Mitochondrial MicroRNAs in Aging and Neurodegenerative Diseases


1
Department of Internal Medicine, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA
2
Department of Pharmacology and Neuroscience, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA
3
Department of Neurology, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA
4
Department of Public Health, Graduate School of Biomedical Sciences, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA
5
Department of Speech, Language, and Hearing Sciences, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA
*
Author to whom correspondence should be addressed.
Cells 2020, 9(6), 1345; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061345
Submission received: 7 April 2020
/
Revised: 26 May 2020
/
Accepted: 27 May 2020
/
Published: 28 May 2020
(This article belongs to the Special Issue Non-Coding RNAs and Neurological Diseases)
Abstract
:MicroRNAs (miRNAs) are important regulators of several biological processes, such as cell growth, cell proliferation, embryonic development, tissue differentiation, and apoptosis. Currently, over 2000 mammalian miRNAs have been reported to regulate these biological processes. A subset of microRNAs was found to be localized to human mitochondria (mitomiRs). Through years of research, over 400 mitomiRs have been shown to modulate the translational activity of the mitochondrial genome. While miRNAs have been studied for years, the function of mitomiRs and their role in neurodegenerative pathologies is not known. The purpose of our article is to highlight recent findings that relate mitomiRs to neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s. We also discuss the involvement of mitomiRs in regulating the mitochondrial genome in age-related neurodegenerative diseases.
1. Introduction
Since their incorporation into eukaryotes billions of years ago, mitochondria have evolved into the source of energy for cell survival [1]. This energy, in the form of adenosine triphosphate (ATP), powers most cellular processes. Given their origin as independent prokaryotes, mitochondria maintain their own circular DNA separate from somatic DNA [1]. However, the proteins that participate in mitochondrial function come from both nuclear and mitochondrial genomes. This interplay that takes place in the symbiotic relationship between cell and mitochondria results in its peculiar structure and subsequent function [1,2,3,4,5]. Mitochondria perform multiple cellular functions, including ATP production, intracellular calcium regulation, apoptotic cell death, free radicals generation and scavenging, and the activation of the caspase family of proteases in apoptosis [1].
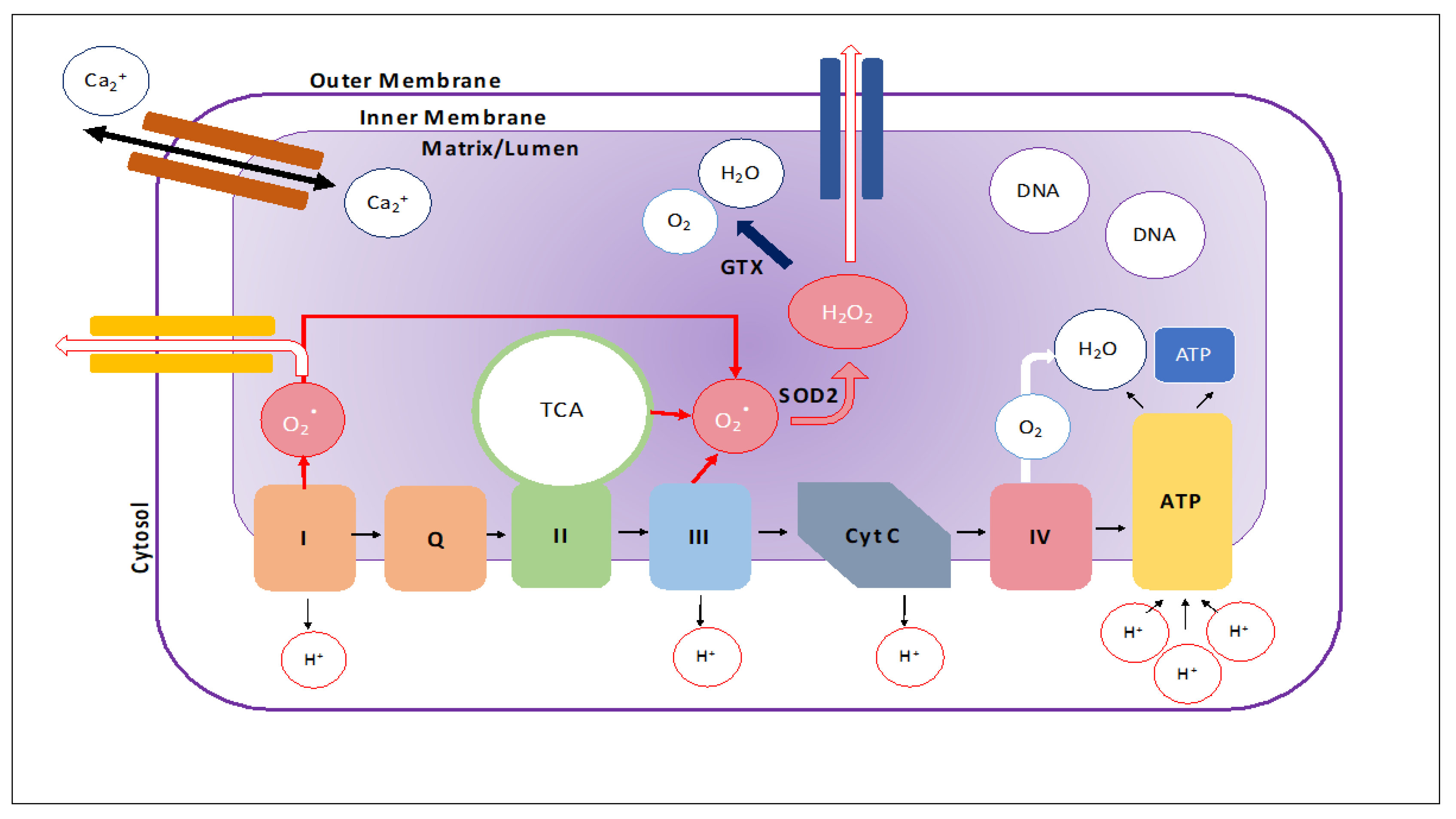
A mitochondrion is comprised of an outer membrane, an inner-membrane, and a matrix where its genome, ribosomes, and other structures lie. The intermembrane space is between the outer and inner membranes (Figure 1). The outer membrane is porous and contains proteins, called porins, that allow for the unfettered movement of ions and small, uncharged molecules into the intermembrane space [1,2,3,4,5]. Larger molecules and proteins must pass through translocase complexes to bypass the outer membrane in order to access the intermembrane space. In contrast, the inner membrane serves as a tight barrier around the matrix of the mitochondrion and only allows molecules and proteins access via membrane-transfer proteins that are selective for specific ions and proteins [6]. Invaginations of the inner membrane, called cristae, allow for increased production of ATP by increasing the surface area of the inner membrane [2].
The division between the matrix and intermembrane space allows the mitochondria to perform its most important function of ATP production [2]. The tightness of the inner membrane allows proteins to pump hydrogen ions out of the matrix and into the intermembrane space to create an electrochemical gradient (Figure 1). ATP synthase utilizes this electrochemical gradient to produce ATP in a process called oxidative phosphorylation [1]. In addition to ATP production, the mitochondria play a key role in cell signaling and cellular differentiation. As a result, mitochondrial dysregulation is largely implicated in aging and neurodegenerative diseases [7]. The purpose of this article is to review the latest developments in mitochondrial microRNAs (miRNAs) research in neurodegenerative diseases. This article also focuses on the role of mitochondrial microRNA in neurodegenerative diseases such as Parkinson’s, Alzheimer’s and Huntington’s.
