, LIG3 (mitochondrial isoform) is the only ligase in mitochondria that supports both its genome replication and repair. Furthermore, although XRCC1 acts as a scaffold for LIG3 in the nuclear genome, studies have reported that XRCC1 is not localized in mitochondria [150]. This raises the question of whether FUS replaces XRCC1 as a stabilizing scaffold for mitochondrial LIG3. Furthermore, PARP-1 also localizes to mitochondria, whereas the role of PARylation in mitochondrial genome repair is still emerging [121,122,123]. Interestingly, recent studies suggested that XRCC4 may localize to mitochondria, where it may collaborate with LIG3 for mitochondria DNA double-strand break repair [151]. In view of our recent discovery of TDP-43′s association with XRCC4, it is likely that TDP-43 function in mitochondrial DNA repair by recruiting the XRCC4-LIG3 complex [149,151]. The potential role of TDP-43 and FUS in maintaining mitochondrial genomic stability requires further investigation.
6. Effect of Mitochondrial Dysfunction beyond Mitochondria in the CNS
Although mitochondria act as major source of energy, other functions performed by these organelles are equally important for the cell. In the CNS, mitostasis assumes greater importance, because the brain consumes a significant amount of oxygen, and thus generates higher levels of ROS, which are reported in many neurological diseases including ALS, AD, PD, and dementia [185,186]. Ca2+ homeostasis is also important for various CNS functions including neurotransmission, along with ER-mitochondrial function, in regulating Ca2+ homeostasis.
Mitochondrial oxidative phosphorylation is believed to be a major source of cellular ROS in most cell types [187]. Particularly in neurons, mitochondria act as a primary site of ROS generation. Outside mitochondria there are other enzymes, which are associated with ROS generation including NADPH oxidase, neural nitric oxide synthase and monoamine oxidase [188,189,190,191,192]. In moderate or low levels, ROS are important for neuronal function and development, but excessive levels are toxic [193,194]. Oxidative stress plays an important role in the development of various diseases ranging from cancer to autoimmune disorders. With increasing age, the redox system becomes imbalanced, leading to elevated levels of reactive oxygen and nitrogen species. It has been shown that different regions of the brain have different levels of vulnerability for oxidative stress. For example the hippocampus, cerebellar granule cells, and amygdala have been shown to be most susceptible to oxidative stress, and are consequently the first to undergo functional decline [195]. Neurons, which carry out dopamine metabolism, are more susceptible to ROS-induced damage; this is also the case for neurons with high metal content [196]. The precise chain of events occurring within the CNS that leads to oxidative stress-induced cognitive or behavioral decline needs to be understood at multiple levels. Apart from affecting several essential mitochondrial functions, including various metabolic functions such as fatty acid oxidation, heme synthesis, amino acid metabolism, and the urea cycle, oxidative damage increases the mitochondrial tendency to release cytochrome c, leading to activation of apoptosis [197].
Overexcitation of NMDA receptors by glutamate is thought to be a key driver of neuronal damage after stroke or neurodegenerative diseases. NMDA receptor activates neurotoxicity by activating nitric oxide synthase which results in nitric oxide (NO) production. Furthermore, peroxynitrites are formed in mitochondria due to presence of superoxides; these lead to DNA damage and PARP-1 activation, which plays a key role in NMDA excitotoxicity [198]. Apoptosis-inducing factor (AIF) is normally localized in mitochondria; following PARP-1 activation, AIF translocates to the nucleus, triggering nuclear shrinkage, chromatin condensation, and DNA fragmentation [199].
Physical and functional coupling between the ER and mitochondria plays an important role in a variety of cellular pathways, and alterations in this coupling are associated with several diseases, including ALS, AD, and PD [200,201]. The ER-mitochondria contact is important in multiple pathways including cell proliferation, death, autophagy, lipid metabolism, Ca2+ signaling, the unfolded protein response, inflammation, and bioenergetics. Motor neurons have multiple properties that make them more vulnerable to Ca2+ dysregulation when compared to other neuronal populations. They have a high number of AMPA receptors which are permeable to Ca2+ at the postsynaptic terminal, resulting in greater vulnerability to excitotoxicity during excitatory neurotransmission events [202]. Motor neurons have decreased cytosolic buffering capacity because they express low levels of Ca2+ buffering proteins such as calbindin and parvalbumin, which render them more dependent on mitochondrial Ca2+ buffering [203,204]. The Ca2+ level in mitochondria regulates ATP production by activating rate limiting enzymes of the Krebs cycle. Decreased ATP levels directly impact the axonal transport of mitochondria and other vesicles important for neuronal function. Elevated cytosolic Ca2+ levels due to defective ER-mitochondria function leads to disruption of mitochondrial axonal transport. Excessive Ca2+ interacts with Miro 1 and prevents its association with mitochondrial KIF5, leading to defects in axonal transport and reduced local ATP levels [79,205].
In ALS, over expression of both wild-type and mutated TDP-43 leads to a decrease in ER-mitochondria physical and functional coupling. TDP-43 perturbs the interaction between the outer mitochondrial membrane protein, protein tyrosine phosphatase-interacting protein-51 (PTPIP51), and resident ER protein vesicle-associated membrane protein associated protein-B (VAPB) by activating GSK-3β (Figure 3) [111]. FUS overexpression is associated with impairment of ER-mitochondria juxtaposition, Ca2+ transfer, and mitochondrial ATP production [120]. Over expression of the ALS-associated SOD1 G93A mutation leads to altered Ca2+ homeostasis. Taken together, these studies indicate that alteration of ER-mitochondrial functional coupling could be an early event in ALS and FTD [126]. Apart from neuronal diseases, alterations in ER-mitochondrial interactions have been shown to be associated with metabolic defects such as obesity and insulin resistance. Ca2+ acts as a mechanistic link between metabolic impairment and ER-mitochondrial interaction. Increasing evidence has been reported on the ability of cancer cells to remodel their intracellular Ca2+ signaling, as it favors their survival and proliferation. Modulation of ER-mitochondrial Ca2+ crosstalk enhances resistance to apoptosis [206].
