All-Trans Retinoic Acid Enhances both the Signaling for Priming and the Glycolysis for Activation of NLRP3 Inflammasome in Human Macrophage

 ,
,  , and
, and
Abstract
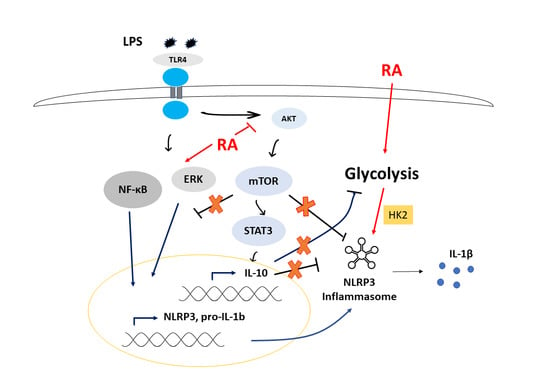
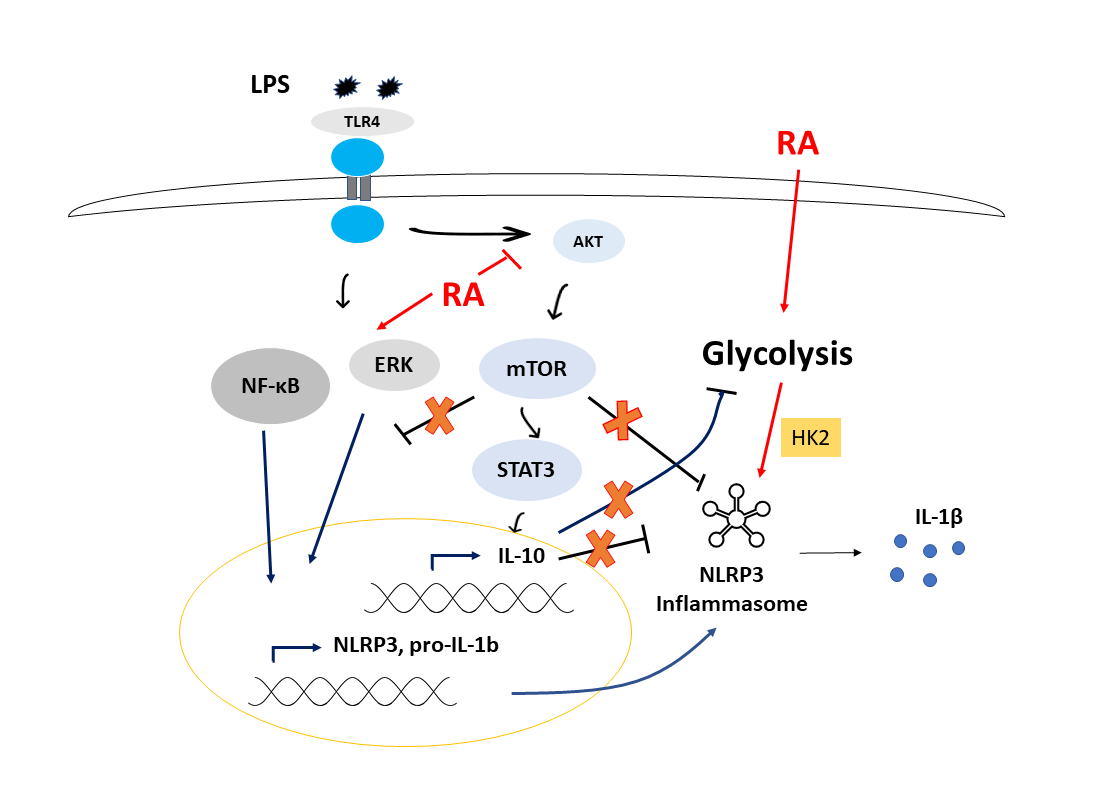
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethics Statement, and Monocyte Isolation and Differentiation
2.3. Monocyte Isolation and Macrophage Differentiation
2.4. Macrophage Treatment
2.5. RNA Preparation, RT-PCR, and Quantitative Real-Time PCR
2.6. Quantitative Real-Time PCR
2.7. Western Blot Analysis
2.8. Metabolic Assays and Extracellular Flux Analysis
2.9. Cytokine Measurements
2.10. Statistical Analysis
3. Results
3.1. ATRA Modifies LPS-Induced Proinflammatory Cytokine Secretion in Human Macrophages
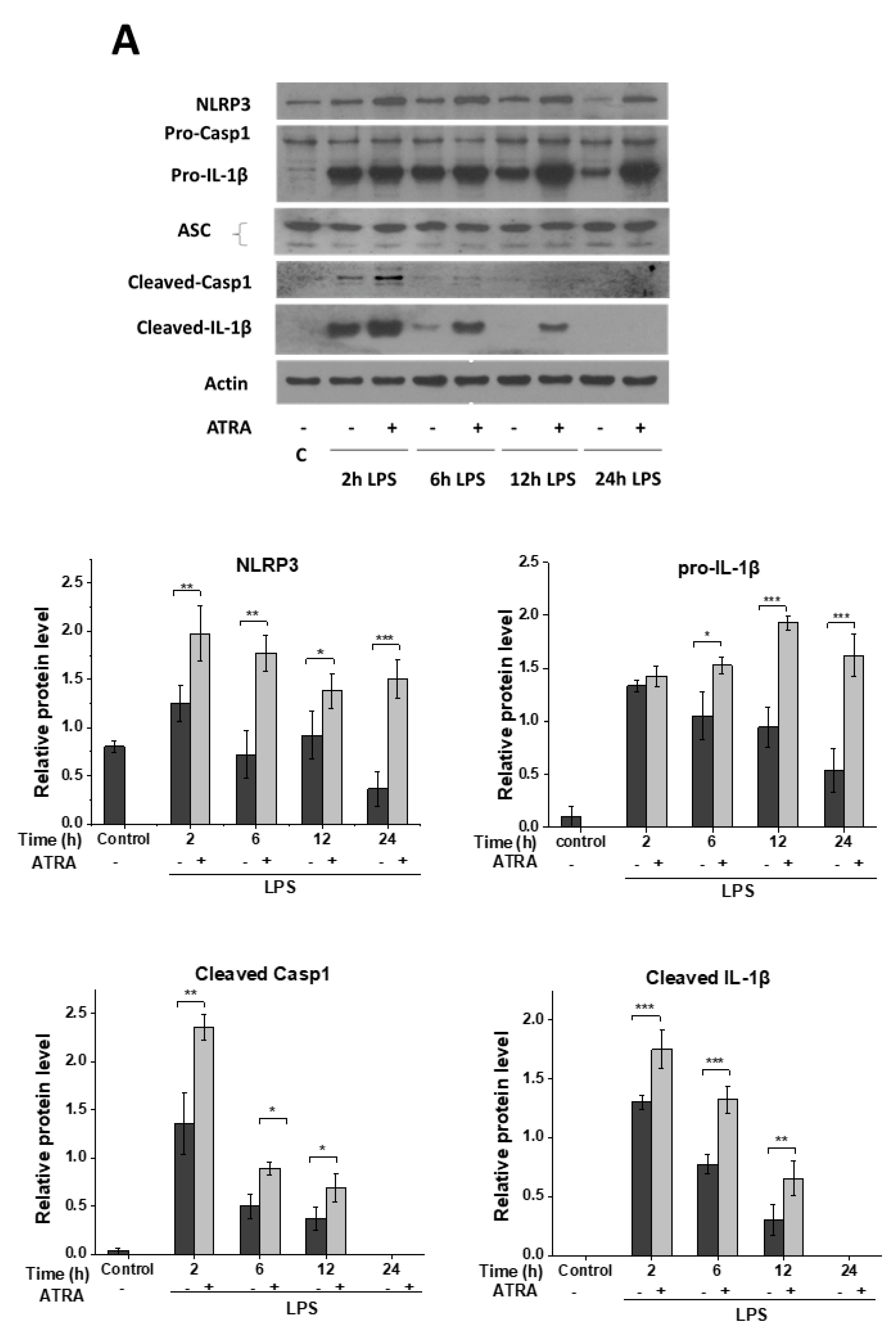
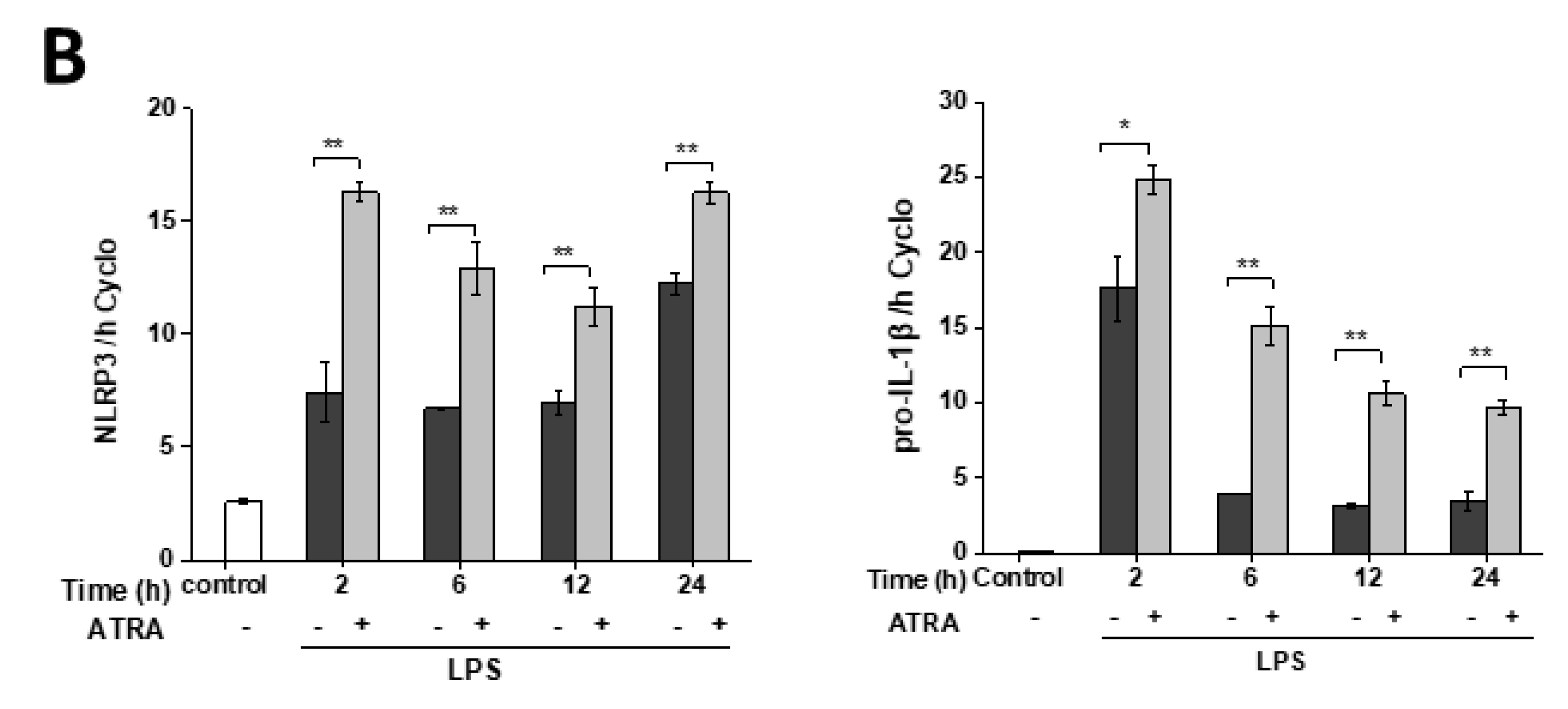
3.2. ATRA Prolongs LPS-Induced IL-1β Cytokine Secretion in Part by Augmenting LPS-Induced NLRP3 and Pro-IL-1β Expression
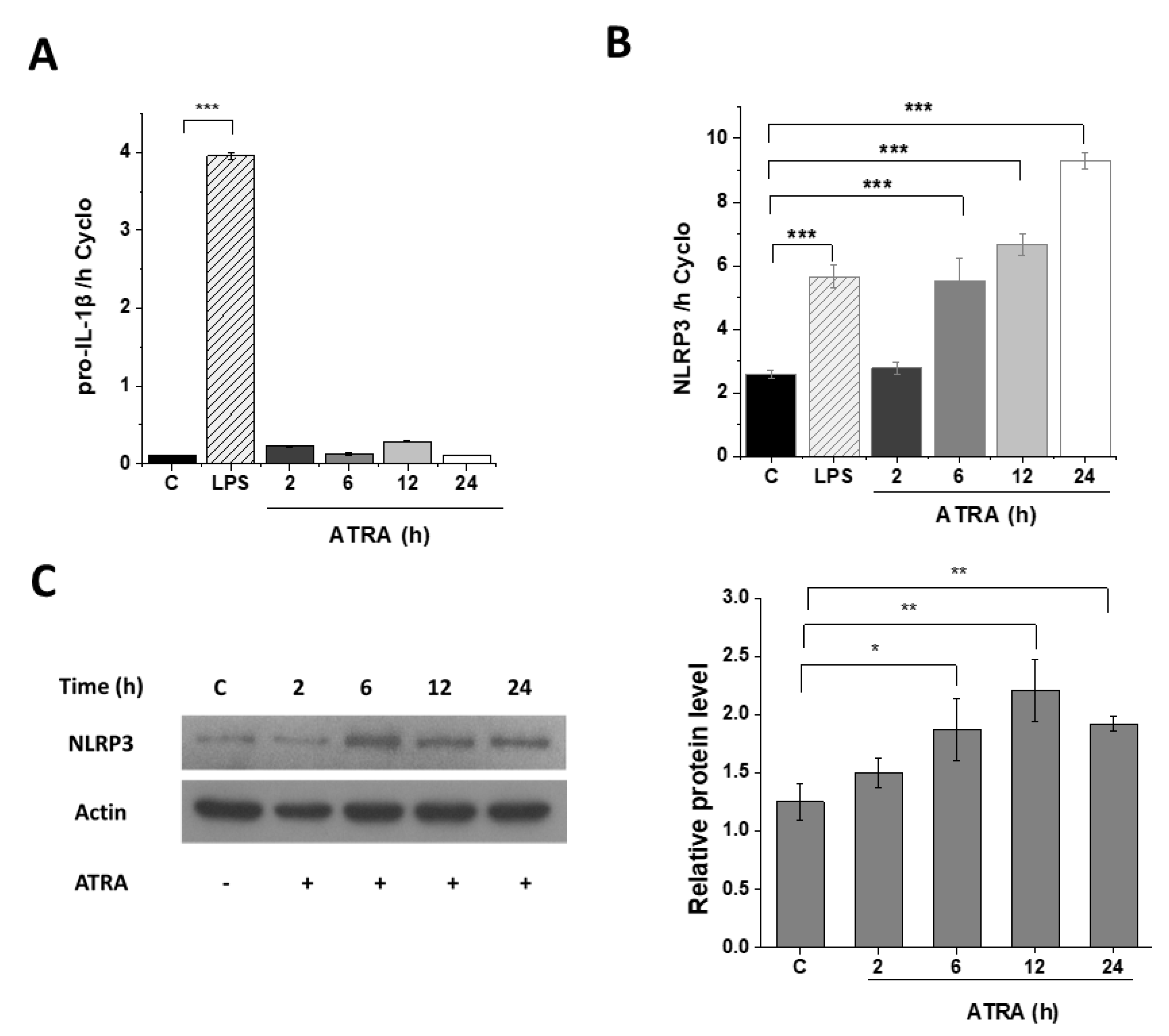
3.3. ATRA Alone Enhances NLRP3 but Not Pro-IL-1β Expression
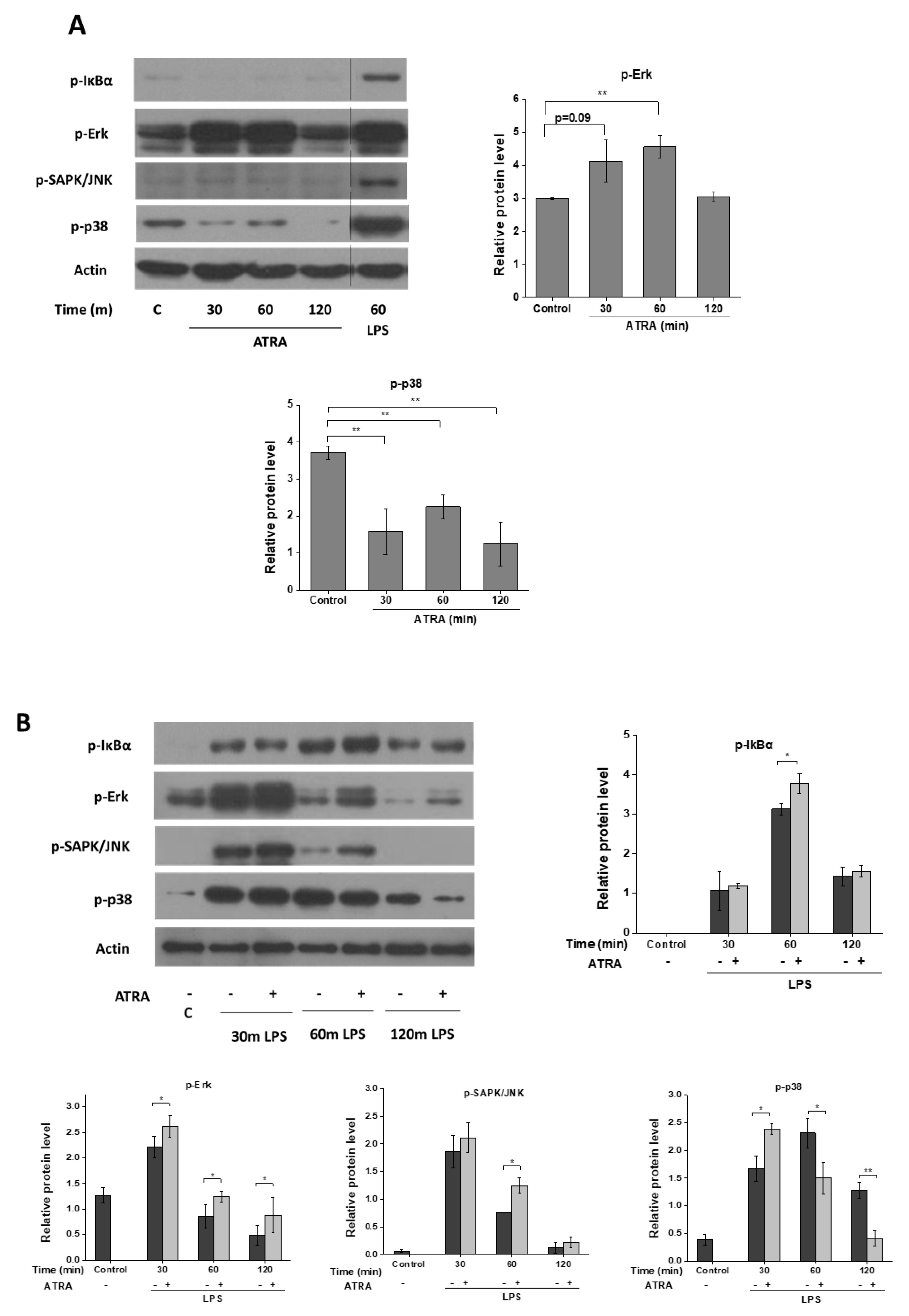
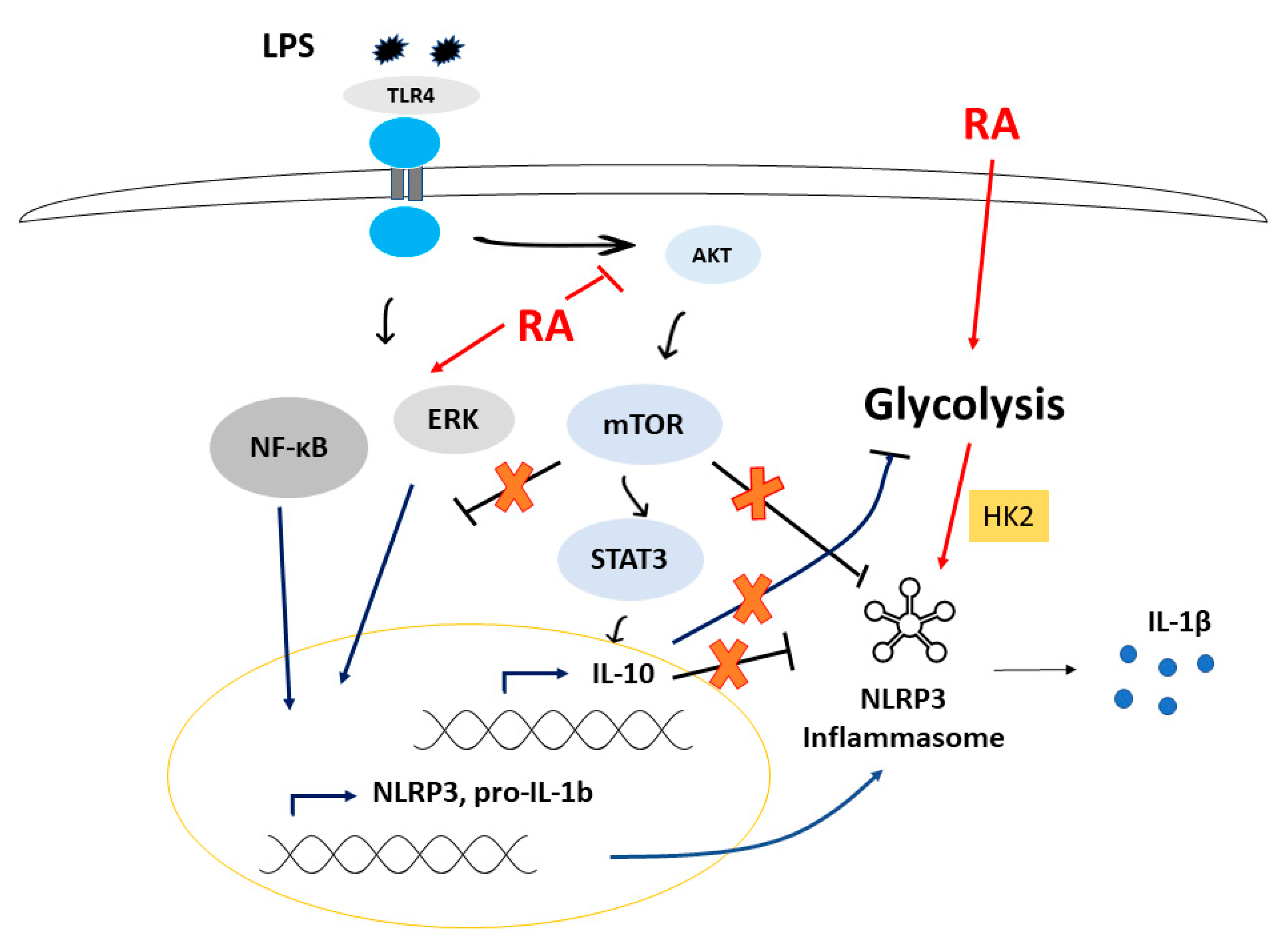
3.4. ATRA Modifies Signal Transduction Pathways Required for Inflammasome Priming
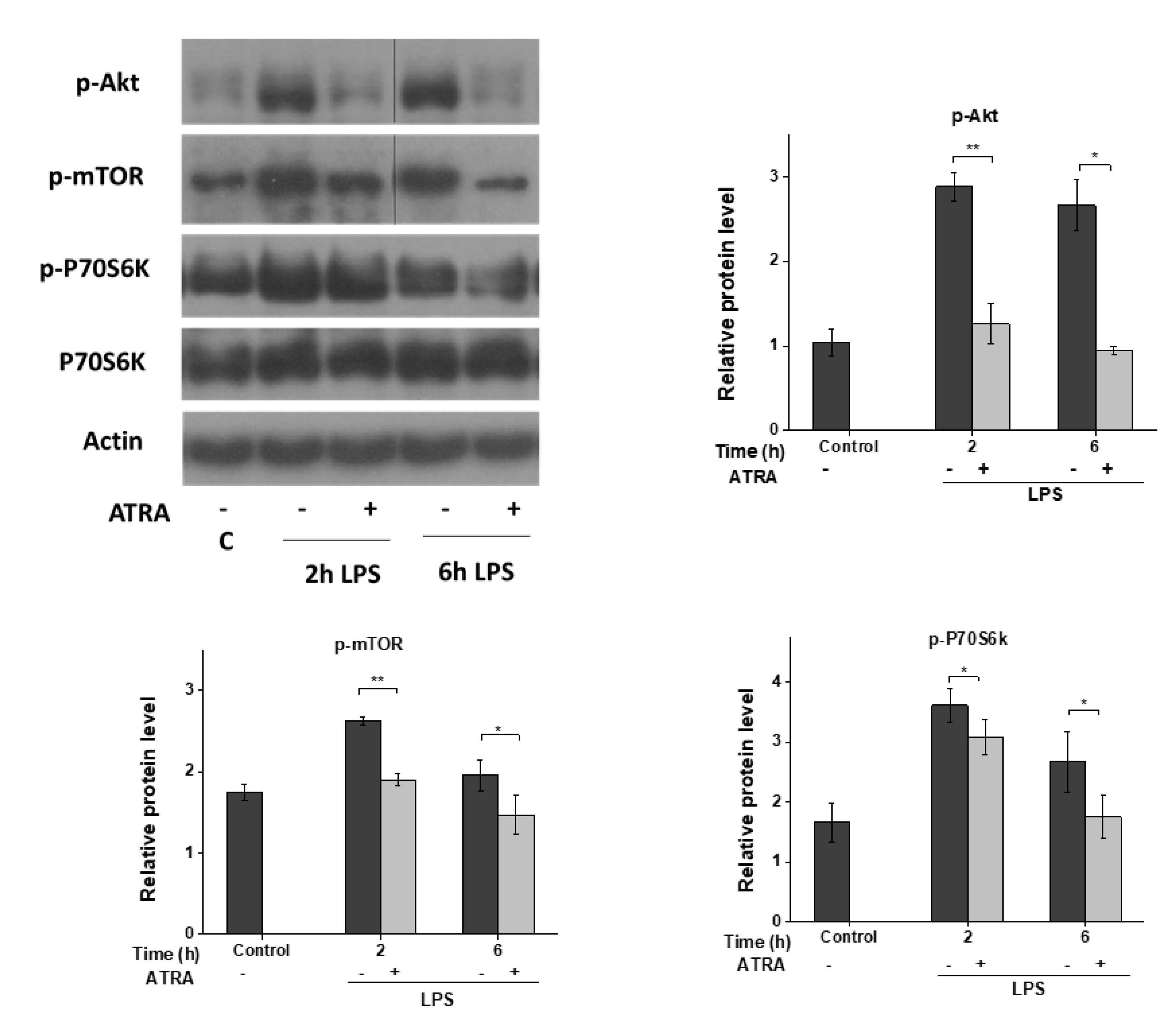
3.5. ATRA Inhibits the LPS-Induced AKT/mTOR Signaling Pathway
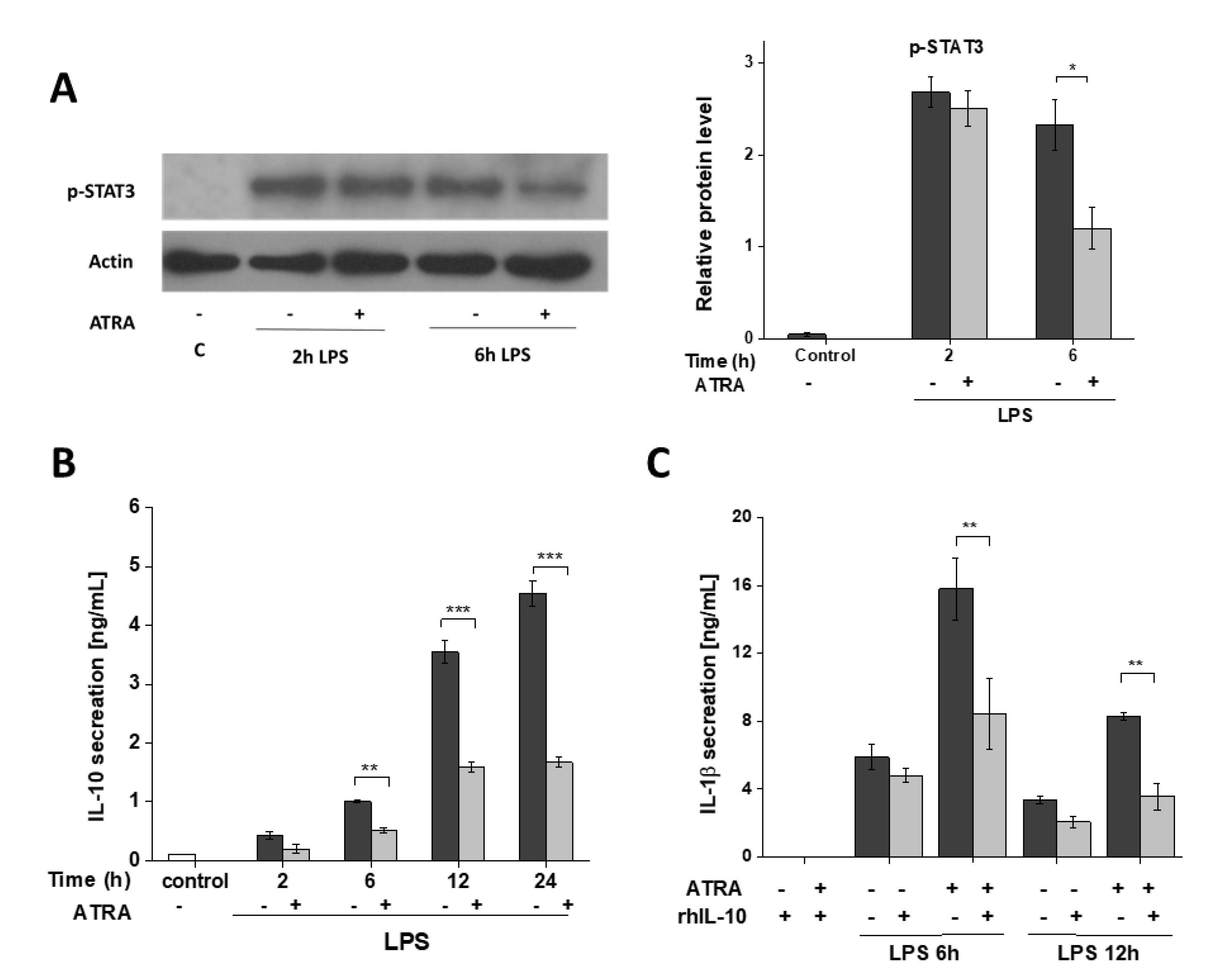
3.6. ATRA Attenuates Secretion of LPS-Induced IL-10
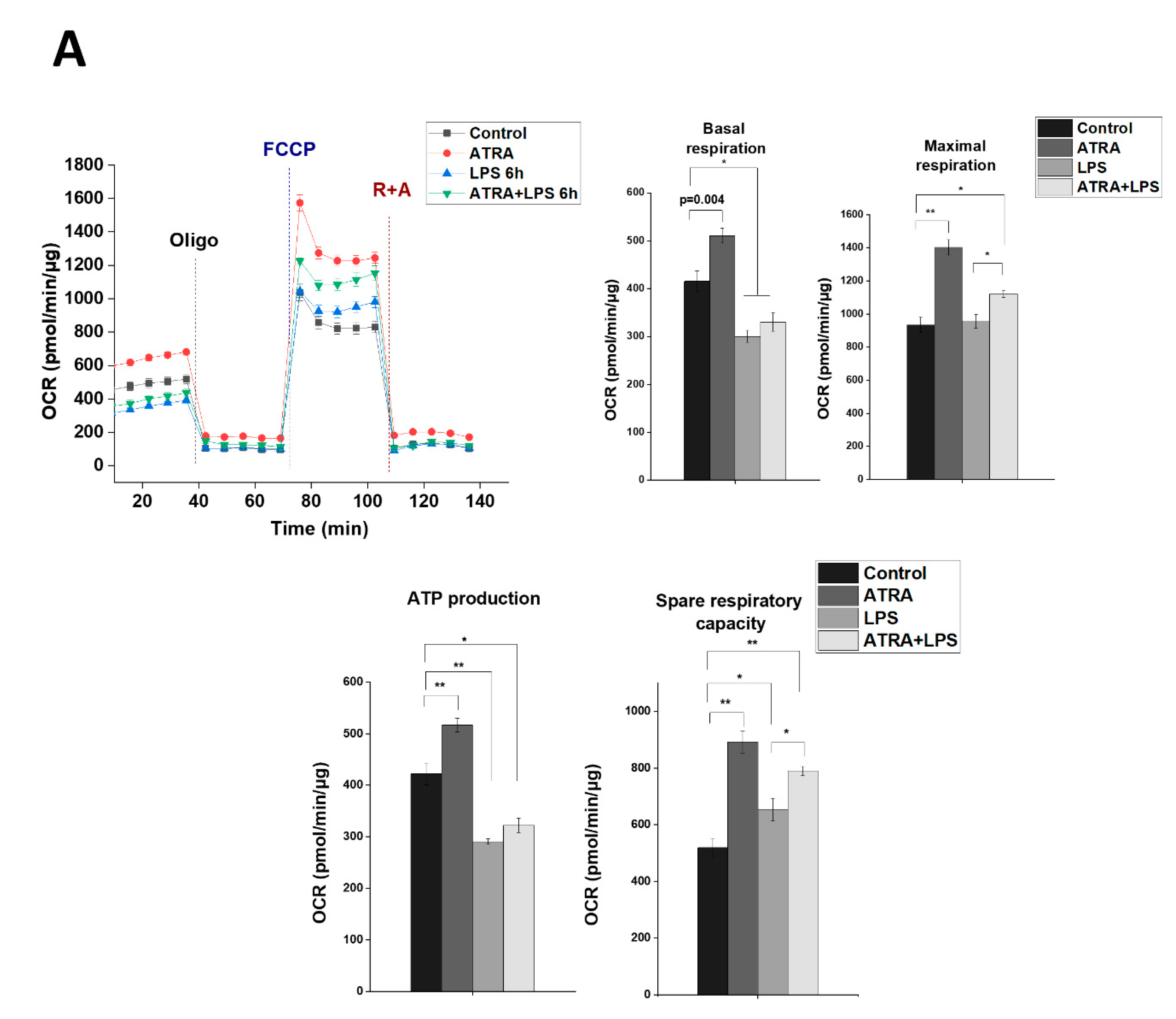
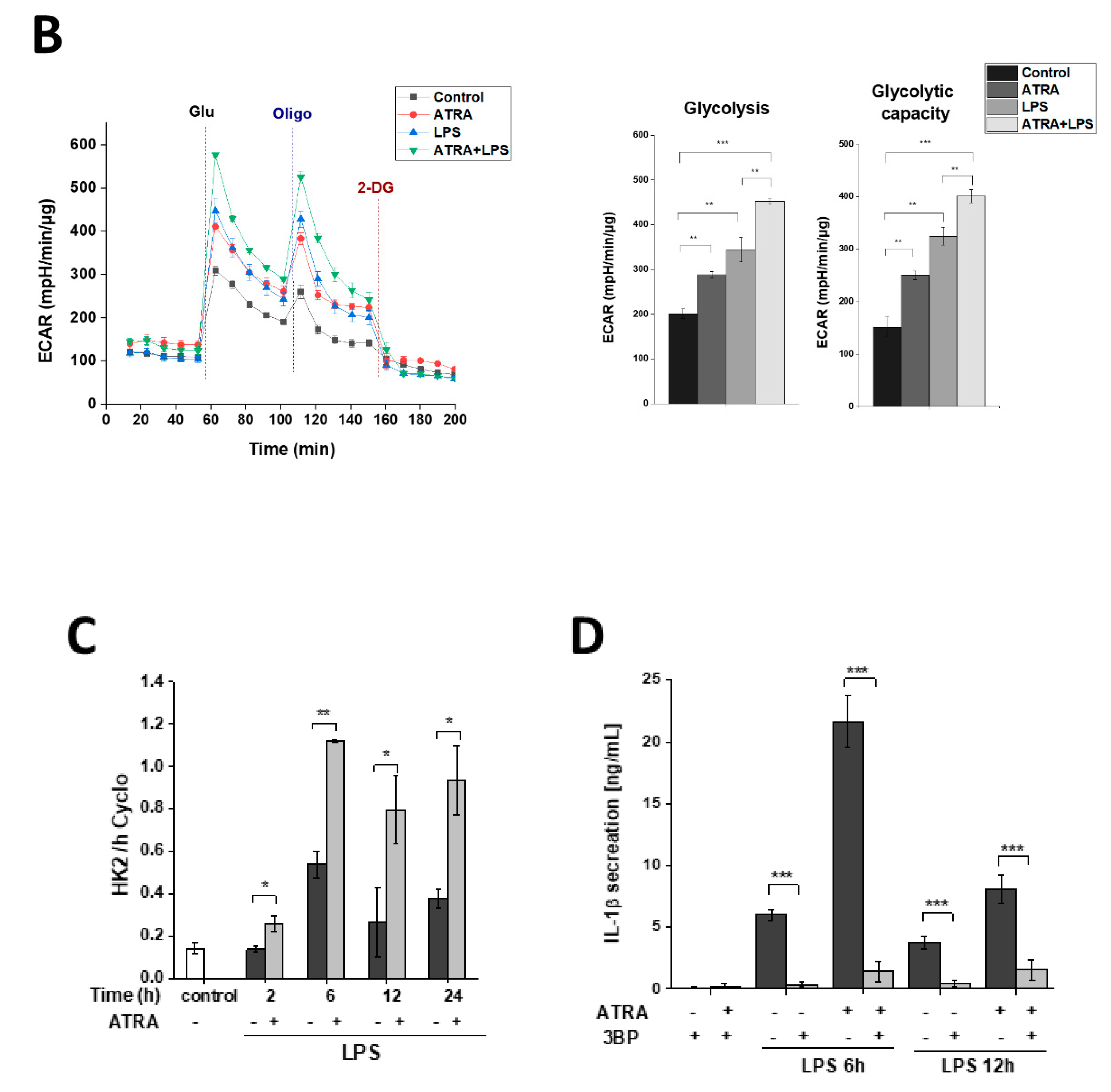
3.7. ATRA Mediates a Metabolic Shift Towards Glycolysis in LPS-Stimulated MΦs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar]
- Szekanecz, Z.; Szamosi, S.; Kovács, G.E.; Kocsis, E.; Benkő, S. The NLRP3 inflammasome-interleukin 1 pathway as a therapeutic target in gout. Arch. Biochem. Biophys. 2019, 670, 82–93. [Google Scholar] [CrossRef]