2. MicroRNAs
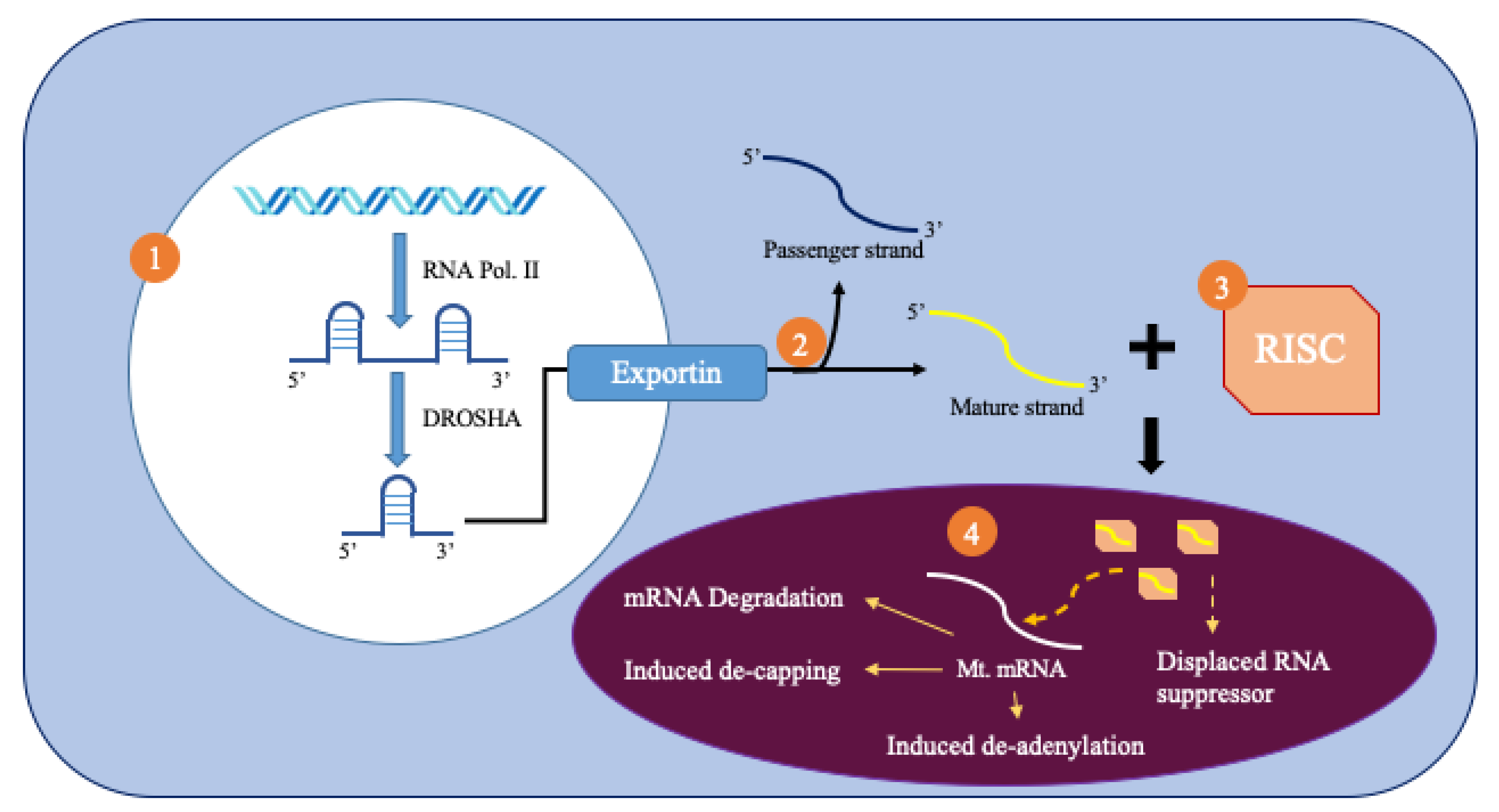
MicroRNAs (miRNAs) are small, noncoding RNA molecules that are 18–25 nucleotides in length and act as post-transcriptional regulators of gene expression. Figure 2 summarizes the biogenesis of miRNAs from the nucleus. RNA polymerase II produces primary miRNAs (pri-miRNAs) [8]. These pri-miRNAs are processed inside the nucleus by the drosha protein that produces a precursor miRNA (pre-miRNA) [8]. Pre-miRNA is more stable than pri-miRNA because of its hairpin structure. The pre-miRNA is then exported to the cytoplasm by exportin 5, which recognizes a 2-nucleotide overhang from the hairpin loop. It is then spliced by the dicer RNAse and one strand is chosen due to its greater thermodynamic stability [9]. The chosen strand, or the mature miRNA, binds to the RNA-induced silencing complex (RISC).
MicroRNAs can be found in multiple organelles. Researchers hypothesized that the region near the 3′ end of the miRNA may contain information on intracellular target location. This finding is not absolute as some miRNAs, such as miR-181, can localize in both the cytoplasm (miR-181a) as well as the mitochondria (miR-181c) despite having the same 3′ motif. Regardless, there are several pathways that miRNA may localize to organelles [9].
MicroRNAs perform multiple important functions in the cell, including cell growth, cell proliferation, embryonic development, tissue differentiation, and apoptosis [10]. MicroRNAs have also been found to improve the gene-regulatory processes in cerebrovascular disease. MicroRNA-mediated gene silencing controls a diverse set of important cellular processes, such as cellular senescence and neurodegeneration. Extensive research has also revealed that microRNAs are implicated in a number of human diseases, including cancer, diabetes, obesity, stroke, Alzheimer’s, Parkinson’s, and Huntington’s, as well as aging [8,10,11,12,13]. These circulatory microRNAs can be used as biomarkers for human diseases [8,10,11,12,13].
3. Mitochondrial MicroRNAs
Mitochondrial microRNAs (mitomiR), named for their location of regulation, must traverse the controlled double membraned system of the mitochondria to regulate mitochondrial gene expression (Figure 2). It has been hypothesized that Ago2 has a mitochondrial targeting sequence near its amino terminal region that helps direct it towards the mitochondria. As a result, Ago2 levels affect the translocation of miRNA into the mitochondria. Furthermore, impairments of Ago2, such as phosphorylation at particular serine residues, can prevent the miRNA complex from binding with its final site of effect, the mRNA. Pathways, such as KRAS-MEK and ANKRD52-PPP6C, separate Ago2 and its associated miRNA from the RISC complex through the inclusion of P-bodies. These P-bodies can help import miRNA into cellular compartments such as the mitochondria [9].
There are multiple postulations as to how microRNA enter the mitochondria. One such postulation is that Ago2 and the miRNA enter the mitochondria via SAM50 and Tom20 pores (outer membrane) and then through translocation of inner membrane (TIM) [9]. Other proteins, such as GW182 and the PNPase enzyme, may also help with the import of miRNA into the mitochondria. The PNPase enzyme, a 3′ to 5′ exoribonuclease and poly-A polymerase found in the mitochondrial intermembrane space, regulates the entry of miRNA into the mitochondria to maintain homeostasis [14].
Once inside the mitochondrial matrix, miRNA can bind to the 3′UTR of targeted mRNAs produced within the mitochondria and affect mitochondrial gene expression and regulation. This altered gene expression is usually observed as a downregulation. This process can occur through mRNA degradation, reduced ribosome occupancy, induced decapping, induced deadenylation, and altered cap-protein binding. This mRNA degradation has been found to prevent mRNA from reaching translational enzymes [8]. However, in some cases, RISC binding increases gene expression by displacing other stronger RNA repressor proteins.
Although miRNAs have been studied for years, researchers are still scratching the surface when it comes to elucidating the role of mitomiRs in disease and aging pathology. One such burgeoning area of research, for example, is the origin of mitomiRs. Though all mitomiRs observed presently are from nuclear DNA, the mitochondrial DNA can also produce mitomiRs. Mitochondrial DNA is a highly coiled circular DNA that holds 37 genes. While difficult to validate due to a lack of DICER and Drosha enzymes, the mitochondrial DNA may also produce miRNAs from its transcripts. These miRNAs may regulate both within the mitochondria as well as at other organelles such as the nucleus [8,9].
Overall, the science of mitomiRs is emerging and important to know the details of. (1) Nuclear-microRNAs importation into mitochondria; (2) a possibility of mitochondrial genome generated microRNAs; and, most importantly, (3) how mitomiRs regulate mitochondrial gene expression and mitochondrial biogenesis and function. Further research is needed to understand these issues.
4. Alzheimer’s Disease
MitomiRs have been associated with Alzheimer’s disease (AD). AD is characterized by a synaptic damage and loss of synapses and an accumulation of amyloid beta and phosphorylated tau in the brain. Such changes alter and destroy synaptic and cognitive functions [15]. Symptoms begin with Mild Cognitive Impairment (MCI) and the inability to form memories. The disease then progresses towards severe cognitive impairment and affects all aspects of intellectual function. The progressive destruction and disruption of synapses culminates in premature cell death. While the complete genesis of the disease is currently unknown, many factors influence its onset and severity.
AD has a profound genetic component. The genes Amyloid Precursor Protein (APP), PSEN1, and PSEN2, which encode proteins involved in APP breakdown and Aβ synthesis, are linked to early onset AD [15]. The APOε4 allele, found on chromosome19q13, is linked to late onset AD in non-Hispanic whites of European ancestry and confers a two- to three-fold risk increase per inherited allele copy [16]. Genome-wide association studies (GWAS) have identified additional genes with varying functions, ranging from APP processing to lipid transportation and immune response, associated with AD pathology [17].
Mitochondrial microRNAs in Alzheimer’s Disease
MicroRNA play a large role in the pathogenesis and progression of AD [8,11,12]. Most of the miRNAs that have been associated with AD onset are also associated with APP processing [8,18,19,20] and phosphorylated tau [11]. For example, Shu et al. (2018) mimicked the symptoms of AD in mice with the injection of amyloid-β 1-42 and noted a decrease in miR-107 levels. Decreased plasma levels of miR-107 seemed to correlate with the abnormal brain cortical anatomy common to AD patients [21]. The subsequent injection of an miR-107 mimic reversed the induced AD symptoms of spatial memory impairment, decreased phosphorylated tau levels, and decreased Aβ neurotoxicity [21]. MiR-107 affects the oxidative abilities of the mitochondria and reductions in miR-107 can decrease mitochondrial volume and cristae. Furthermore, decreased miR-107 levels can cause mitochondrial dysregulation via a reduction in mitochondrial membrane potential and ETC activity (complexes 1, 3, 4, 5 protein levels decrease) [22].