7. Conclusions and Perspectives
Although MNDs were first reported more than a century ago, there is yet no effective treatment. Two drugs, namely Riluzole and Edaravone, are currently approved by FDA for use in ALS. The mechanism of action of Riluzole is not very clear, but some studies point to its antioxidant activity by induction of glutathione synthesis, which can only extend survival by 3 months. Edaravone acts as a free radical scavenger [207]. Overwhelming evidence suggests that mitochondrial dysfunction plays a significant role in proteinopathies of neurodegenerative diseases. In ALS, TDP-43 and FUS are reported to be recruited in mitochondria; toxic or mutant forms of these proteins lead to defects in mitochondrial function and mitostasis. Although recent studies provide some insights into how these proteins may affect mitochondrial function, a comprehensive understanding of how these proteins are recruited to mitochondria, their differential regulation in normal versus affected neurons, and the physiological as well as pathological importance of these proteins in mitochondria are required to develop effective mechanism-based therapeutics. Many of the strategies used to develop therapeutic interventions against imbalances in mitochondria focus on antioxidant therapy. Metabolic intermediates such as creatine and Coenzyme Q10, although promising in animal studies, were not effective in clinical trials [208,209,210,211]. Many drugs and gene therapy-based approaches, targeted to improve mitochondrial function, have also not shown promise in trials. Free radical scavengers such as Ederavone only had a marginal effect on disease progression. The inconsistency between mouse models and clinical trials raises an essential need to generate clinically relevant disease models [212,213]. Furthermore, there are variations in the type of mitochondrial dysfunction caused by each protein, suggesting that a single form of therapy will not effectively attenuate all these different aspects. Thus, it is important to identify critical pathways and mechanisms of mitochondrial dysfunction during disease initiation and progression. Future studies should also focus on common mechanisms among various neurodegenerative conditions, which will enhance our understanding of essential requirements for neuronal survival.
Author Contributions
M.K. and H.W. prepared the original draft and contributed to final formatting. M.L.H. developed the concept, edited and finalized the manuscript. M.L.H. is responsible for funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
Research in the Hegde laboratory is supported by grants from the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute of Ageing (NIA) of the National Institutes of Health (NIH) (award numbers R01NS088645, RF1NS112719, R03AG064266), and Houston Methodist Research Institute funds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Acknowledgments
The authors thank other members of the Hegde laboratory, Joy Mitra, and Pavana Hegde for assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Motor Neuron Disease Collaborators. Global, regional, and national burden of motor neuron diseases 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1083–1097. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Robberecht, W. Recent advances in motor neuron disease. Curr. Opin. Neurol. 2009, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, J.; de Visser, M.; Linssen, W.H.; de Haan, R.J.; Schmand, B. The cognitive profile of amyotrophic lateral sclerosis: A meta-analysis. Amyotroph. Lateral Scler 2010, 11, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Lomen-Hoerth, C.; Murphy, J.; Langmore, S.; Kramer, J.H.; Olney, R.K.; Miller, B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 2003, 60, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Mendez, E.F.; Sattler, R. Biomarker development for C9orf72 repeat expansion in ALS. Brain Res. 2015, 1607, 26–35. [Google Scholar] [CrossRef]
- Manjaly, Z.R.; Scott, K.M.; Abhinav, K.; Wijesekera, L.; Ganesalingam, J.; Goldstein, L.H.; Janssen, A.; Dougherty, A.; Willey, E.; Stanton, B.R.; et al. The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotroph. Lateral Scler 2010, 11, 439–442. [Google Scholar] [CrossRef]
- Mitsuyama, Y. Presenile dementia with motor neuron disease. Dementia 1993, 4, 137–142. [Google Scholar] [CrossRef]
- Hudson, A.J. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: A review. Brain 1981, 104, 217–247. [Google Scholar] [CrossRef]
- Achi, E.Y.; Rudnicki, S.A. ALS and Frontotemporal Dysfunction: A Review. Neurol. Res. Int. 2012, 2012, 806306. [Google Scholar] [CrossRef]
- Kuzuhara, S.; Kokubo, Y.; Sasaki, R.; Narita, Y.; Yabana, T.; Hasegawa, M.; Iwatsubo, T. Familial amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Kii Peninsula of Japan: Clinical and neuropathological study and tau analysis. Ann. Neurol. 2001, 49, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Cudkowicz, M.; Ascherio, A. Prospective study of cigarette smoking and amyotrophic lateral sclerosis. Am. J. Epidemiol. 2004, 160, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Vergara, X.; Kheifets, L.; Greenland, S.; Oksuzyan, S.; Cho, Y.S.; Mezei, G. Occupational exposure to extremely low-frequency magnetic fields and neurodegenerative disease: A meta-analysis. J. Occup. Environ. Med. 2013, 55, 135–146. [Google Scholar] [CrossRef]
- Kamel, F.; Umbach, D.M.; Lehman, T.A.; Park, L.P.; Munsat, T.L.; Shefner, J.M.; Sandler, D.P.; Hu, H.; Taylor, J.A. Amyotrophic lateral sclerosis, lead, and genetic susceptibility: Polymorphisms in the delta-aminolevulinic acid dehydratase and vitamin D receptor genes. Environ. Health Perspect 2003, 111, 1335–1339. [Google Scholar] [CrossRef]
- Mattsson, P.; Lonnstedt, I.; Nygren, I.; Askmark, H. Physical fitness, but not muscle strength, is a risk factor for death in amyotrophic lateral sclerosis at an early age. J. Neurol. Neurosurg. Psychiatry 2012, 83, 390–394. [Google Scholar] [CrossRef]
- Morozova, N.; Weisskopf, M.G.; McCullough, M.L.; Munger, K.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Diet and amyotrophic lateral sclerosis. Epidemiology 2008, 19, 324–337. [Google Scholar] [CrossRef]
