K+ Channel Tetramerization Domain 5 (KCTD5) Protein Regulates Cell Migration, Focal Adhesion Dynamics and Spreading through Modulation of Ca2+ Signaling and Rac1 Activity

 , and
, and
Abstract
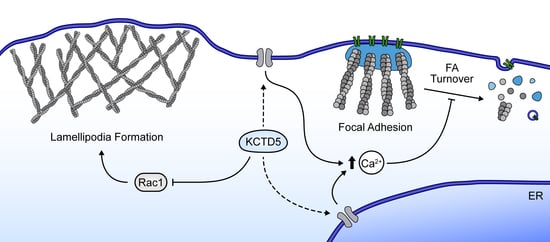
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plasmids
2.3. Antibodies
2.4. Cell Culture and Transfection
2.5. KCTD5 Knockout B16-F10 (B16-F10kctd5-/-) and Control B16-F10 (B16-F10Control) Cells Generation
2.6. Immunoblot Analyses
2.7. mRNA Expression Analyses by RT-qPCR
2.8. Boyden Chamber Transwell Migration Assays
2.9. Spreading Assay
2.10. Focal Adhesion Analyses
2.11. Focal Adhesions Dynamics Analyses
2.12. Intracellular Calcium Measurements
2.13. Statistical Analyses
3. Results
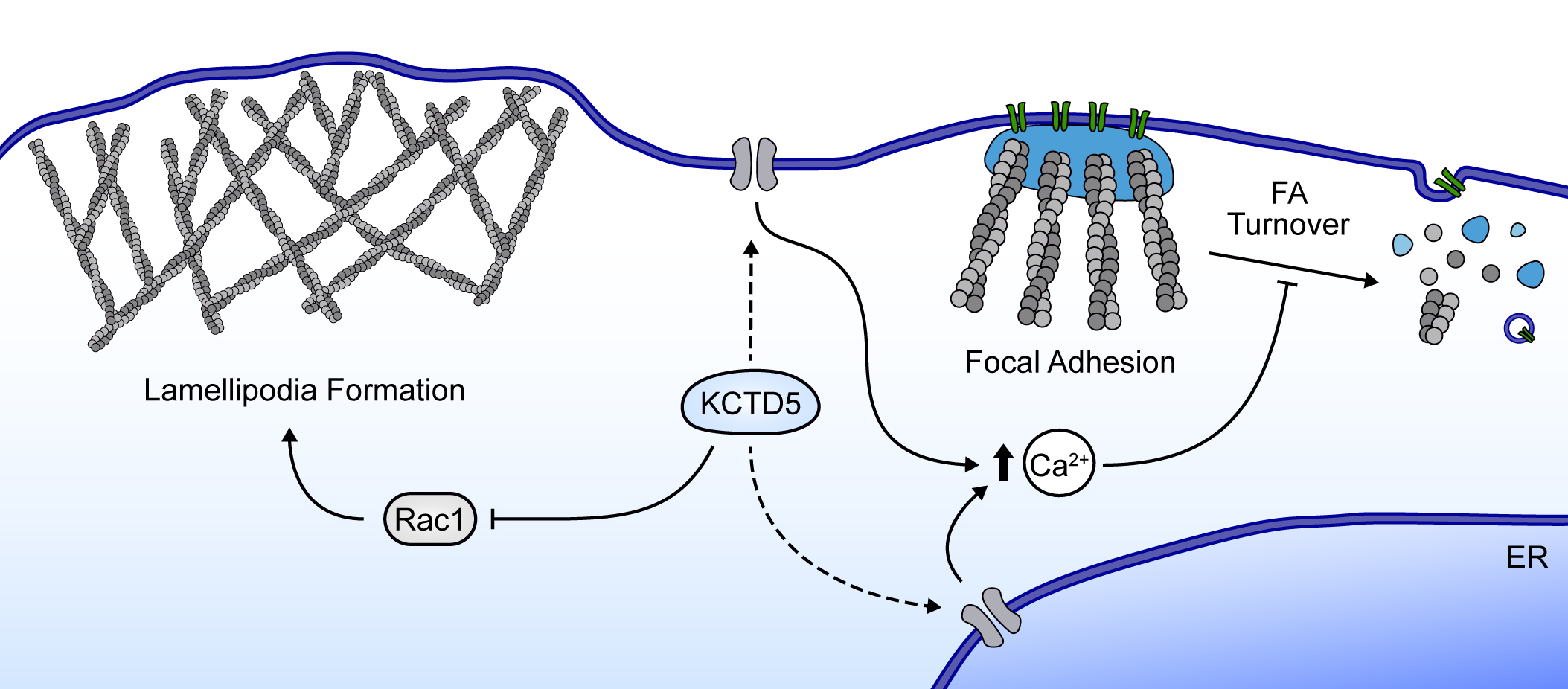
3.1. KCTD5 Depletion Increases Cell Migration of B16-F10 Cells