Li et al. (2017) discovered that miR92a plays an important role in tau-related inducement of anxiety in AD [23]. Tau accumulation in the brain selectively suppresses the expression of the intra-vesicular gamma-aminobutyric acid (GABA) transporter (vGAT) by increasing levels of miR92a, which binds to the vGAT mRNA 3′UTR and inhibits translation. Intervention at any step of the pathway (injection of a GABA antagonist, upregulating vGAT synthesis, or injection of a miR92a inhibitor) showed attenuation of the mice’s anxiety [23].
Other mitomiRs have been implicated in tau hyperphosphorylation and cognitive deficits in AD. Ma et al. (2017) showed that miR-125b was higher in the MCI and AD tissue. Using a terminal deoxynucleotide transferase dUTP nick and end labeling (TUNEL) say, the researchers demonstrated that transfecting cells with miR-125b induced neural cell apoptosis. Western Blot analysis showed that the pro-apoptotic protein Bax and the antiapoptotic protein Bcl-2 were upregulated and downregulated, respectively, suggesting that miR-125b induces cellular apoptosis. Bcl-2 and Bax are proteins found in the cytoplasm that can have a regulatory role in the mitochondrial membrane permeability via the induction of transition pores and the release of cytochrome c. Bcl-2 and Bax work antagonistically such that Bcl-2 suppresses apoptosis while Bax encourages it [24]. This same study showed that miR-125b, when targeting the gene FOXQI, also plays a role in enhancing the expression of phosphorylated tau, which is known to be pathologically related to AD [25].
Weinberg et al. (2015) found evidence of neuronal protection via the downregulation of miR-23a and miR-23b [26]. They performed mRNA expression analysis on brain tissues harvested from subjects with amnestic mild cognitive impairment (aMCI), a phase that precedes AD. They found decreased levels of miR23a and miR23b. Both of these miRNAs are thought to decrease the cellular translation of SIRT1, a protein involved in protective neuronal cell stress responses. Quantitative RT-PCR (qRT-PCR) revealed an upregulation of SIRT1 in the tissue samples of the aMCI subjects relative to the control. In vitro studies confirmed this relation between downregulation of these mRNA and increased expression of SIRT1 [27] leading to inhibition of caspase activation and apoptosis.
Russell et al. (2016) report that oligomeric Aβ42, stimulates TNF-α production, which in turn increases levels of miR-34a, a regulator of phosphorylation complexes I-IV, in the mitochondria [28]. The increased mitochondrial dysfunction results in an increase in cellular APP and an increase in Aβ42. This vicious cycle repeats itself and spreads throughout the neurons of the brain. Shi et al. (2011) demonstrated that miR-743a targets mdh2, malate dehydrogenase, which is an enzyme of the citric acid cycle that is elevated in AD brains [29]. Zhang et al. (2016) showed that miR-195, which is downregulated in AD, partially regulates Mitofusion2 (Mfn2), a protein that plays a role in the fusion of mitochondria [30].
Therefore, there is much evidence that suggests that mitomiRs play a critical role in regulating mitochondrial function under the physiological and pathological conditions of AD. The precise relationship between any abnormal location and distribution of miRNAs in normal functioning/dysfunction of mitochondria in AD still needs to be established. Details of reported miRNAs that regulate mitochondrial functions are given in Table 1. It is possible that an abnormal location and distribution of mitomiRs are involved with impaired mitochondrial dynamics, defective biogenesis, and defective mitophagy. The precise involvement of mitomiRs in these mitochondrial processes in healthy and AD states still needs to be investigated.
5. Parkinson’s Disease
Parkinson’s disease (PD) is characterized by resting tremors, muscular rigidity, bradykinesia, and postural inability (citation). Patients with PD do not show any changes in the brain structure unless they also develop dementia. PD-associated dementia results in cerebral atrophy, characterized by the loss of pigmentation in the substantia nigra and locus coeruleus. The loss of dopaminergic neurons in the substantia nigra as well as the formation of Lewy bodies in neurons are the anatomical manifestations of PD. Cell loss is usually found in the lateral ventral tier of the pars-compacta [57]. More than half of the nigral neurons degenerate before symptoms of PD start to show. PD leaves most of its patients with a drastic reduction of up to 80% of their nigral neurons.
Alpha-synuclein is the protein that leads to the formation of Lewy bodies in the neurons of PD patients with dementia. Lewy bodies are spherical inclusions made up of neurofilament proteins and ubiquitin and are around 8 to 30 µm in diameter [58]. Ubiquitin is a cell stress protein that marks damaged or unwanted cellular proteins for degradation [58].
Among all neurodegenerative diseases, PD is most affected by mitochondrial dysfunction. Mitochondrial dysfunction is an early event in PD progression, and mitochondrial abnormalities are involved in almost all PD cases [59]. These abnormalities include: (1) defects in the mitochondrial respiratory chain, (2) defects in mitochondrial DNA, particularly large DNA deletions in nigral cells, (3) increased levels of reactive oxygen species (ROS) (4) calcium dys-homeostasis, particularly in affected nigral cells, (5) defective mitochondrial biogenesis, (6) impaired mitochondrial dynamics (increased fission and reduced fusion), (7) defective mitophagy, and(8) defective axonal transport and distribution of mitochondria in PD neurons [59].
Mitochondrial MicroRNAs in Parkinson’s Disease
MitomiRs play a key role in the pathogenesis of PD (Table 2). Changes in mitomiR expression trigger mitochondrial abnormalities in mitochondrial activity biogenesis and function. For example, mitomiRs can affect PTEN-induced kinase 1 (PINK1). Cell culture studies revealed that the PINK1-induced impairment of mitochondrial membrane polarization triggers the translocation of parkin from the cytosol to mitochondria [60], promoting the damaged mitochondria to form autophagosomes.
MiR-7 targets Bax and SIRT2, thereby inhibiting the production of pro-apoptotic molecules and reducing neuronal cell death in PD [87]. Mir-7 also downregulates the outer membrane of the protein VDAC1, which aids in maintaining the mitochondrial membrane potential. When stress signals activate VDAC1, VDAC1 opens the pore and destroys the membrane potential, leading to the activation of the apoptotic pathway. The downregulation of VDAC1 by miR-7 increases mitochondrial stability. When miR-7 is downregulated, as is in PD, mitochondrial dysfunction can lead to increased ROS production and the upregulation of VDAC1 [62].
MiR-21 is another microRNA involved in PD pathogenesis and is associated with tumors in PD. In some human tumors, miR-21 levels are significantly increased, which corresponds to the down-regulation of PTEN, a gene that has been found to regulate PINK1, that is involved in the mitophagy pathway [66,88]. Further research is still needed to determine the mechanism by which miRNAs control autophagy and mitophagy in PD.
In PD, the downregulation of miR-34b/c is an early event, but it is not known whether this downregulation triggers mitochondrial dysfunction [70]. Researchers have noted a ballooning of the mitochondria, a decrease in ATP production, and an increase in ROS production [70]. Further research is needed to determine the underlying mechanism by which miR-34b/c misexpression contributes to PD pathogenesis.
6. Huntington’s Disease
Huntington’s disease (HD) is a fatal, genetic neurodegenerative disease that presents with a combination of psychiatric, motor, and cognitive impairments [90] HD is caused by expanded polyglutamine (CAG) repeats in the first exon of the HD gene. The CAG trinucleotide repeats of the HD gene mapped to the p arm of chromosome 4. It is an autosomal dominant disorder that leads to the selective death of striatal neurons and the degeneration of the cerebral cortex that impairs communication between the basal ganglia and cerebral cortex [91]. This disease, named after George Huntington, is most prevalent in Western nations.
Research points to a possible role of the HD gene in the development of the brain, synaptic transmission, vesicle transport, and the regulation of transcription [92]. The expanded CAG repeats a result in a defective gene that can lead to striatal cell death as a result of impaired synaptic transmission of glutamate. However, to compound the defect in glutamate, there is also a defect in GABA release, an important neurotransmitter in the basal ganglia and striatum. Ion influx of calcium and potassium are also impaired in HD [92].
Several cellular changes caused by mutant huntingtin have been reported in HD, including: transcriptional regulation, calcium homeostasis, axonal transport, bioenergetics, oxidative stress, and mitochondrial dysfunction [93,94,95]. Furthermore, mutant huntingtin inhibits the activity of histone acetyltransferase and increases the activity of CREB binding protein (CBP). Mutant huntingtin can also bind to the p53 tumor suppressor protein and increases its activity [91]. PGC-1α, a transcriptional co-activator, is repressed by huntingtin through its stimulation of CREB. PGC-1α is important in regulating mitochondrial gene expression and can activate genes that detoxify reactive oxidative species. The down-regulation of PGC-1α will make the cell vulnerable to increasing oxidative stress. Mitochondrial function is also impaired as a result of defective complex 2. Other complexes of the electron transport chain, such as complex 3 and 4, have also been observed to have diminished activity and function [91].