- Berger, M.M.; Kopp, N.; Vital, C.; Redl, B.; Aymard, M.; Lina, B. Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS. Neurology 2000, 54, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Charcot, J.M. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere. Archives Pathologie Normale Pathologique 1869, 2, 744–760. [Google Scholar]
- Pochet, R. Genetics and ALS: Cause for Optimism. Cerebrum 2017, 2017, cer-05-17. [Google Scholar]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Lill, C.M.; Abel, O.; Bertram, L.; Al-Chalabi, A. Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotroph. Lateral Scler 2011, 12, 238–249. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- van der Zee, J.; Gijselinck, I.; Dillen, L.; Van Langenhove, T.; Theuns, J.; Engelborghs, S.; Philtjens, S.; Vandenbulcke, M.; Sleegers, K.; Sieben, A.; et al. A pan-European study of the C9orf72 repeat associated with FTLD: Geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 2013, 34, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Nilsson, P.; Forsgren, L.; Marklund, S.L. CuZn-superoxide dismutase, extracellular superoxide dismutase, and glutathione peroxidase in blood from individuals homozygous for Asp90Ala CuZu-superoxide dismutase mutation. J. Neurochem. 1998, 70, 715–720. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Zetterstrom, P.; Stewart, H.G.; Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brannstrom, T.; Oliveberg, M.; Marklund, S.L. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc. Natl. Acad. Sci. USA 2007, 104, 14157–14162. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Broustal, O.; Camuzat, A.; Guillot-Noel, L.; Guy, N.; Millecamps, S.; Deffond, D.; Lacomblez, L.; Golfier, V.; Hannequin, D.; Salachas, F.; et al. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. 2010, 22, 765–769. [Google Scholar]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Hegde, M.L. New Mechanisms of DNA Repair Defects in Fused in Sarcoma-Associated Neurodegeneration: Stage Set for DNA Repair-Based Therapeutics? J. Exp. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Sama, R.R.; Ward, C.L.; Bosco, D.A. Functions of FUS/TLS from DNA repair to stress response: Implications for ALS. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Adv. Psychopharmacol 2018, 8, 33–48. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Pathogenesis of the tauopathies. J. Mol. Neurosci. 2011, 45, 425–431. [Google Scholar] [CrossRef]
- van Swieten, J.C.; Heutink, P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008, 7, 965–974. [Google Scholar] [CrossRef]
- Gass, J.; Cannon, A.; Mackenzie, I.R.; Boeve, B.; Baker, M.; Adamson, J.; Crook, R.; Melquist, S.; Kuntz, K.; Petersen, R.; et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006, 15, 2988–3001. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.; Lippa, C.F.; Rosso, A. Progranulin (PGRN) expression in ALS: An immunohistochemical study. J. Neurol. Sci. 2009, 276, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; De Muynck, L.; Thu, H.N.; Weynants, B.; Vanacker, P.; Dhondt, J.; Sleegers, K.; Schelhaas, H.J.; Verbeek, M.; Vandenberghe, R.; et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 1191–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereson, S.; Wils, H.; Kleinberger, G.; McGowan, E.; Vandewoestyne, M.; Van Broeck, B.; Joris, G.; Cuijt, I.; Deforce, D.; Hutton, M.; et al. Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J. Pathol. 2009, 219, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu Rev. Genom. Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Atteia, A.; Adrait, A.; Brugiere, S.; Tardif, M.; van Lis, R.; Deusch, O.; Dagan, T.; Kuhn, L.; Gontero, B.; Martin, W.; et al. A proteomic survey of Chlamydomonas reinhardtii mitochondria sheds new light on the metabolic plasticity of the organelle and on the nature of the alpha-proteobacterial mitochondrial ancestor. Mol. Biol. Evol. 2009, 26, 1533–1548. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Genova, M.L. Structural and functional organization of the mitochondrial respiratory chain: A dynamic super-assembly. Int. J. Biochem. Cell Biol. 2009, 41, 1750–1772. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Davies, K.M.; Wurm, C.A.; Spahr, H.; Bonekamp, N.A.; Kuhl, I.; Joos, F.; Polosa, P.L.; Park, C.B.; Posse, V.; et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, 11288–11293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflügers Archiv 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takuma, K.; Yan, S.S.; Stern, D.M.; Yamada, K. Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J. Pharm. Sci. 2005, 97, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Toescu, E.C.; Verkhratsky, A. The importance of being subtle: Small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 2007, 6, 267–273. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Berthold, C.H.; Fabricius, C.; Rydmark, M.; Andersen, B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J. Neurocytol. 1993, 22, 925–940. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burte, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Beckervordersandforth, R.; Ebert, B.; Schaffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 560–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993, 104, 917–927. [Google Scholar] [PubMed]
- Kaczmarek, J.S.; Riccio, A.; Clapham, D.E. Calpain cleaves and activates the TRPC5 channel to participate in semaphorin 3A-induced neuronal growth cone collapse. Proc. Natl. Acad. Sci. USA 2012, 109, 7888–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, S.B.; Pineda, F.J.; Hardwick, J.M. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008, 15, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapaport, D.; Brunner, M.; Neupert, W.; Westermann, B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 20150–20155. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Isobe, K.; Nakada, K.; Hayashi, J.I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 2001, 28, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Morsci, N.S.; Hall, D.H.; Driscoll, M.; Sheng, Z.H. Age-Related Phasic Patterns of Mitochondrial Maintenance in Adult Caenorhabditis elegans Neurons. J. Neurosci. 2016, 36, 1373–1385. [Google Scholar] [CrossRef]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Hubley, M.J.; Locke, B.R.; Moerland, T.S. The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim. Biophys. Acta 1996, 1291, 115–121. [Google Scholar] [CrossRef]