- Shao, B.-Z.; Xu, Z.-Q.; Han, B.-Z.; Su, D.-F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Fusco, R.; D’amico, R.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Absence of formyl peptide receptor 1 causes endometriotic lesion regression in a mouse model of surgically-induced endometriosis. Oncotarget 2018, 9, 31355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, R.; Fusco, R.; Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Impellizzeri, D.; Perretti, M.; Cuzzocrea, S. Formyl peptide receptor 1 signalling promotes experimental colitis in mice. Pharmacol. Res. 2019, 141, 591–601. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, A.; Li, B.; Kombe, J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, R.; Fusco, R.; Cordaro, M.; Siracusa, R.; Peritore, A.F.; Gugliandolo, E.; Crupi, R.; Scuto, M.; Cuzzocrea, S.; Di Paola, R. Modulation of NLRP3 Inflammasome through Formyl Peptide Receptor 1 (Fpr-1) Pathway as a New Therapeutic Target in Bronchiolitis Obliterans Syndrome. Int. J. Mol. Sci. 2020, 21, 2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawana, N.; Yamamoto, Y.; Kino, Y.; Satoh, J.-I. Molecular Network of NLRP3 Inflammasome Activation-Responsive Genes in a Human Monocyte Cell Line. Austin J. Clin. Immunol. 2014, 1, 1017. [Google Scholar]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wager, C.M.L.; Arnett, E.; Schlesinger, L.S. Macrophage nuclear receptors: Emerging key players in infectious diseases. PLoS Pathog. 2019, 15. [Google Scholar]
- Benko, S.; Love, J.D.; Beládi, M.; Schwabe, J.W.; Nagy, L. Molecular determinants of the balance between co-repressor and co-activator recruitment to the retinoic acid receptor. J. Biol. Chem. 2003, 278, 43797–43806. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological relevance. Atertio. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef]
- Le Maire, A.; Teyssier, C.; Balaguer, P.; Bourguet, W.; Germain, P. Regulation of RXR-RAR Heterodimers by RXR-and RAR-Specific Ligands and Their Combinations. Cells 2019, 8, 1392. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.d.M.; Teixeira, F.M.E.; Sato, M.N. Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediators Inflamm. 2018, 2018, 3067126. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular metabolism and macrophage functional polarization. Int. Rev. Immunol. 2015, 34, 82–100. [Google Scholar] [CrossRef]