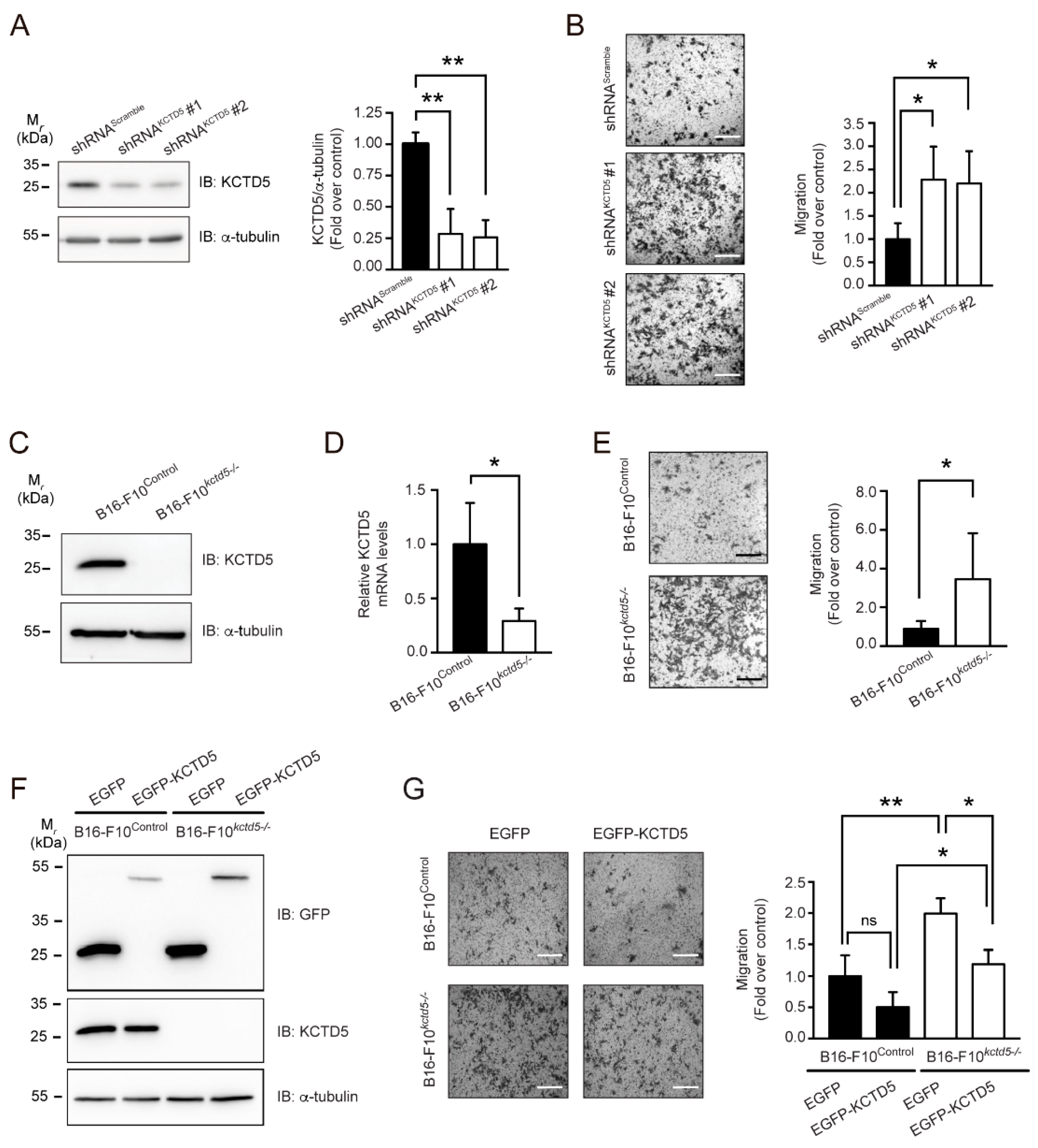
3.2. KCTD5 Depletion Enhances the Spreading of B16-F10 Cells
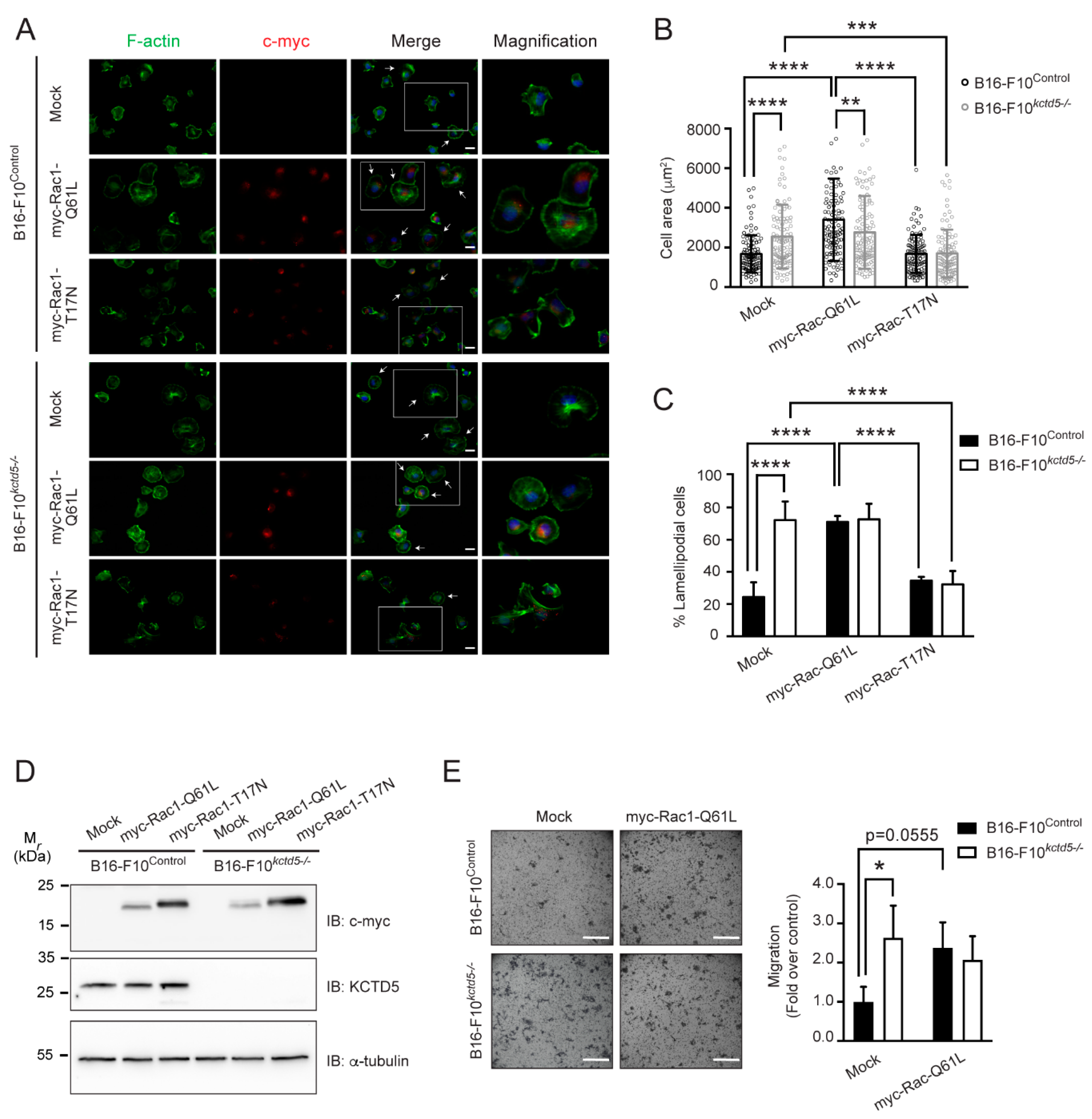
3.3. Cell Spreading and Migration Promoted by KCTD5-Depletion Is Dependent on Rac1 Activity
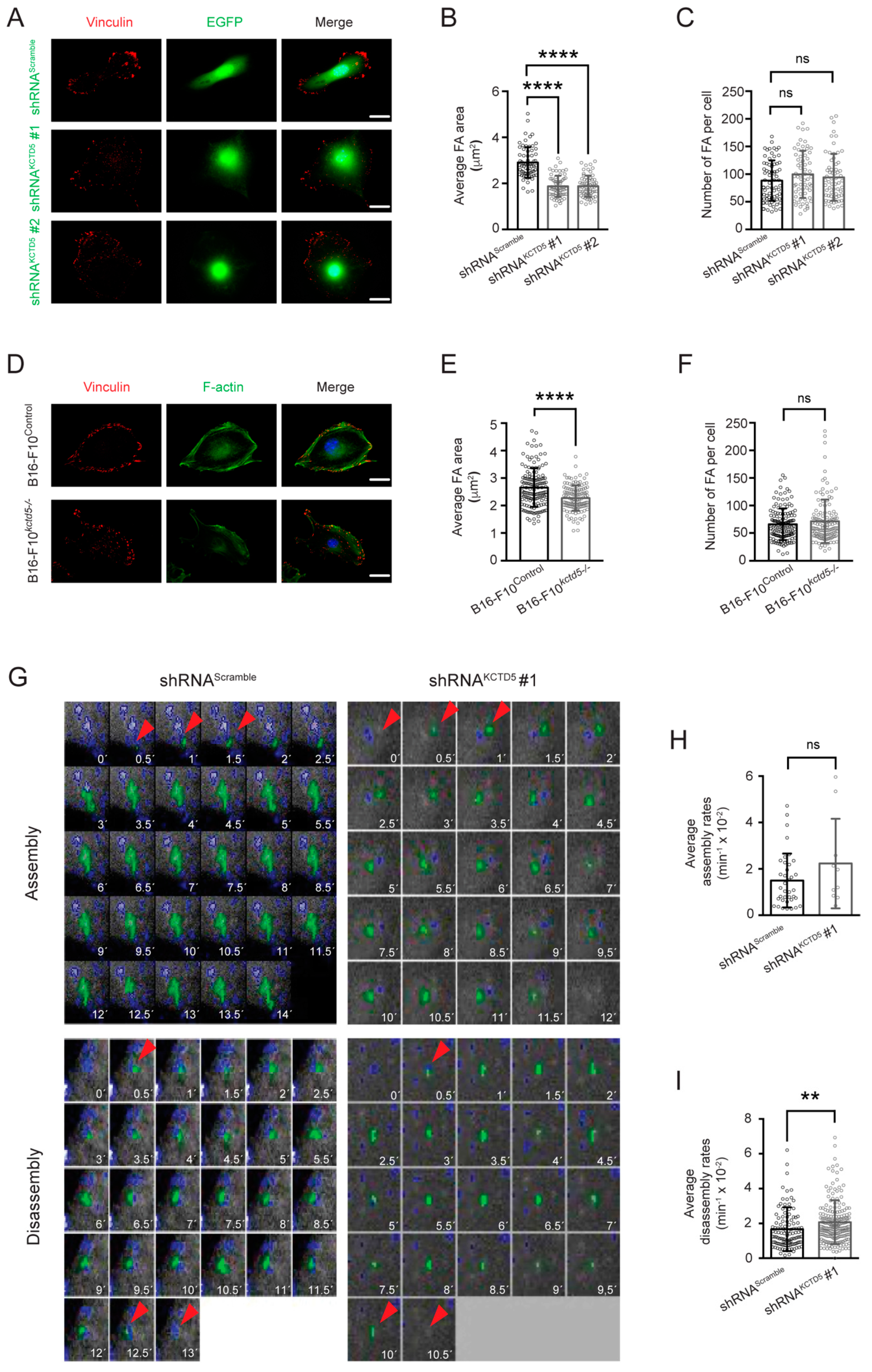