- Yi, M.; Weaver, D.; Hajnoczky, G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Malenka, R.C.; Nicoll, R.A. Long-term potentiation—A decade of progress? Science 1999, 285, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Liu, D. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem. Biophys. Res. Commun. 2003, 304, 539–549. [Google Scholar] [CrossRef]
- Wieraszko, A. Changes in the hippocampal slices energy metabolism following stimulation and long-term potentiation of Schaffer collaterals-pyramidal cell synapses tested with the 2-deoxyglucose technique. Brain Res. 1982, 237, 449–457. [Google Scholar] [CrossRef]
- Stanton, P.K.; Schanne, F.A. Hippocampal long-term potentiation increases mitochondrial calcium pump activity in rat. Brain Res. 1986, 382, 185–188. [Google Scholar] [CrossRef]
- Williams, J.M.; Thompson, V.L.; Mason-Parker, S.E.; Abraham, W.C.; Tate, W.P. Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Brain Res. Mol. Brain Res. 1998, 60, 50–56. [Google Scholar] [CrossRef]
- Alnaes, E.; Rahamimoff, R. On the role of mitochondria in transmitter release from motor nerve terminals. J. Physiol. 1975, 248, 285–306. [Google Scholar] [CrossRef]
- Mattson, M.P.; Partin, J. Evidence for mitochondrial control of neuronal polarity. J. Neurosci. Res. 1999, 56, 8–20. [Google Scholar] [CrossRef]
- Bigler, R.L.; Kamande, J.W.; Dumitru, R.; Niedringhaus, M.; Taylor, A.M. Messenger RNAs localized to distal projections of human stem cell derived neurons. Sci. Rep. 2017, 7, 611. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, P.K.; Smith, D.S.; Perrone-Bizzozero, N.; Twiss, J.L. Axonal mRNA transport and translation at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Aschrafi, A.; Natera-Naranjo, O.; Gioio, A.E.; Kaplan, B.B. Regulation of axonal trafficking of cytochrome c oxidase IV mRNA. Mol. Cell Neurosci. 2010, 43, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, G.M.; Harris, K.M. Three-dimensional structure and composition of CA3→CA1 axons in rat hippocampal slices: Implications for presynaptic connectivity and compartmentalization. J. Neurosci. 1998, 18, 8300–8310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Overly, C.C.; Rieff, H.I.; Hollenbeck, P.J. Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J. Cell Sci. 1996, 109, 971–980. [Google Scholar] [PubMed]
- Ligon, L.A.; Steward, O. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 340–350. [Google Scholar] [CrossRef]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2011, 67, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Browne, S.E.; Beal, M.F. The energetics of Huntington’s disease. Neurochem. Res. 2004, 29, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 2012, 530, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Stribl, C.; Samara, A.; Trumbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; Hrabe de Angelis, M.; Rathkolb, B.; Wolf, E.; et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Kawamata, H.; Peixoto, P.; Konrad, C.; Palomo, G.; Bredvik, K.; Gerges, M.; Valsecchi, F.; Petrucelli, L.; Ravits, J.M.; Starkov, A.; et al. Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol. Neurodegener 2017, 12, 37. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta 2009, 1787, 1324–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Zhang, J.; Geser, F.; Trojanowski, J.Q.; Strober, J.B.; Dickson, D.W.; Brown, R.H., Jr.; Shapiro, B.E.; Lomen-Hoerth, C. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 2010, 20, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.C.; Durham, H.D.; Richard, S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012, 21, 136–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.; Cho, Y.; Collins, L.B.; Swenberg, J.A.; Di Giulio, R.T. Effects of benzo[a]pyrene on mitochondrial and nuclear DNA damage in Atlantic killifish (Fundulus heteroclitus) from a creosote-contaminated and reference site. Aquat. Toxicol. 2009, 95, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Horne, D.W.; Holloway, R.S.; Wagner, C. Transport of S-adenosylmethionine in isolated rat liver mitochondria. Arch. Biochem. Biophys. 1997, 343, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, D.M.A.; Reddy, P.H. Molecular Basis of Alzheimer’s Disease: Focus on Mitochondria. J. Alzheimers Dis. 2019, 72, S95–S116. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Kasparek, T.R.; Humphrey, T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011, 22, 886–897. [Google Scholar] [CrossRef]
- Bailey, L.J.; Doherty, A.J. Mitochondrial DNA replication: A PrimPol perspective. Biochem. Soc. Trans. 2017, 45, 513–529. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef] [Green Version]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem. Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, U.; Campbell, C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999, 27, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyagarajan, B.; Padua, R.A.; Campbell, C. Mammalian mitochondria possess homologous DNA recombination activity. J. Biol. Chem. 1996, 271, 27536–27543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, G.; Lakshmipathy, U.; Campbell, C. Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic Acids Res. 1999, 27, 3348–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009, 37, 4218–4226. [Google Scholar] [CrossRef] [Green Version]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dolle, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernandez, A.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [Green Version]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.K.; Van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef]
- Wisnovsky, S.; Jean, S.R.; Kelley, S.O. Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Chem. Biol. 2016, 12, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Crivellato, E.; Pines, A.; Paron, I.; Pucillo, C.; Manzini, G.; Bandiera, A.; Kelley, M.R.; Di Loreto, C.; Damante, G. Mitochondrial localization of APE/Ref-1 in thyroid cells. Mutat. Res. 2001, 485, 143–152. [Google Scholar] [CrossRef]