- Masiá, S.; Alvarez, S.; de Lera, A.R.; Barettino, D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol. Endocrinol. 2007, 21, 2391–2402. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, W.; Zhang, X.; Bai, J.; Chen, G.; Li, L.; Li, M. All-trans retinoic acid-induced deficiency of the Wnt/β-catenin pathway enhances hepatic carcinoma stem cell differentiation. PLoS ONE 2015, 10, e0143255. [Google Scholar] [CrossRef] [Green Version]
- García-Regalado, A.; Vargas, M.; García-Carrancá, A.; Aréchaga-Ocampo, E.; González-De la Rosa, C.H. Activation of Akt pathway by transcription-independent mechanisms of retinoic acid promotes survival and invasion in lung cancer cells. Mol. Cancer 2013, 12, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, S.-K.; Park, M.-K.; Cho, M.-L.; Oh, H.-J.; Park, E.-M.; Lee, D.-G.; Lee, J.; Kim, H.-Y.; Park, S.-H. Retinoic acid attenuates rheumatoid inflammation in mice. J. Immunol. 2012, 189, 1062–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnewski, P.; Das, S.; Parigi, S.M.; Villablanca, E. Retinoic acid and its role in modulating intestinal innate immunity. Nutrients 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semba, R.D. Vitamin A and immunity to viral, bacterial and protozoan infections. Proc. Nutr. Soc. 1999, 58, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Larange, A.; Cheroutre, H. Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu. Rev. Immunol. 2016, 34, 369–394. [Google Scholar] [CrossRef]
- Wheelwright, M.; Kim, E.W.; Inkeles, M.S.; De Leon, A.; Pellegrini, M.; Krutzik, S.R.; Liu, P.T. All-trans retinoic acid–triggered antimicrobial activity against Mycobacterium tuberculosis Is Dependent on NPC2. J. Immunol. 2014, 192, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Babina, M.; Guhl, S.; Motakis, E.; Artuc, M.; Hazzan, T.; Worm, M.; Forrest, A.R.; Zuberbier, T. Retinoic acid potentiates inflammatory cytokines in human mast cells: Identification of mast cells as prominent constituents of the skin retinoid network. Mol. Cell. Endocrinol. 2015, 406, 49–59. [Google Scholar] [CrossRef]