Mitochondrial MicroRNAs in Huntington’s Disease
As shown in Table 3, mitomiRs may be involved in regulating mitochondrial dynamics, biogenesis, and mitophagy in HD. However, further research is needed to understand the precise role of mitomiRs in HD.
7. Aging
Aging, also considered a neurodegenerative process, is the natural and gradual decline of cellular function. Hallmarks of aging include genomic instability (both nuclear and mitochondrial), telomere attrition, epigenetic alterations, the loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intracellular communication. Cellular senescence, in particular, results from and also drives cellular aging [112]. Senescence is characterized by the induction of the stable arrest of cellular growth and phenotypic changes, including alterations to chromatin remodeling, increased autophagy, the production of an inflammatory cytokine, and increased growth factor of heavy senescence-messaging secrotome. Throughout the lifespan of the cell, various stress factors damage its DNA. The cell initiates senescence to prevent damaged genomes from replicating. Age-induced changes in cellular physiology and structure have been associated with cardiovascular disease, diabetes, cancer, hypertension, and neurodegenerative diseases, such as AD and PD [112].
Mitochondria have been known to be implicated in aging since the 1950s, when the mitochondrial free radical theory of aging (MFRTA) was developed. The reactive oxygen species (ROS) found in the body are primarily produced by mitochondrial damage over time. The accumulation of mitochondrial DNA (mtDNA) damage leads to impaired mitochondrial functionality and cell death. Recent studies cast doubt on the validity of MFRTA, suggesting that neither increased ROS nor upregulating the mitochondrial enzymes that dispose of ROS lead to progeroid syndromes. However, it is clear that mitochondria play an important role in aging and age-related chronic diseases.
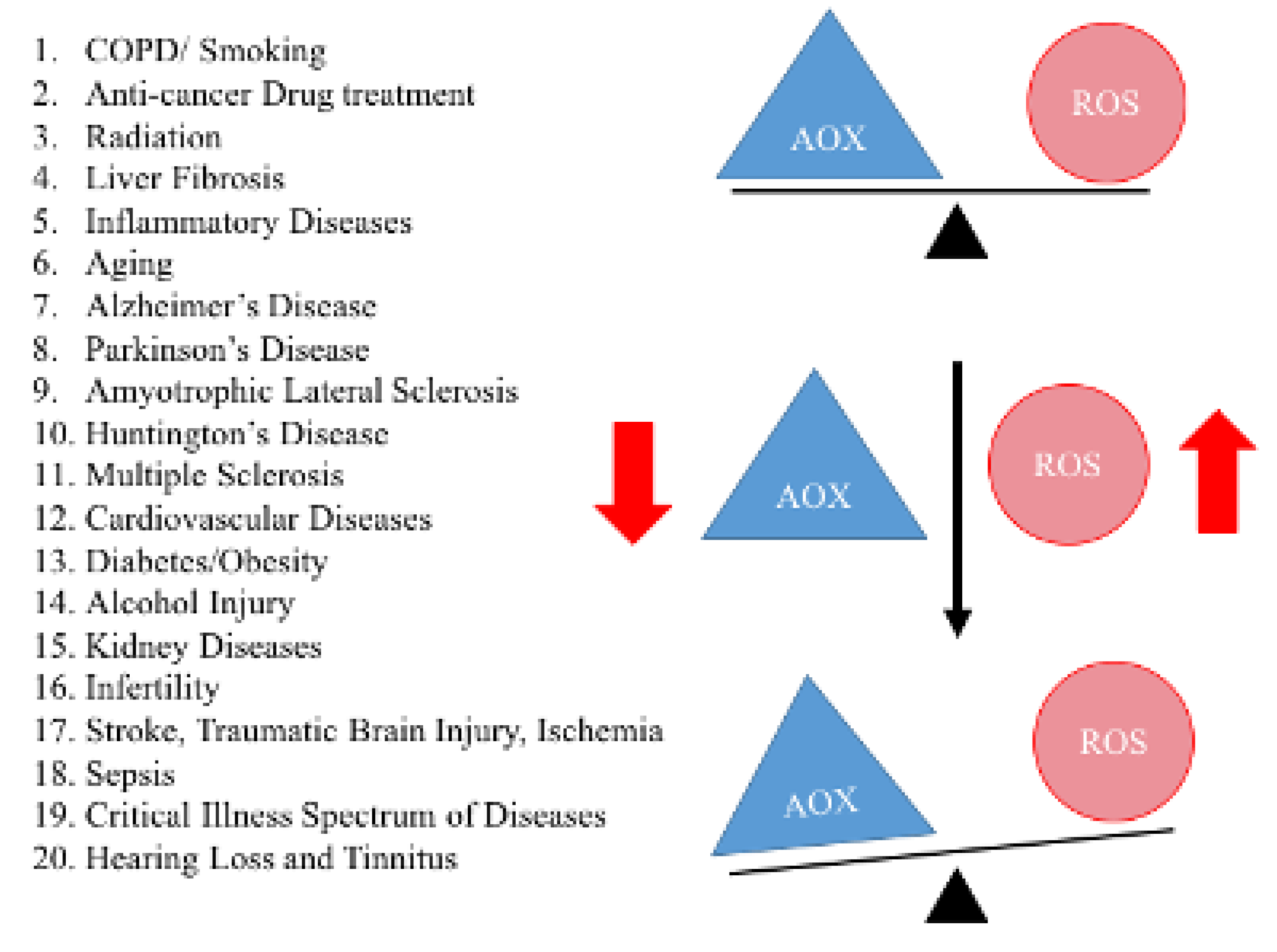
Oxidative stress and mitochondrial dysfunction are key players in age-related chronic diseases. As shown in Figure 3, healthy cells can balance the production of ROS with a concomitant production of antioxidant enzymes in the mitochondria. However, under extreme oxidative stress, there may be a combination of increased levels of ROS production and decreased antioxidant enzyme production. The imbalance between ROS production and endogenous antioxidant enzymes, or oxidative stress, is a characteristic feature of the diseases listed in Figure 3.
The increased production of mitochondrial ROS has been hypothesized to be responsible for mitochondrial DNA (mtDNA) mutations. Deletions and single nucleotide changes can reduce mitochondrial function and ATP production. These changes occur cyclically: increased ROS induces changes and ultimately reduces ATP, and vice versa.
To determine other roles of mtDNA in mice, researchers produced mtDNA mutator mice [113], which possess a proofreading-deficient version of the mtDNA polymerase. The mutator mice allow more specific studies of mitochondria and mtDNA damage [113]. Findings from these mutator mice suggest that, in the mitochondria of stem cells, mtDNA mutations alter oxidative phosphorylation and ROS production.
Aging phenotypes have also been associated with aberrant mitochondrial biogenesis that results from signals dependent on mitochondrial metabolism, particularly the insulin/IGF-1 and target of rapamycin pathways nutrient-sensing pathways [113]. The inhibition of these pathways has been shown to extend the lifespan of worms, flies, and some mammals [2]. Reduced nutrient availability has shown the same life-extending properties, although by an unknown mechanism.
MicroRNAs play a role in affecting the mitochondria’s function in aging. Mitochondria in skeletal muscles show aged phenotypes, such as low mitochondrial mass and impaired tolerance to exercise. They present with low levels of miR-133a [114]. MiR-133a interacts with the mitochondrial protein Nix during the regulation of mitochondrial function by controlling mitochondrial membrane potential in muscles. While low levels of mir-133a may cause exercise intolerance, high levels of mir-133a are also deleterious and are seen in heart failure patients [115]. Furthermore, miR-133a is upregulated in aged internal anal sphincter muscles, which has been found to restore tonicity to the muscle fibers [116].
MiR-181a is upregulated in aging dermal fibroblasts and plays a role in extracellular matrix remodeling and tissue senescence. MiR-181a also seems to play a role in age-associated decline in pancreatic function, being under expressed in the β-cells of aged rats [117]. MiR-181a can target NRF1, COX11, COQ10B and other genes by silencing them. The silencing of these genes reduces mitochondrial biogenesis; it also regulates mitophagy via Atg5 and Park2 [118].
Another example of mitomiR misexpression is that of miR-193. miR-193b is upregulated in aging cartilage [119]. Cells with inhibited miR-193b expression showed increased proliferation, suggesting miR-193b involvement in chondrocyte senescence. Furthermore, it can affect the E2F6 pathway, and SOX5 and SOX9 expression [120]. SOX5 can affect mitochondrial membrane permeability via its activity with Bim, Bax and caspases 9 and 3 [121].