- Tsuchimoto, D.; Sakai, Y.; Sakumi, K.; Nishioka, K.; Sasaki, M.; Fujiwara, T.; Nakabeppu, Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001, 29, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykora, P.; Croteau, D.L.; Bohr, V.A.; Wilson, D.M., 3rd. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 7437–7442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalifa, L.; Beutner, G.; Phadnis, N.; Sheu, S.S.; Sia, E.A. Evidence for a role of FEN1 in maintaining mitochondrial DNA integrity. DNA Repair 2009, 8, 1242–1249. [Google Scholar] [CrossRef] [Green Version]
- van Loon, B.; Samson, L.D. Alkyladenine DNA glycosylase (AAG) localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (mtSSB). DNA Repair 2013, 12, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000, 28, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.M.; Hegde, M.L.; Chatterjee, A.; Hegde, P.M.; Szczesny, B.; Banerjee, D.; Boldogh, I.; Gao, R.; Falkenberg, M.; Gustafsson, C.M.; et al. Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 2012, 287, 2819–2829. [Google Scholar] [CrossRef] [Green Version]
- Luna, L.; Bjoras, M.; Hoff, E.; Rognes, T.; Seeberg, E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. 2000, 460, 95–104. [Google Scholar] [CrossRef]
- Kang, D.; Nishida, J.; Iyama, A.; Nakabeppu, Y.; Furuichi, M.; Fujiwara, T.; Sekiguchi, M.; Takeshige, K. Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J. Biol. Chem. 1995, 270, 14659–14665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.N.; Carbone, M.; Mostocotto, C.; Mancone, C.; Tripodi, M.; Maione, R.; Amati, P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009, 284, 31616–31624. [Google Scholar] [CrossRef] [Green Version]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Gray, H.; Wong, T.W. Purification and identification of subunit structure of the human mitochondrial DNA polymerase. J. Biol. Chem. 1992, 267, 5835–5841. [Google Scholar]
- Das, B.B.; Dexheimer, T.S.; Maddali, K.; Pommier, Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 19790–19795. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997, 25, 750–755. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Li, X.; Owens, K.M.; Vanniarajan, A.; Liang, P.; Singh, K.K. Human REV3 DNA Polymerase Zeta Localizes to Mitochondria and Protects the Mitochondrial Genome. PLoS ONE 2015, 10, e0140409. [Google Scholar] [CrossRef] [Green Version]
- Dahal, S.; Dubey, S.; Raghavan, S.C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell Mol. Life Sci. 2018, 75, 1641–1655. [Google Scholar] [CrossRef]
- Sage, J.M.; Gildemeister, O.S.; Knight, K.L. Discovery of a novel function for human Rad51: Maintenance of the mitochondrial genome. J. Biol. Chem. 2010, 285, 18984–18990. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva, N.I.; Malide, D.; Burg, M.B. Mre11 is expressed in mammalian mitochondria where it binds to mitochondrial DNA. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R632–R640. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Goldstine, J.V.; Gatti, R.A. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum. Mol. Genet. 2007, 16, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, G.; Campbell, C. An alternate form of Ku80 is required for DNA end-binding activity in mammalian mitochondria. Nucleic Acids Res. 2000, 28, 3793–3800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumbo, A.C.; Cortez, E.; Rodrigues, C.A.; Henriques, M.; Porto, L.C.; Barbosa, H.S.; Carvalho, L. Mitochondrial localization of non-histone protein HMGB1 during human endothelial cell-Toxoplasma gondii infection. Cell Biol. Int. 2008, 32, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Rashid, S.; Freitas, M.O.; Cucchi, D.; Bridge, G.; Yao, Z.; Gay, L.; Williams, M.; Wang, J.; Suraweera, N.; Silver, A.; et al. MLH1 deficiency leads to deregulated mitochondrial metabolism. Cell Death Dis. 2019, 10, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Fang, H.; Chi, Z.; Wu, Z.; Wei, D.; Mo, D.; Niu, K.; Balajee, A.S.; Hei, T.K.; Nie, L.; et al. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res. 2015, 43, 5476–5488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Du, W.; Wilson, A.F.; Namekawa, S.H.; Andreassen, P.R.; Meetei, A.R.; Pang, Q. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci. Rep. 2017, 7, 45626. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.S.; Leung, K.S.; Hicks, M.J.; Hastings, P.J.; Youssoufian, H.; Plon, S.E. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 2006, 175, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, M.; Yang, X.; Lisby, M.; Mazon, G. The FANCM family Mph1 helicase localizes to the mitochondria and contributes to mtDNA stability. DNA Repair 2019, 82, 102684. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Saxena, S.; Kaushal, A.; Nagaraju, G. RAD51C/XRCC3 Facilitates Mitochondrial DNA Replication and Maintains Integrity of the Mitochondrial Genome. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Coene, E.D.; Hollinshead, M.S.; Waeytens, A.A.; Schelfhout, V.R.; Eechaute, W.P.; Shaw, M.K.; Van Oostveldt, P.M.; Vaux, D.J. Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 2005, 16, 997–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1704. [Google Scholar] [CrossRef] [Green Version]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef]
- Riederer, P.; Konradi, C.; Schay, V.; Kienzl, E.; Birkmayer, G.; Danielczyk, W.; Sofic, E.; Youdim, M.B. Localization of MAO-A and MAO-B in human brain: A step in understanding the therapeutic action of L-deprenyl. Adv. Neurol. 1987, 45, 111–118. [Google Scholar]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48. [Google Scholar] [CrossRef]
- Zhu, X.; Smith, M.A.; Honda, K.; Aliev, G.; Moreira, P.I.; Nunomura, A.; Casadesus, G.; Harris, P.L.; Siedlak, S.L.; Perry, G. Vascular oxidative stress in Alzheimer disease. J. Neurol. Sci. 2007, 257, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, M.C.W.; Garnham, N.; Sweeney, S.T.; Landgraf, M. Regulation of neuronal development and function by ROS. FEBS Lett. 2018, 592, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of Mitochondrial Reactive Oxygen Species in the Activation of Cellular Signals, Molecules, and Function. Handb. Exp. Pharm. 2017, 240, 439–456. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Lin, T.K.; Cheng, C.H.; Chen, S.D.; Liou, C.W.; Huang, C.R.; Chuang, Y.C. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef] [Green Version]