- Yamada, H.; Mizuno, S.; Ross, A.C.; Sugawara, I. Retinoic acid therapy attenuates the severity of tuberculosis while altering lymphocyte and macrophage numbers and cytokine expression in rats infected with Mycobacterium tuberculosis. J. Nut. 2007, 137, 2696–2700. [Google Scholar] [CrossRef] [Green Version]
- Trechsel, U.; Evêquoz, V.; Fleisch, H. Stimulation of interleukin 1 and 3 production by retinoic acid in vitro. Biochem. J. 1985, 230, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Matikainen, S.; Serkkola, E.; Hurme, M. Retinoic acid enhances IL-1 beta expression in myeloid leukemia cells and in human monocytes. J. Immunol. 1991, 147, 162–167. [Google Scholar]
- Hayashi, S.; Hashimoto, S.; Kitamura, N.; Hanazawa, S.; Horie, T. Retinoic acid regulates differentially the expression of IL-1β and IL-1 receptor antagonist (IL-1ra) in PMA-activated human monocytes. Biochm. Bophys. Res. Commun. 1996, 224, 574–578. [Google Scholar] [CrossRef]
- Anand, P.K.; Malireddi, R.; Kanneganti, T.-D. Role of the nlrp3 inflammasome in microbial infection. Front. Microbiol. 2011, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Budai, M.M.; Tőzsér, J.; Benkő, S. Different dynamics of NLRP3 inflammasome-mediated IL-1β production in GM-CSF–and M-CSF–differentiated human macrophages. J. Leukocyte Biol. 2017, 101, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Piskunov, A.; Rochette-Egly, C. A retinoic acid receptor RARα pool present in membrane lipid rafts forms complexes with G protein αQ to activate p38MAPK. Oncogene 2012, 31, 3333–3345. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Säemann, M.D. The multiple facets of mTOR in immunity. Trends Immunol. 2009, 30, 218–226. [Google Scholar] [CrossRef]
- Schmitz, F.; Heit, A.; Dreher, S.; Eisenächer, K.; Mages, J.; Haas, T.; Krug, A.; Janssen, K.P.; Kirschning, C.J.; Wagner, H. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur. J. Immunol. 2008, 38, 2981–2992. [Google Scholar] [CrossRef]