8. Conclusions and Future Directions
We present recent findings on mitomiRs in aging and age-related neurodegenerative diseases, with a particular emphasis on AD, PD, and HD. MitomiRs have been shown to regulate the translational activity of the nuclear and mitochondrial genomes. Overall, the science of mitomiRs is emerging and may improve our understanding of mitochondrial genome regulation, particularly in disease states such as AD, PD and HD. Current research suggests that over 400 microRNAs are localized to mitochondria and may be involved in mitochondrial genome regulation. However, several important issues need to be addressed: (1) the details of how nuclear-microRNAs import into mitochondria; (2) the details of how mitomiRs regulate mitochondrial gene expression and mitochondrial biogenesis and mitochondrial function; (3) the precise role of each mitomiRs in AD, PD, HD and other neurodegenerative diseases; (4) clarification on whether mitomiRs individually and/or collectively regulate mitochondrial function in all neurodegenerative diseases. Addressing these issues may offer novel solutions to protect against mitochondrial dysfunction and to have more precise control over the transcription of protein products. Further research is still needed to better understand the details of mitomiRs and their role in mitochondrial biogenesis, function and mitophagy.
Author Contributions
P.H.R. contributed to conceptualization and formatting of the article. A.J., A.K. and P.H.R. are responsible for writing, original draft preparation, and finalization of manuscript. P.H.R. is responsible for funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
The research presented in this article was supported by NIH grants AG042178, AG047812, NS105473, AG060767 (to PHR).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Mitochondrial dysfunction in aging and Alzheimer’s disease: Strategies to protect neurons. Antioxid. Redox Signal. 2007, 9, 1647–1658. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Macgregor-Das, A.M.; Das, S. A microRNA’s journey to the center of the mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H206–H215. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, M.; Reddy, P.H. Non-Coding RNAs Based Molecular Links in Type 2 Diabetes, Ischemic Stroke, and Vascular Dementia. J. Alzheimers Dis. 2020. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tonk, S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Reddy, A.P. A critical evaluation of neuroprotective and neurodegenerative MicroRNAs in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1156–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Vijayan, M.; Bhatti, J.S.; Reddy, P.H. MicroRNAs as Peripheral Biomarkers in Aging and Age-Related Diseases. Prog. Mol. Biol. Transl. Sci. 2017, 146, 47–94. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Reddy, P.H. Peripheral biomarkers of stroke: Focus on circulatory microRNAs. Biochim. Biophys. Acta 2016, 1862, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S499–S512. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, A.P.; Yin, X.; Reddy, P.H. Novel MicroRNA-455-3p and its protective effects against abnormal APP processing and amyloid beta toxicity in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2428–2440. [Google Scholar] [CrossRef]
- Amakiri, N.; Kubosumi, A.; Tran, J.; Reddy, P.H. Amyloid Beta and MicroRNAs in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 430. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Qin, L.; Tang, B. MicroRNAs in Alzheimer’s Disease. Front. Genet. 2019, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Shu, B.; Zhang, X.; Du, G.; Fu, Q.; Huang, L. MicroRNA-107 prevents amyloid-β-induced neurotoxicity and memory impairment in mice. Int. J. Mol. Med. 2018, 41, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Rech, M.; Kuhn, A.R.; Lumens, J.; Carai, P.; van Leeuwen, R.; Verhesen, W.; Verjans, R.; Lecomte, J.; Liu, Y.; Luiken, J.J.F.P.; et al. AntagomiR-103 and -107 Treatment Affects Cardiac Function and Metabolism. Mol. Nucleic Acids 2019, 14, 424–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Z.; Tan, L.; Wang, Y.; Lu, C.; Chen, R.; Zhang, S.; Gao, Y.; Liu, Y.; Yin, Y.; et al. Correcting miR92a-vGAT-Mediated GABAergic Dysfunctions Rescues Human Tau-Induced Anxiety in Mice. Mol. Ther. 2017, 25, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisichella, V.; Giurdanella, G.; Platania, C.B.; Romano, G.L.; Leggio, G.M.; Salomone, S.; Drago, F.; Caraci, F.; Bucolo, C. TGF-β1 prevents rat retinal insult induced by amyloid-β (1-42) oligomers. Eur. J. Pharm. 2016, 787, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, L.; Meng, J. MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer’s disease. Neurosci. Lett. 2017, 661, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.B.; Mufson, E.J.; Counts, S.E. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front. Neurosci. 2015, 9, 430. [Google Scholar] [CrossRef] [Green Version]
- Jovicic, A.; Zaldivar Jolissaint, J.F.; Moser, R.; Silva Santos, M.e.F.; Luthi-Carter, R. MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS ONE 2013, 8, e54222. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.E.; Doll, D.N.; Sarkar, S.N.; Simpkins, J.W. TNF-α and Beyond: Rapid Mitochondrial Dysfunction Mediates TNF-α-Induced Neurotoxicity. J. Clin. Cell Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Shi, Q.; Gibson, G.E. Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by miR-743a. J. Neurochem. 2011, 118, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhou, H.; Jiang, L.; Mao, Y.; Cui, X.; Xie, B.; Cui, D.; Wang, H.; Zhang, Q.; Xu, S. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016, 1652, 135–143. [Google Scholar] [CrossRef]
- Sørensen, S.S.; Nygaard, A.B.; Christensen, T. miRNA expression profiles in cerebrospinal fluid and blood of patients with Alzheimer’s disease and other types of dementia—An exploratory study. Transl. Neurodegener. 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Jiao, J.; Gao, G.; Prabhakar, B.S. Control of mitochondrial activity by miRNAs. J. Cell Biochem. 2012, 113, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, G.L.; Platania, C.B.M.; Drago, F.; Salomone, S.; Ragusa, M.; Barbagallo, C.; Di Pietro, C.; Purrello, M.; Reibaldi, M.; Avitabile, T.; et al. Retinal and Circulating miRNAs in Age-Related Macular Degeneration: An In vivo Animal and Human Study . Front. Pharm. 2017, 8, 168. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.X.; Kim, C.; Nelson, P.T.; Springer, W. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Li, Y.; Su, B. Identification of Circulating miR-125b as a Potential Biomarker of Alzheimer’s Disease in APP/PS1 Transgenic Mouse. J. Alzheimers Dis. 2017, 59, 1449–1458. [Google Scholar] [CrossRef]
- Li, S.P.; Liu, B.; Song, B.; Wang, C.X.; Zhou, Y.C. miR-28 promotes cardiac ischemia by targeting mitochondrial aldehyde dehydrogenase 2 (ALDH2) in mus musculus cardiac myocytes. Eur. Rev. Med. Pharm. Sci. 2015, 19, 752–758. [Google Scholar]
- Sarkar, S.; Engler-Chiurazzi, E.B.; Cavendish, J.Z.; Povroznik, J.M.; Russell, A.E.; Quintana, D.D.; Mathers, P.H.; Simpkins, J.W. Over-expression of miR-34a induces rapid cognitive impairment and Alzheimer’s disease-like pathology. Brain Res. 2019, 1721, 146327. [Google Scholar] [CrossRef]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging (Albany NY) 2018, 10, 2855–2873. [Google Scholar] [CrossRef]
- Chen, J.; Qi, Y.; Liu, C.F.; Lu, J.M.; Shi, J.; Shi, Y. MicroRNA expression data analysis to identify key miRNAs associated with Alzheimer’s disease. J. Gene Med. 2018, 20, e3014. [Google Scholar] [CrossRef]
- Frankel, L.B.; Wen, J.; Lees, M.; Høyer-Hansen, M.; Farkas, T.; Krogh, A.; Jäättelä, M.; Lund, A.H. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011, 30, 4628–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Noh, H.; Lee, Y.; Jeon, J.; Shanmugavadivu, A.; McPhie, D.L.; Kim, K.S.; Cohen, B.M.; Seo, H.; Sonntag, K.C. MiR-126 Regulates Growth Factor Activities and Vulnerability to Toxic Insult in Neurons. Mol. Neurobiol. 2016, 53, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 suppresses mesothelioma malignancy by targeting IRS1 and interfering with the mitochondrial function. Antioxid. Redox Signal. 2014, 21, 2109–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhter, R.; Shao, Y.; Shaw, M.; Formica, S.; Khrestian, M.; Leverenz, J.B.; Bekris, L.M. Regulation of ADAM10 by miR-140-5p and potential relevance for Alzheimer’s disease. Neurobiol. Aging 2018, 63, 110–119. [Google Scholar] [CrossRef]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Z. Downregulation of miR143/145 gene cluster expression promotes the aortic media degeneration process via the TGF-β1 signaling pathway. Am. J. Transl. Res. 2019, 11, 370–378. [Google Scholar]
- Rippo, M.R.; Olivieri, F.; Monsurrò, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol 2014, 56, 154–163. [Google Scholar] [CrossRef]