- Dawson, V.L.; Dawson, T.M. Deadly conversations: Nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 2004, 36, 287–294. [Google Scholar] [CrossRef]
- Cande, C.; Cohen, I.; Daugas, E.; Ravagnan, L.; Larochette, N.; Zamzami, N.; Kroemer, G. Apoptosis-inducing factor (AIF): A novel caspase-independent death effector released from mitochondria. Biochimie 2002, 84, 215–222. [Google Scholar] [CrossRef]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; Vandenberghe, W.; Klaassen, H.; Van Houtte, E.; Robberecht, W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Ho, B.K.; Mohamed, A.H.; La Bella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1994, 36, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Ince, P.; Stout, N.; Shaw, P.; Slade, J.; Hunziker, W.; Heizmann, C.W.; Baimbridge, K.G. Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol. Appl. Neurobiol. 1993, 19, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Sheng, Z.H. Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron 2009, 61, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta 2014, 1843, 2184–2194. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, Z.F.; Liu, W.; Xu, B.; Yang, H.B.; Wei, Y.G. Riluzole-triggered GSH synthesis via activation of glutamate transporters to antagonize methylmercury-induced oxidative stress in rat cerebral cortex. Oxid. Med. Cell Longev. 2012, 2012, 534705. [Google Scholar] [CrossRef] [Green Version]
- Klivenyi, P.; Ferrante, R.J.; Matthews, R.T.; Bogdanov, M.B.; Klein, A.M.; Andreassen, O.A.; Mueller, G.; Wermer, M.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat. Med. 1999, 5, 347–350. [Google Scholar] [CrossRef]
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. A clinical trial of creatine in ALS. Neurology 2004, 63, 1656–1661. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J. Bioenerg. Biomembr. 2004, 36, 381–386. [Google Scholar] [CrossRef]
- Reddy, A.P.; Reddy, P.H. Mitochondria-Targeted Molecules as Potential Drugs to Treat Patients With Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 173–201. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Iwasaki, Y. Edaravone, a Free Radical Scavenger, Delayed Symptomatic and Pathological Progression of Motor Neuron Disease in the Wobbler Mouse. PLoS ONE 2015, 10, e0140316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benatar, M. Lost in translation: Treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 2007, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mitochondrial fission and fusion defects in motor neuron disease and plausible mechanisms. Mitochondrial fusion is maintained at outer mitochondrial membrane by Mfn1/2 and Opa1 mediates inner mitochondrial membrane fusion. Mutated C9ORF72 affects mitochondrial fusion by increasing Mfn1 levels leading to elongated mitochondria, mutated SOD1 affects both fusion and fission by causing increase in DRP1 levels and decreasing Mfn1 and Opa1 levels, contributing to fragmented mitochondrial morphology. TDP-43 and FUS pathology cause increase in Fis1 levels leading to fragmented mitochondrial morphology. Fis1: Mitochondrial fission 1 protein, Mfn1/2: Mitofusin1 and 2, Opa1: Opa1 Mitochondrial dynamin-like GTPase, DRP1: Dynamin-1-lrelated protein, TDP-43: TAR DNA-binding protein 43, FUS: Fused in sarcoma, SOD1: Superoxide dismutase 1.
Figure 1.
Mitochondrial fission and fusion defects in motor neuron disease and plausible mechanisms. Mitochondrial fusion is maintained at outer mitochondrial membrane by Mfn1/2 and Opa1 mediates inner mitochondrial membrane fusion. Mutated C9ORF72 affects mitochondrial fusion by increasing Mfn1 levels leading to elongated mitochondria, mutated SOD1 affects both fusion and fission by causing increase in DRP1 levels and decreasing Mfn1 and Opa1 levels, contributing to fragmented mitochondrial morphology. TDP-43 and FUS pathology cause increase in Fis1 levels leading to fragmented mitochondrial morphology. Fis1: Mitochondrial fission 1 protein, Mfn1/2: Mitofusin1 and 2, Opa1: Opa1 Mitochondrial dynamin-like GTPase, DRP1: Dynamin-1-lrelated protein, TDP-43: TAR DNA-binding protein 43, FUS: Fused in sarcoma, SOD1: Superoxide dismutase 1.

Figure 2.
Motor neuron disease factors are linked to mitochondrial unfolded protein response (UPRmt) and defective electron transport chain. For ATP generation, proper functioning of electron transport chain is important. Mutated SOD1 (mtSOD1) was shown to cause inhibition of VDAC channel leading to polarity defects. FUS and TDP-43 pathology were reported to inhibit electron transport chain leading to decrease in ATP production and UPRmt. Accumulation of both wild-type FUS and the amyotrophic lateral sclerosis (ALS)-associated P525L mutant disrupts the formation of ATP synthase complex and suppresses the activity of ATP synthase by interacting with ATP5B, leading to loss of mitochondrial cristae, and thereby causing mitochondrial fragmentation. ATP5B: ATP synthase subunit beta, VDAC: Voltage-dependent anion-selective channel protein, ANT1: ADP/ATP translocase 1.
Figure 2.
Motor neuron disease factors are linked to mitochondrial unfolded protein response (UPRmt) and defective electron transport chain. For ATP generation, proper functioning of electron transport chain is important. Mutated SOD1 (mtSOD1) was shown to cause inhibition of VDAC channel leading to polarity defects. FUS and TDP-43 pathology were reported to inhibit electron transport chain leading to decrease in ATP production and UPRmt. Accumulation of both wild-type FUS and the amyotrophic lateral sclerosis (ALS)-associated P525L mutant disrupts the formation of ATP synthase complex and suppresses the activity of ATP synthase by interacting with ATP5B, leading to loss of mitochondrial cristae, and thereby causing mitochondrial fragmentation. ATP5B: ATP synthase subunit beta, VDAC: Voltage-dependent anion-selective channel protein, ANT1: ADP/ATP translocase 1.

Figure 3.