- Jones, R.G.; Pearce, E.J. MenTORing immunity: mTOR signaling in the development and function of tissue-resident immune cells. Immunity 2017, 46, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Hörl, W.H.; Hengstschläger, M. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32. [Google Scholar]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Res. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.b.; Gu, J.d.; Zhou, Q.h. Review of aerobic glycolysis and its key enzymes–new targets for lung cancer therapy. Thoracic Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Cassani, B.; Villablanca, E.J.; De Calisto, J.; Wang, S.; Mora, J.R. Vitamin A and immune regulation: Role of retinoic acid in gut-associated dendritic cell education, immune protection and tolerance. Mol. Asp. Med. 2012, 33, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Mora, J.R.; Iwata, M.; Von Andrian, U.H. Vitamin effects on the immune system: Vitamins A and D take centre stage. Nat. Rev. Immunol. 2008, 8, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.L.; Amory, J.K.; Walsh, T.J.; Isoherranen, N. A sensitive and specific method for measurement of multiple retinoids in human serum with UHPLC-MS/MS. J. Lipid Res. 2012, 53, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.M.; Basdeo, S.A.; Coleman, A.M.; Cheallaigh, C.N.; Peral de Castro, C.; McLaughlin, A.M.; Dunne, P.J.; Harris, J.; Keane, J. All-trans retinoic acid augments autophagy during intracellular bacterial infection. Am. J. Respir. Cell Mol. Biol. 2018, 59, 548–556. [Google Scholar] [CrossRef]
- Na, S.-Y.; Kang, B.Y.; Chung, S.W.; Han, S.-J.; Ma, X.; Trinchieri, G.; Im, S.-Y.; Lee, J.W.; Kim, T.S. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFκB. J. Biol. Chem. 1999, 274, 7674–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, B.; Chung, S.; Kim, S.; Kang, S.; Choe, Y.; Kim, T.S. Retinoid-mediated inhibition of interleukin-12 production in mouse macrophages suppresses Th1 cytokine profile in CD4+ T cells. Br. J. Pharmacol. 