- Wang, W.X.; Springer, J.E. Role of mitochondria in regulating microRNA activity and its relevance to the central nervous system. Neural. Regen Res. 2015, 10, 1026–1028. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, C.J.; Prieto, G.A.; Martini, A.C.; Forner, S.; Trujillo-Estrada, L.; LaFerla, F.M.; Baglietto-Vargas, D.; Cotman, C.W.; Kitazawa, M. miR-181a negatively modulates synaptic plasticity in hippocampal cultures and its inhibition rescues memory deficits in a mouse model of Alzheimer’s disease. Aging Cell 2020, 19, e13118. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Yan, K.; An, T.; Zhai, M.; Huang, Y.; Wang, Q.; Wang, Y.; Zhang, R.; Wang, T.; Liu, J.; Zhang, Y.; et al. Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siedlecki-Wullich, D.; Català-Solsona, J.; Fábregas, C.; Hernández, I.; Clarimon, J.; Lleó, A.; Boada, M.; Saura, C.A.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimers Res. 2019, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Z.F.; Li, W.; Hong, H.; Chen, J.; Tian, Y.; Liu, Z.Y. Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J. Cell Biochem. 2018, 119, 5437–5448. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. 2011, 121, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.J.; Zhang, Y.F.; Dammer, E.B.; Zhou, Y.; Wang, L.L.; Liu, X.H.; Feng, B.L.; Jiang, G.X.; Chen, S.D.; Wang, G.; et al. Peripheral Blood MicroRNA Expression Profiles in Alzheimer’s Disease: Screening, Validation, Association with Clinical Phenotype and Implications for Molecular Mechanism. Mol. Neurobiol. 2016, 53, 5772–5781. [Google Scholar] [CrossRef]
- Hu, Y.B.; Zhang, Y.F.; Wang, H.; Ren, R.J.; Cui, H.L.; Huang, W.Y.; Cheng, Q.; Chen, H.Z.; Wang, G. miR-425 deficiency promotes necroptosis and dopaminergic neurodegeneration in Parkinson’s disease. Cell Death Dis. 2019, 10, 589. [Google Scholar] [CrossRef] [Green Version]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar]
- Pahapill, P.A.; Lozano, A.M. The pedunculopontine nucleus and Parkinson’s disease. Brain 2000, 123, 1767–1783. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Cheng, Y. miR-16-1 promotes the aberrant α-synuclein accumulation in parkinson disease via targeting heat shock protein 70. Sci. World J. 2014, 2014, 938348. [Google Scholar] [CrossRef] [Green Version]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [Green Version]
- Martinez, B.; Peplow, P.V. MicroRNAs in Parkinson’s disease and emerging therapeutic targets. Neural. Regen. Res. 2017, 12, 1945–1959. [Google Scholar] [CrossRef]
- Zhang, J.G.; Wang, J.J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.H. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef]
- Lungu, G.; Stoica, G.; Ambrus, A. MicroRNA profiling and the role of microRNA-132 in neurodegeneration using a rat model. Neurosci. Lett. 2013, 553, 153–158. [Google Scholar] [CrossRef]
- Li, D.; Yang, H.; Ma, J.; Luo, S.; Chen, S.; Gu, Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum. Cell 2018, 31, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Gong, X.; He, X.; Lu, G.; Lu, F.; et al. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of Parkinson’s Disease by Targeting to Bim. Brain Pathol. 2016, 26, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, Y.; McKenna, N.D.; Yi, M.; Simunovic, F.; Wang, Y.; Kong, B.; Rooney, R.J.; Seo, H.; Stephens, R.M.; et al. miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol. Aging 2014, 35, 1712–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Pan, X.; Zhang, J.; Ma, A.; Yang, S.; Ma, J.; Xie, A. Plasma levels of miR-137 and miR-124 are associated with Parkinson’s disease but not with Parkinson’s disease with depression. Neurol. Sci. 2017, 38, 761–767. [Google Scholar] [CrossRef]
- Cardo, L.F.; Coto, E.; Ribacoba, R.; Mata, I.F.; Moris, G.; Menéndez, M.; Alvarez, V. The screening of the 3’UTR sequence of LRRK2 identified an association between the rs66737902 polymorphism and Parkinson’s disease. J. Hum. Genet. 2014, 59, 346–348. [Google Scholar] [CrossRef]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Li, K.; Zhang, J.; Ji, C.; Wang, L. MiR-144-3p and Its Target Gene β-Amyloid Precursor Protein Regulate 1-Methyl-4-Phenyl-1,2-3,6-Tetrahydropyridine-Induced Mitochondrial Dysfunction. Mol. Cells 2016, 39, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Caggiu, E.; Paulus, K.; Mameli, G.; Arru, G.; Sechi, G.P.; Sechi, L.A. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. eNeurologicalSci. 2018, 13, 1–4. [Google Scholar] [CrossRef]
- Du, C.; Yao, F.; Ren, Y.; Du, Y.; Wei, J.; Wu, H.; Duan, H.; Shi, Y. SOCS-1 is involved in TNF-α-induced mitochondrial dysfunction and apoptosis in renal tubular epithelial cells. Tissue Cell 2017, 49, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; van der Walt, J.M.; Mayhew, G.; Li, Y.J.; Züchner, S.; Scott, W.K.; Martin, E.R.; Vance, J.M. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am. J. Hum. Genet. 2008, 82, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gao, C.; Sun, Q.; Pan, H.; Huang, P.; Ding, J.; Chen, S. MicroRNA-4639 Is a Regulator of DJ-1 Expression and a Potential Early Diagnostic Marker for Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 232. [Google Scholar] [CrossRef]
- Li, S.; Lv, X.; Zhai, K.; Xu, R.; Zhang, Y.; Zhao, S.; Qin, X.; Yin, L.; Lou, J. MicroRNA-7 inhibits neuronal apoptosis in a cellular Parkinson’s disease model by targeting Bax and Sirt2. Am. J. Transl. Res. 2016, 8, 993–1004. [Google Scholar]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef]
- Yhnell, E. Huntington’s disease: Of mice and men. Oncotarget 2017, 8, 12552–12553. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Johnson, G.V. Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res. Bull. 2009, 80, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepeda, C.; Tong, X.P. Huntington’s disease: From basic science to therapeutics. CNS Neurosci. 2018, 24, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Shirendeb, U.P. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Mao, P.; Manczak, M. Mitochondrial structural and functional dynamics in Huntington’s disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, N.; Reddy, P.H. Role of Phosphorylated Tau and Glucose Synthase Kinase 3 Beta in Huntington’s Disease Progression. J. Alzheimers Dis. 2019, 72, S177–S191. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F.; MacDonald, M.E.; Chen, J.F.; Akbarian, S.; Weng, Z.; et al. miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genom. 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Shi, X.; Wei, S.; Ma, D.; Oyinlade, O.; Lv, S.Q.; Ying, M.; Zhang, Y.A.; Claypool, S.M.; Watkins, P.; et al. Krüppel-like factor 4 (KLF4) induces mitochondrial fusion and increases spare respiratory capacity of human glioblastoma cells. J. Biol. Chem. 2018, 293, 6544–6555. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, T.; Zhu, F.; Deng, S.; Li, X.; He, Y. Regulatory roles of miR-22/Redd1-mediated mitochondrial ROS and cellular autophagy in ionizing radiation-induced BMSC injury. Cell Death Dis. 2019, 10, 227. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Chu, K.; Im, W.S.; Yoon, H.J.; Im, J.Y.; Park, J.E.; Park, K.H.; Jung, K.H.; Lee, S.K.; Kim, M.; et al. Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 2011, 227, 172–179. [Google Scholar] [CrossRef]
- Holley, A.K.; St Clair, D.K. Watching the watcher: Regulation of p53 by mitochondria. Future Oncol. 2009, 5, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Im, W.; Mook-Jung, I.; Kim, M. MicroRNA-124 slows down the progression of Huntington’s disease by promoting neurogenesis in the striatum. Neural Regen. Res. 2015, 10, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.N.; Johnson, G.V. The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. J. Bioenerg. Biomembr. 2010, 42, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocerha, J.; Xu, Y.; Prucha, M.S.; Zhao, D.; Chan, A.W. microRNA-128a dysregulation in transgenic Huntington’s disease monkeys. Mol. Brain 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, Â. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, M.; Takahashi, M.; Fujita, H.; Chiyo, T.; Popiel, H.A.; Watanabe, S.; Furuya, H.; Murata, M.; Wada, K.; Okada, T.; et al. Supplemental Treatment for Huntington’s Disease with miR-132 that Is Deficient in Huntington’s Disease Brain. Mol. Nucleic Acids 2018, 11, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Ghose, J.; Sinha, M.; Das, E.; Jana, N.R.; Bhattacharyya, N.P. Regulation of miR-146a by RelA/NFkB and p53 in STHdh(Q111)/Hdh(Q111) cells, a cell model of Huntington’s disease. PLoS ONE 2011, 6, e23837. [Google Scholar] [CrossRef]
- Zhang, X.; Li, C.F.; Zhang, L.; Wu, C.Y.; Han, L.; Jin, G.; Rezaeian, A.H.; Han, F.; Liu, C.; Xu, C.; et al. TRAF6 Restricts p53 Mitochondrial Translocation, Apoptosis, and Tumor Suppression. Mol. Cell 2016, 64, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Kunkanjanawan, T.; Carter, R.L.; Prucha, M.S.; Yang, J.; Parnpai, R.; Chan, A.W. miR-196a Ameliorates Cytotoxicity and Cellular Phenotype in Transgenic Huntington’s Disease Monkey Neural Cells. PLoS ONE 2016, 11, e0162788. [Google Scholar] [CrossRef]