TDP-43, FUS, and SOD1 toxicities cause impaired Endoplasmic reticulum- mitochondrial contacts and defective mitochondrial transport. Exchange of Ca2+ between ER and mitochondria is important for maintaining Ca2+ homeostasis. Mutant FUS and TDP-43 pathology activate GSK3β, which inhibits VAPB-PTPIP51 interaction leading to decrease in contact between ER-mitochondria. SOD1 mutant inhibits VDAC channels and inhibit mitochondrial import of Ca2+. Contact disruption leads to increase in cytoplasmic Ca2+ levels which leads to disruption of various cellular pathways including mitochondrial transport. IP3R: Inositol triphosphate receptor, VAPB: Vesicle-associated membrane protein-associated protein B/C, MFN: Mitofusin, VDAC: Voltage-dependent anion channel, PTPIP51: Protein tyrosine phosphatase interacting protein 51, GSK3β: Glycogen Synthase Kinase 3 Beta, ER: Endoplasmic reticulum, OMM: Outer mitochondrial membrane.
Figure 3.
TDP-43, FUS, and SOD1 toxicities cause impaired Endoplasmic reticulum- mitochondrial contacts and defective mitochondrial transport. Exchange of Ca2+ between ER and mitochondria is important for maintaining Ca2+ homeostasis. Mutant FUS and TDP-43 pathology activate GSK3β, which inhibits VAPB-PTPIP51 interaction leading to decrease in contact between ER-mitochondria. SOD1 mutant inhibits VDAC channels and inhibit mitochondrial import of Ca2+. Contact disruption leads to increase in cytoplasmic Ca2+ levels which leads to disruption of various cellular pathways including mitochondrial transport. IP3R: Inositol triphosphate receptor, VAPB: Vesicle-associated membrane protein-associated protein B/C, MFN: Mitofusin, VDAC: Voltage-dependent anion channel, PTPIP51: Protein tyrosine phosphatase interacting protein 51, GSK3β: Glycogen Synthase Kinase 3 Beta, ER: Endoplasmic reticulum, OMM: Outer mitochondrial membrane.


{kind=link}
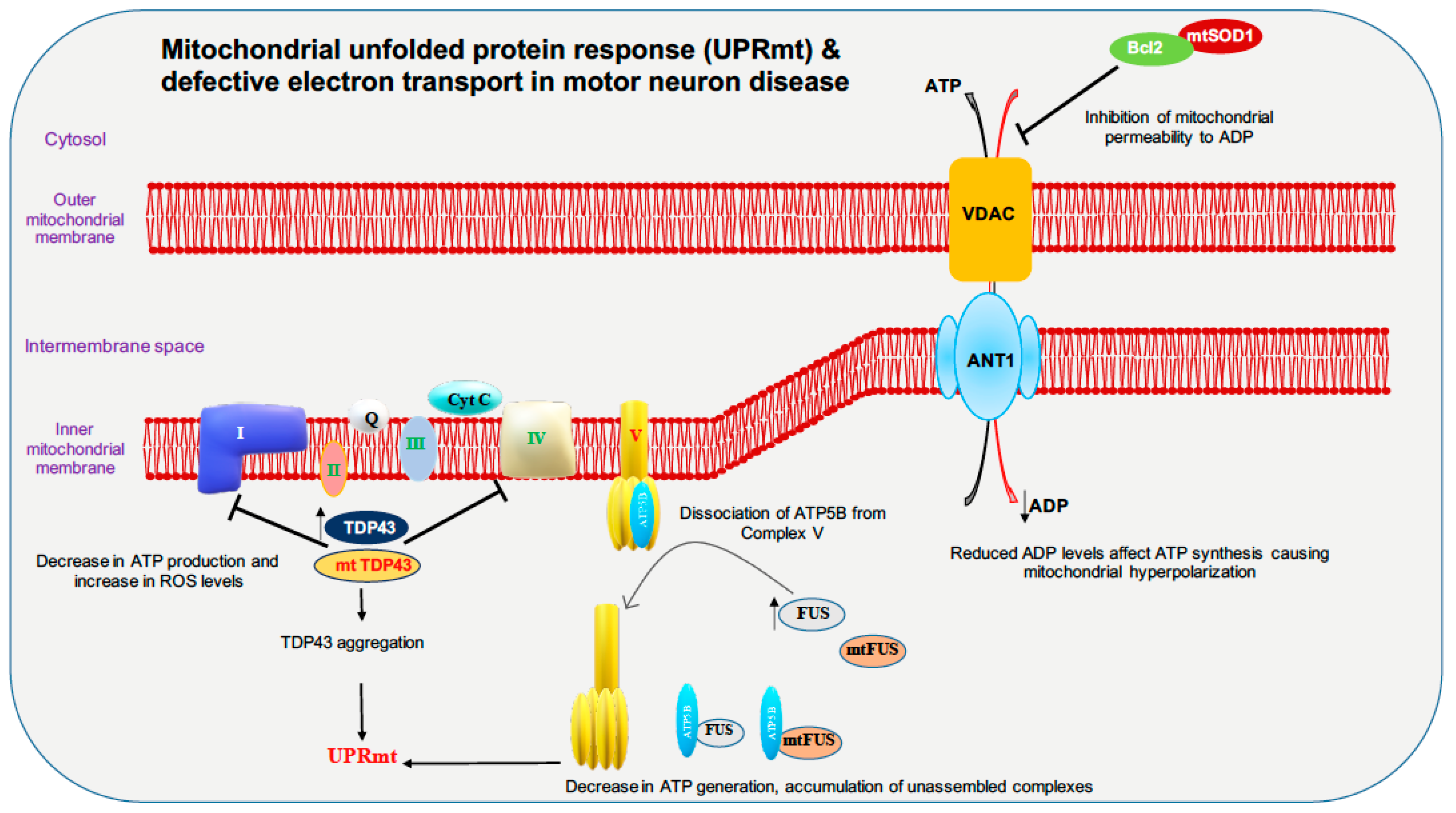{kind=link}
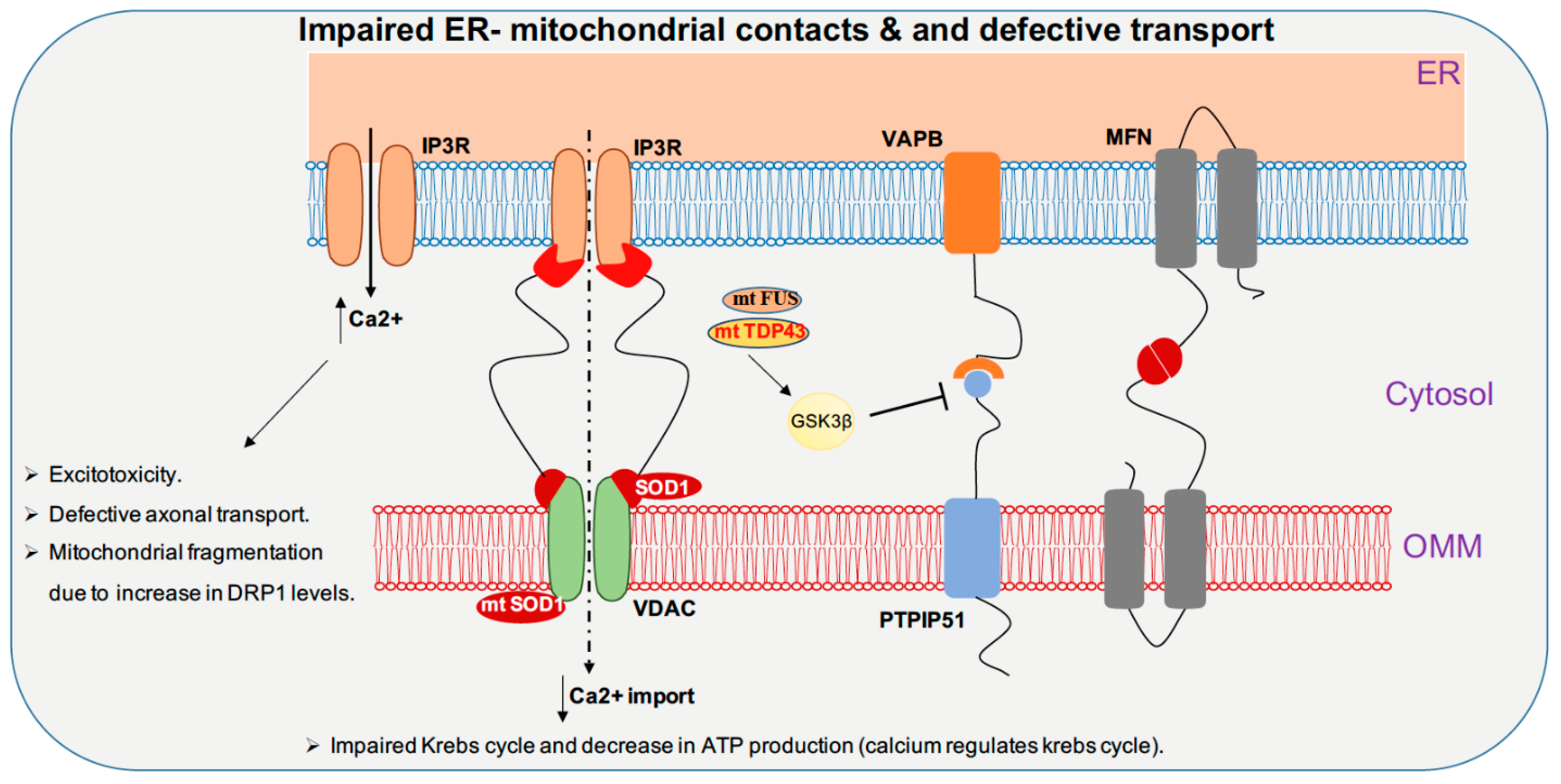{kind=link}
Table 1.