2000, 130, 581–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Allen, C.; Ballow, M. Retinoic acid enhances the production of IL-10 while reducing the synthesis of IL-12 and TNF-α from LPS-stimulated monocytes/macrophages. J. Clin. Immunol. 2007, 27, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Torres, R. Role of interleukin-1β during pain and inflammation. Brain Res. Rev. 2009, 60, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siggers, T.; Gordaˆn, R. Protein–DNA binding: Complexities and multi-protein codes. Nucleic Acids Res. 2014, 42, 2099–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.; Whitfield, T.W.; Greven, M.C.; Pierce, B.G.; Dong, X.; Kundaje, A.; Cheng, Y. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012, 22, 1798–1812. [Google Scholar] [CrossRef] [Green Version]
- Nagy, G.; Daniel, B.; Cuaranta-Monroy, I.; Nagy, L. Unraveling the hierarchy of cis and trans factors that determine the DNA binding by PPARγ. Mol. Cell. Biol. 2020, 40, e00547-19. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 inflammasome by phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yao, J.; Zou, C.; Zhang, H.; Zhang, S.; Liu, J.; Ma, G.; Jiang, P.; Zhang, W. Asiatic acid protects against hepatic ischemia/reperfusion injury by inactivation of Kupffer cells via PPARγ/NLRP3 inflammasome signaling pathway. Oncotarget 2017, 8, 86339. [Google Scholar] [CrossRef]
- Tulk, S.E.; Liao, K.C.; Muruve, D.A.; Li, Y.; Beck, P.L.; MacDonald, J.A. Vitamin D3 Metabolites Enhance the NLRP3-Dependent Secretion of IL-1β From Human THP-1 Monocytic Cells. J. Cell. Biochem. 2015, 116, 711–720. [Google Scholar] [CrossRef]
- Billon, C.; Murray, M.H.; Avdagic, A.; Burris, T. RORγ regulates the NLRP3 inflammasome. J. Biol. Chem. 2019, 294, 10–19. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekedereli, I.; Akar, U.; Alpay, S.N.; Lopez-Berestein, G.; Ozpolat, B. Autophagy is Required to Regulate Mitochondria Renewal, Cell Attachment, and All-trans–Retinoic Acid–Induced Differentiation in NB4 Acute Promyelocytic Leukemia Cells. J. Environ. Pathol. Toxicol. Oncol. 2019, 38, 13–20. [Google Scholar] [CrossRef]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Moon, J.-S.; Hisata, S.; Park, M.-A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897. [Google Scholar] [CrossRef]