- Di Rita, A.; Maiorino, T.; Bruqi, K.; Volpicelli, F.; Bellenchi, G.C.; Strappazzon, F. miR-218 Inhibits Mitochondrial Clearance by Targeting PRKN E3 Ubiquitin Ligase. Int. J. Mol. Sci. 2020, 21, 355. [Google Scholar] [CrossRef] [Green Version]
- Díez-Planelles, C.; Sánchez-Lozano, P.; Crespo, M.C.; Gil-Zamorano, J.; Ribacoba, R.; González, N.; Suárez, E.; Martínez-Descals, A.; Martínez-Camblor, P.; Álvarez, V.; et al. Circulating microRNAs in Huntington’s disease: Emerging mediators in metabolic impairment. Pharm. Res. 2016, 108, 102–110. [Google Scholar] [CrossRef]
- Zhao, X.; Song, X.; Bai, X.; Tan, Z.; Ma, X.; Guo, J.; Zhang, Z.; Du, Q.; Huang, Y.; Tong, D. microRNA-222 Attenuates Mitochondrial Dysfunction During Transmissible Gastroenteritis Virus Infection. Mol. Cell Proteom. 2019, 18, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.; Smith, F.; Kumar, S.; Vijayan, M.; Reddy, P.H. Are microRNAs true sensors of ageing and cellular senescence? Ageing Res. Rev. 2017, 35, 350–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Sato, Y.; Wang, C.; Yue, F.; Kuang, S.; Gavin, T.P. Impaired exercise tolerance, mitochondrial biogenesis, and muscle fiber maintenance in miR-133a-deficient mice. Faseb J. 2016, 30, 3745–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Luo, J.; Zhao, J.; Shang, D.; Lv, Q.; Zang, P. Combined Use of Circulating miR-133a and NT-proBNP Improves Heart Failure Diagnostic Accuracy in Elderly Patients. Med. Sci. Monit. 2018, 24, 8840–8848. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Boopathi, E.; Addya, S.; Phillips, B.; Rigoutsos, I.; Penn, R.B.; Rattan, S. Aging-associated changes in microRNA expression profile of internal anal sphincter smooth muscle: Role of microRNA-133a. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G964–G973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Sanchez, A.; Rutter, G.A.; Latreille, M. MiRNAs in β-Cell Development, Identity, and Disease. Front. Genet. 2016, 7, 226. [Google Scholar] [CrossRef] [Green Version]
- Indrieri, A.; Carrella, S.; Romano, A.; Spaziano, A.; Marrocco, E.; Fernandez-Vizarra, E.; Barbato, S.; Pizzo, M.; Ezhova, Y.; Golia, F.M.; et al. miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Meng, F.; Li, Z.; Zhang, Z.; Yang, Z.; Kang, Y.; Zhao, X.; Long, D.; Hu, S.; Gu, M.; He, S.; et al. MicroRNA-193b-3p regulates chondrogenesis and chondrocyte metabolism by targeting HDAC3. Theranostics 2018, 8, 2862–2883. [Google Scholar] [CrossRef]
- Ukai, T.; Sato, M.; Akutsu, H.; Umezawa, A.; Mochida, J. MicroRNA-199a-3p, microRNA-193b, and microRNA-320c are correlated to aging and regulate human cartilage metabolism. J. Orthop. Res. 2012, 30, 1915–1922. [Google Scholar] [CrossRef]
- Kurtsdotter, I.; Topcic, D.; Karlén, A.; Singla, B.; Hagey, D.W.; Bergsland, M.; Siesjö, P.; Nistér, M.; Carlson, J.W.; Lefebvre, V.; et al. SOX5/6/21 Prevent Oncogene-Driven Transformation of Brain Stem Cells. Cancer Res. 2017, 77, 4985–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mitochondrial structure. Schematic of the mitochondrion showing the electron transport chain, flow of ions, and generation reactive oxidative species. These reactive oxidative species can be reduced to hydrogen peroxide via the SOD2 enzyme and further broken down into H2O and O2 by the GPX enzyme. However, the reduction process is not perfect and when under high stress, reactive oxidative species can leak out of the mitochondria. The mitochondria are also important regulators of intracellular calcium concentration.
Figure 1.
Mitochondrial structure. Schematic of the mitochondrion showing the electron transport chain, flow of ions, and generation reactive oxidative species. These reactive oxidative species can be reduced to hydrogen peroxide via the SOD2 enzyme and further broken down into H2O and O2 by the GPX enzyme. However, the reduction process is not perfect and when under high stress, reactive oxidative species can leak out of the mitochondria. The mitochondria are also important regulators of intracellular calcium concentration.

Figure 2.
Production and import of mitomiRNA.

Figure 3.
Demonstrates oxidative stress in aging and other age-related diseases. A healthy cell can balance the production of reactive oxidative species and antioxidant enzymes. However, in disease states, such as Alzheimer’s, Parkinson’s, a combination of increased levels of reactive oxidative species production and decreased antioxidant enzymes production have been observed. This imbalance is known as oxidative stress, observed in a large number of human diseases.
Figure 3.
Demonstrates oxidative stress in aging and other age-related diseases. A healthy cell can balance the production of reactive oxidative species and antioxidant enzymes. However, in disease states, such as Alzheimer’s, Parkinson’s, a combination of increased levels of reactive oxidative species production and decreased antioxidant enzymes production have been observed. This imbalance is known as oxidative stress, observed in a large number of human diseases.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Mitochondrial microRNAs related to Alzheimer’s disease.
| miRNA | Change | Potential Role | Reference(s) |
|---|---|---|---|
| miR-15a | Upregulation | Targets BACE1 | Sørensen et al. (2016) [31] |
| increases mitochondrial dysfunction and unbalances mitochondrial membrane potential | Li et al. (2012) [32] | ||
| miR-23a/23b | Downregulation | Promote SIRT1 | Weinberg et al. (2015) [26] |
| Increased SIRT1 affects mitochondrial biogenesis and turnover. | Tang (2016) [33] | ||
| miR-27a | Upregulation | Targets the TGF-β pathway | Romano et al. (2017) [34] |
| Negatively regulates PINK1 mediated mitochondrial clearance | Kim et al. (2016) [35] | ||
| miR-28-3p | Upregulation | APP/PS1 mice | Hong et al. (2017) [36] |
| Inhibition of Aldehyde dehydrogenase 2 | Li et al. (2015) [37] | ||
| miR-34a | Upregulation | Targets ADAM10, NMDAR 2B, and SIRT1 | Sarkar et al. (2019) [38] |
| Localizes in the mitochondria and downregulates Bcl-2 which increases casp-1 activity. Activates casp-3. Promotes apoptosis and dysfunction of mitochondria | Giuliani et al. (2018) [39] | ||
| miR-101 | Upregulation | Transcription regulation | Chen et al. (2018) [40] |
| Targets STMN1, RAB5A and ATG4D. Inhibits autophagy thus persistence of damaged mitochondria. | Frankel et al. (2011) [41] | ||
| miR-107 | Downregulation | Reduced expression in the hippocampus | Shu et al. (2018) [21] |
| Decreased mitochondrial ETC function and morphological changes | Rech et al. (2019) [22] | ||
| miR-125b | Upregulation | Increased Bax, decreased Bcl-2 | Ma et al. (2017) [25] |
| miR-126 | Upregulation | GF/PI3K/AKT and ERK signaling cascades | Kim et al. (2016) [42] |
| Inhibits complex 1 of mitochondria and reduces aerobic respiration. | Tomasetti et al. (2014) [43] | ||
| miR-132 | Downregulation | Inhibits complex 1 of mitochondria and reduces aerobic respiration. | Weinberg et al. (2015) [26] |
| miR-140 | Upregulation | ADAM10 | Akhter et al. (2018) [44] |
| Promotes mitochondrial fission via Mfn1 | Duarte et al. (2014) [45] | ||
| miR-143 | Downregulation | Increased activation of TGF-β | Zhang and Wang (2019) [46] |
| Increased mitochondrial death as decreased ERK5 pathway | Li et al. (2012) [32] | ||
| miR-146a | Upregulation | Associated with mTOR, TNF α | Romano et al. (2017) [34] |
| Modulates Bcl-2 | Rippo et al. (2014) [47] | ||
| miR-155 | Upregulation | Associated with mTOR, TNF α | Romano et al. (2017) [34] |
| Localizes in the mitochondria | Wang and Springer (2015) [48] | ||
| miR-181a | Upregulation | Upregulation of GluA2 | Rodriguez-Ortiz et al. (2020) [49] |
| Localizes in the mitochondria and downregulates Bcl-2 which increases casp-1 activity. Activates casp-3. Promotes apoptosis and dysfunction of mitochondria | Giuliani et al. (2018) [39] | ||
| miR-181c | Upregulation | Down-regulates Bcl-2 and leads to apoptosis | Fisichella et al. (2016) [24] |
| miR-195 | Downregulation | Reduced targeting of BACE1 leads to an increased Aβ levels. | Zhu et al. (2012) [50] |
| Reduced mitochondrial ATP production | Yan et al. (2019) [51] | ||
| miR-210-3p | Upregulation | Clinical marker for MCI and AD | Siedlecki-Wullich et al. (2019) [52] |
| Targets mitochondrial iron sulfur cluster homologue. Decreasing these clusters can reduce activity of mitochondrial enzymes that require iron sulfur clusters. miR-210 can affect aconitase. | Li et al. (2012) [32] | ||
| miR-212 | Downregulation | Increase SIRT1 in aMCI in the frontal cortex. While it may be protective, sustained downregulation can lead to FOX03a mediated apoptosis. | Weinberg et al. (2015) [26] |
| miR-330 | Downregulation | Affects VAV1 and affects mitochondria through MAPK signaling | Zhou et al. (2018) [53] |
| miR-424 | Upregulation | Cortex white matter | Wang et al. (2011) [54] |
| Suppression of ATP levels and mitochondrial integrity through ADP-ribosylation factor-like 2 mRNA. | Duarte et al. (2014) [45] | ||
| miR-425 | Upregulation | BACE1 protein inhibition | Ren et al. (2016) [55] |
| Via RIPK1 causes mitochondrial dysfunction and increased ROS production. Involved in necroptosis. | Hu et al. (2019) [56] |
Table 2.