DNA repair proteins and their associated pathways reported to be localized in mitochondria.
Table 1.
DNA repair proteins and their associated pathways reported to be localized in mitochondria.
| DNA Repair Pathway | Target Lesions | Repair Protein(s) Localized in Mitochondria |
|---|---|---|
| Base excision repair (BER) | Base modification | APE1 [152], APE2 [153], APTX [154], DNA2 [155], FEN1 [156], LIG3 [157], MPG [157], MUTYH [158], NEIL1 [159], NEIL2 [160], NTHL1 [161], NUDT1 [162], OGG1 [163], PARP1 [164], PNKP [160], POLB [165], POLG [166], TDP1 [167], UNG [168]. |
| Translesion synthesis (TLS) | Lesion causing replication fork stalling | POLZ [169], POLQ [151]. |
| Homologous recombination (HR) | Double-strand breaks containing strong sequence homology | RAD50 [170], RAD51 [171], MRE11 [172], NIBRIN [170], LIG3 [157]. |
| Non-homologous end joining (NHEJ) | Double-strand breaks with little or no sequence homology/blunt ends | ATM [173], ATR [174], KU80 [175], XRCC4 [151]. |
| Microhomology-mediated end joining (MMEJ) | Double-strand breaks with micro homology 3–25 bp in length | LIG3 [150], PARP1 [164], CtIP [145], MRE11 [172], RAD50 [170]. |
| Mismatch repair (MMR) | Sequence mismatches | HMBG1 [176], MLH1 [177], YB-1 [178]. |
| Nucleotide excision repair | Helix distorting and bulky lesions | CSA [147], CSB [147], LIG3 [157], ERCC2 [179]. |
| Fanconi anemia pathway (FA) | DNA interstrand cross links | FANCC [148], FANCD2 [180], FANCG [181], FANCM [182], FANCO [183], FANCS [184]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041065
AMA Style
Kodavati M, Wang H, Hegde ML. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells. 2020; 9(4):1065. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041065
Chicago/Turabian StyleKodavati, Manohar, Haibo Wang, and Muralidhar L. Hegde. 2020. "Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective" Cells 9, no. 4: 1065. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041065
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.