- Tannahill, G.; Curtis, A.; Adamik, J.; Palsson-McDermott, E.; McGettrick, A.; Goel, G.; Frezza, C.; Bernard, N.; Kelly, B.; Foley, N. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238. [Google Scholar] [CrossRef]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Sanman, L.E.; Qian, Y.; Eisele, N.A.; Ng, T.M.; van der Linden, W.A.; Monack, D.M.; Weerapana, E.; Bogyo, M. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 2016, 5, e13663. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; O’Neill, L.A. Metabolic regulation of NLRP 3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Hexokinase-2 Glycolytic Overload in Diabetes and Ischemia–Reperfusion Injury. Trends Endocrinol. Metab. 2019, 30, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D.E. MondoA senses non-glucose sugars regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136. [Google Scholar] [CrossRef]
- Rhee, E.-J.; Plutzky, J. Retinoid metabolism and diabetes mellitus. Diabetes Metab. J. 2012, 36, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.M.; Lee, J.O.; Jung, J.-H.; Kim, J.H.; Park, S.-H.; Park, J.M.; Kim, E.-K.; Suh, P.-G.; Kim, H.S. Retinoic acid leads to cytoskeletal rearrangement through AMPK-Rac1 and stimulates glucose uptake through AMPK-p38 MAPK in skeletal muscle cells. J. Biol. Chem. 2008, 283, 33969–33974. [Google Scholar] [CrossRef] [Green Version]
- Ip, W.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, Y.; Qi, G.; Brand, D.; Zheng, S.G. Role of vitamin A in the immune system. J. Clin. Med. 2018, 7, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aibana, O.; Franke, M.F.; Huang, C.