Mitochondrial microRNAs related to Parkinson’s disease.
| miRNA | Change | Potential Role | References |
|---|---|---|---|
| miR-7 | Downregulation | Increased a-SYN/Substantia Nigra | Junn et al. (2009) [61] |
| Reduced binding to 3′UTR of VDAC1 thus upregulation of anion channel and increased ROS production. | Chaudhuri et al. (2016) [62] | ||
| miR-16-1 | Upregulation | Decrease HSP70 leading to an increased a-SYN | Zhang et al. (2014) [63] |
| HSP70 blocks mitochondrial translocation of Bax, membrane permeabilization, and apoptosis. | Radons (2016) [64] | ||
| miR-21 | Upregulation | Upregulated in midbrain and directly targets 3′UTR of LAMP2A. | Martinez and Peplow (2017) [65] |
| Downregulates PTEN and PINK1, key regulators of mitophagy. | Zhang et al. (2010) [66] | ||
| miR-27a/b | Unknown | Increased accumulation and decreased suppression of PINK1 at 3′UTR. Decreased degradation of damaged mitochondria. | Kim et al. (2016) [35] |
| miR-29a | Upregulation | Mitochondrial voltage dependent anion channel | Lungu et al. (2013) [67] |
| miR-29b | Upregulation | Loss of mitochondrial membrane potential | Lungu et al. (2013) [67] |
| miR-30e | Downregulation | Reduced suppression of Nlrp3 | Li et al. (2018) [68] |
| NLRP3 resides in ER but upon stimulation can interact with mitochondria and cause loss of mitochondrial membrane potential, increase ROS production, and calcium dys-homeostasis. | Liu (2018) [69] | ||
| miR-34b/c | Downregulation | Decreased Parkin/DJ-1 leads to increased ROS production in the mitochondria. Decreased ability to reduce MTT. Ballooning of the mitochondria with decreased ATP production. | Miñones-Moyano et al. (2011) [70] |
| miR-124 | Upregulated | Reduced translocation of Bax into mitochondria due to inhibition of Bim. | Wang et al. (2016) [71] |
| miR-126 | Upregulated | Downregulation of IGF-1/PI3K signaling. It targets TOM1, p85beta, insulin receptor substrate 1, CRK | Kim et al. (2014) [72] |
| miR-137 | Slightly Upregulated | Involved in expression of mitophagy receptors FUNDC1 and NIX. Inhibits mitophagy | Li et al. (2014) [73] |
| Li et al. (2017) [74] | |||
| miR138-2-3p | Downregulation | Increased LRRK2 (lysosomal function in astrocytes) | Cardo et al. (2014) [75] |
| LRRK2 localizes in mitochondria and has regulatory function on mitochondrial fission and fusion. Mutations of LRRK2 leads to increased oxidative stress. | Singh et al. (2019) [76] | ||
| miR-144-3p | Downregulation | Besides inhibiting the expression of APP, miR-144-3p is involved in the mitochondrial gene expression of PGC-1α, NRF-1, TFAM. | Li et al. (2016) [77] |
| miR-153 | Downregulation | Decreased mTOR signaling | Doxakis et al. (2010) [78] |
| Decreased mTOR signaling can lead to reduced clearance of dysfunctional mitochondria (ROS producing) and reduces mitochondrial biogenesis. | Weichhart (2018) [79] | ||
| miR-155 | Upregulation | Suppression of SOCS-1 and SOC-3 (anti-inflammatory molecules) | Caggiu et al. (2018) [80] |
| SOCS-1 suppresses damage to mitochondrial membrane and oxidative stress. | Du et al. (2017) [81] | ||
| miR-205 | Downregulation | Increased LRRK2 | Cho et al. (2013) [82] |
| LRRK2 localizes in mitochondria and has regulatory function on mitochondrial fission and fusion. Mutations of LRRK2 leads to increased oxidative stress. | Singh et al. (2019) [76] | ||
| miR-494 | Upregulation | Decreased DJ-1 (mitochondrial damage) | Xiong et al. (2014) [83] |
| miR-433 | Binding inhibited | Increasing FGF20 (cell death) | Wang et al. (2008) [84] |
| FGF20 increases translation of alpha-synuclein, which in turn disrupts calcium exchange between mitochondria and ER. There is a reduction in mitochondrial ATP production. | Paillusson et al. (2017) [85] | ||
| miR-4639-5p | Upregulation | Decreased DJ-1 leads to mitochondrial fragmentation | Chen et al. (2017) [86] |
Table 3.
MicroRNAs and their role in Huntington’s disease.
| miRNA | Change | Potential Role | References |
|---|---|---|---|
| miR-10b-5p | Upregulation | Targets HOXD10, NF1, KLF4. | Hoss et al. (2015) [96] |
| Overexpression of KLF4 can lead to increased ROS production in mitochondria in already impaired mitochondria. KLF4 can also induce mitochondrial fusion. | Wang et al. (2018) [97] | ||
| miR-22 | Downregulation | Reduced caspase activation and targets HDAC4, Redd1, Rcor1, and Rgs2. | Jovicic et al. (2013) [27] |
| Increased Bcl-xl expression leads to decreased pro-apoptotic proteins thus possibly allowing damaged mitochondria to survive. | Liu et al. (2019) [98] | ||
| miR-29c | Downregulation | Normally upregulates p53 levels by suppressing p85 alpha. | Lee et al. (2011) [99] |
| Reduced levels of p53 may not lead to apoptotic events of damaged mitochondria that have increased ROS production. | Holley and Clair (2009) [100] | ||
| miR-124 | Downregulation | Decreased PGC-1α | Liu et al. (2015) [101] |
| Decreased mitochondrial biogenesis and dysfunction (increased ROS, decreased ATP synthesis) | Jin and Johnson (2010) [102] | ||
| miR-128a | Downregulation | Tumor repression, apoptosis, epileptic seizure repression | Kocerha et al. (2014) [103] |
| Targets FADD. Decreased FADD can prevent apoptosis in damaged mitochondria. | Cavalcante et al. (2019) [104] | ||
| miR-132 | Downregulation | Decreased AGO2 function | Fukuoka et al. (2018) [105] |
| miR-146a | Downregulation | HTT gene downregulates miR-146a | Ghose et al. (2011) [106] |
| Affect IRAK-1 and TRAF-6 which are inflammation mediators. | Rippo et al. (2014) [47] | ||
| TRAF-6 restricts mitochondrial translocation | Zhang et al. (2016) [107] | ||
| miR-196a | Upregulation | Increased BDNF and ANX1A. Increased mitochondrial fusion. Decreased PGC-1α and CBP. | Kunkanjanawan et al. (2016) [108] |
| miR-218 | Downregulation | Normally downregulates PRKN. However, in diseased state upregulates PRKN. There is increased mitochondrial degradation and mitophagy as a result. | Di Rita et al. (2019) [109] |
| miR-222-3p | Upregulation | Targets MMP1, PTEN, SOD2, and other targets | Díez-Planelles et al. (2016) [110] |
| Increase THBS1 thus increasing mitochondrial calcium level. Reduces mitochondrial membrane | Zhao et al. (2019) [111] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
John, A.; Kubosumi, A.; Reddy, P.H. Mitochondrial MicroRNAs in Aging and Neurodegenerative Diseases. Cells 2020, 9, 1345. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061345
AMA Style
John A, Kubosumi A, Reddy PH. Mitochondrial MicroRNAs in Aging and Neurodegenerative Diseases. Cells. 2020; 9(6):1345. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061345
Chicago/Turabian StyleJohn, Albin, Aaron Kubosumi, and P. Hemachandra Reddy. 2020. "Mitochondrial MicroRNAs in Aging and Neurodegenerative Diseases" Cells 9, no. 6: 1345. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061345
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.