-C.; Galea, J.T.; Calderon, R.; Zhang, Z.; Becerra, M.C.; Smith, E.R.; Ronnenberg, A.G.; Contreras, C. Impact of vitamin A and carotenoids on the risk of tuberculosis progression. Clin. Infect. Dis. 2017, 65, 900–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, A.; Foster, A.; Sommer, A. Vitamin A supplements and mortality related to measles: A randomised clinical trial. Br. Med. J. (Clin. Res. Ed.) 1987, 294, 294–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussey, G.D.; Klein, M. A randomized, controlled trial of vitamin A in children with severe measles. N. Engl. J. Med. 1990, 323, 160–164. [Google Scholar] [CrossRef]
- Friis, H.; Mwaniki, D.; Omondi, B.; Muniu, E.; Magnussen, P.; Geissler, W.; Thiong’o, F.; Michaelsen, K.F. Serum retinol concentrations and Schistosoma mansoni, intestinal helminths, and malarial parasitemia: A cross-sectional study in Kenyan preschool and primary school children. Am. J. Clin.Nutr. 1997, 66, 665–671. [Google Scholar] [CrossRef]
- Dreno, B.; Gollnick, H.; Kang, S.; Thiboutot, D.; Bettoli, V.; Torres, V.; Leyden, J.; Acne, G.A.T.I.O.I. Understanding innate immunity and inflammation in acne: Implications for management. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, S.; Werner, L.; Watson, R.R. High dietary vitamin A (retinyl palmitate) and cellular immune functions in mice. Immunology 1985, 56, 169. [Google Scholar]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alatshan, A.; Kovács, G.E.; Aladdin, A.; Czimmerer, Z.; Tar, K.; Benkő, S. All-Trans Retinoic Acid Enhances both the Signaling for Priming and the Glycolysis for Activation of NLRP3 Inflammasome in Human Macrophage. Cells 2020, 9, 1591. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9071591
Alatshan A, Kovács GE, Aladdin A, Czimmerer Z, Tar K, Benkő S. All-Trans Retinoic Acid Enhances both the Signaling for Priming and the Glycolysis for Activation of NLRP3 Inflammasome in Human Macrophage. Cells. 2020; 9(7):1591. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9071591
Chicago/Turabian StyleAlatshan, Ahmad, Gergő E. Kovács, Azzam Aladdin, Zsolt Czimmerer, Krisztina Tar, and Szilvia Benkő. 2020. "All-Trans Retinoic Acid Enhances both the Signaling for Priming and the Glycolysis for Activation of NLRP3 Inflammasome in Human Macrophage" Cells 9, no. 7: 1591. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9071591