3.4. KCTD5 Depletion Affects Focal Adhesion Dynamics of B16-F10 Cells
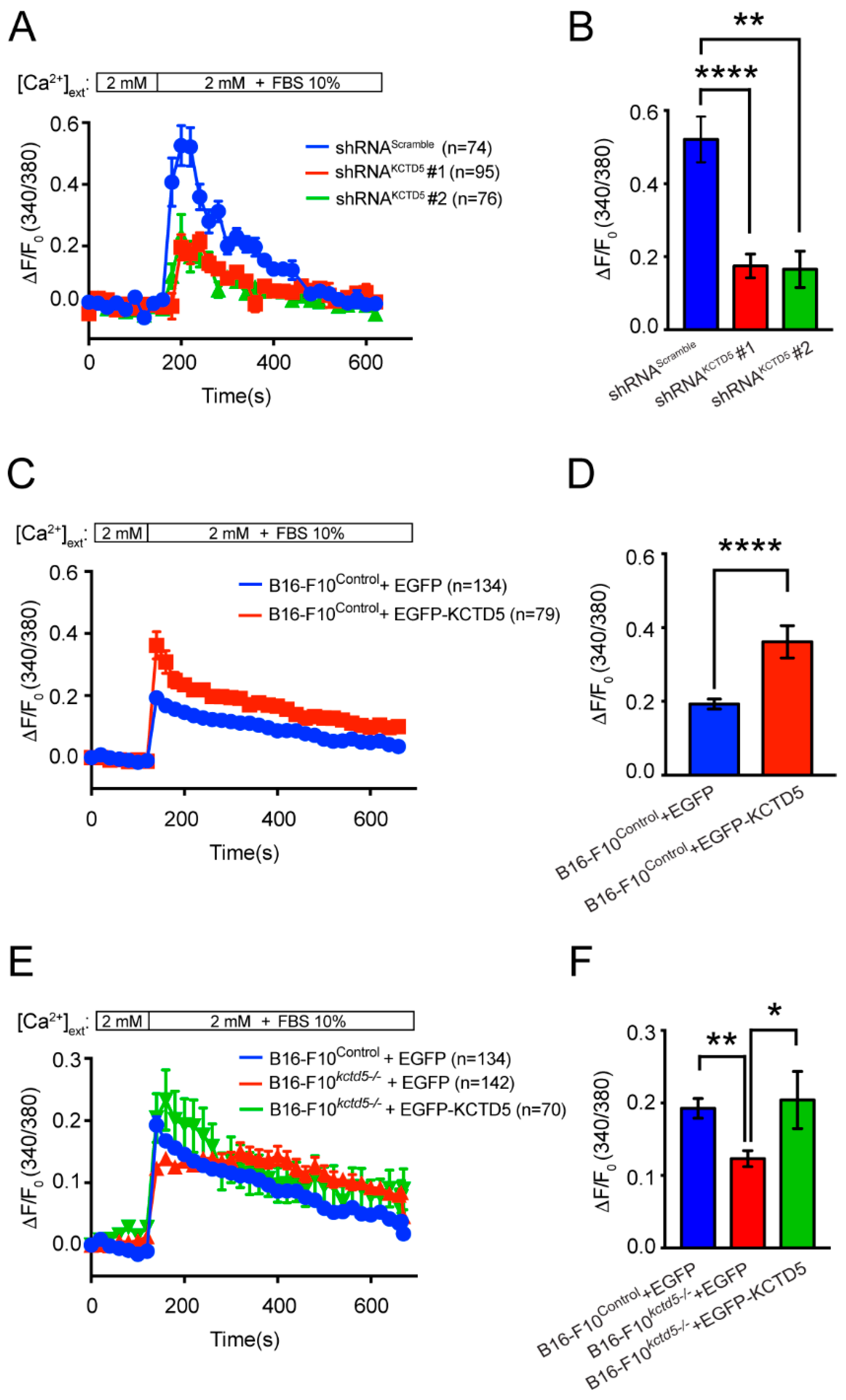
3.5. KCTD5 Depletion Impacts in Serum-Induced Ca2+ Signaling
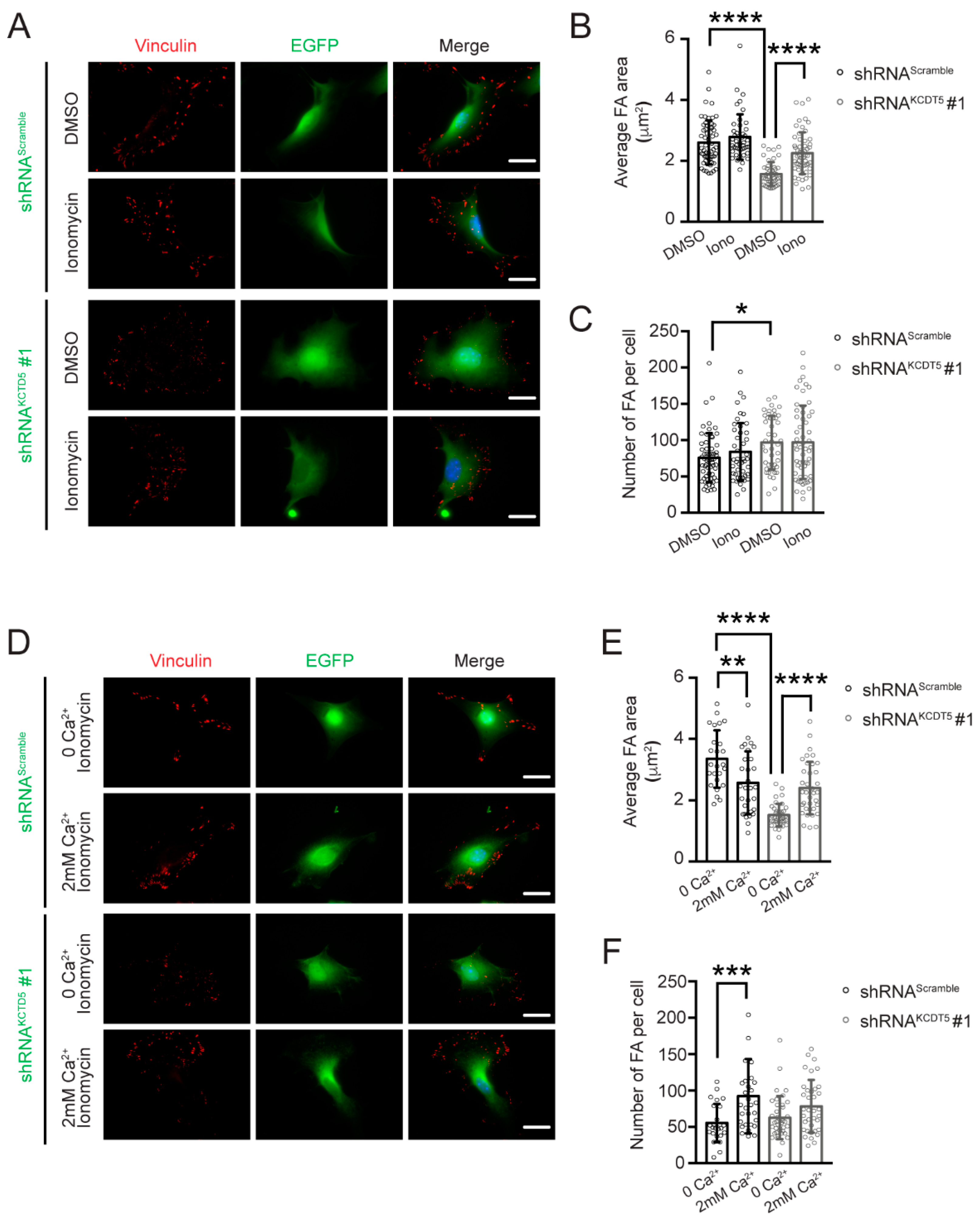
3.6. Serum-Induced Ca2+ Rise Is Necessary for KCTD5-Mediated Focal Adhesion Regulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Locascio, A.; Nieto, M.A. Cell movements during vertebrate development: Integrated tissue behaviour versus individual cell migration. Curr. Opin. Genet. Dev. 2001, 11, 464–469. [Google Scholar] [CrossRef]
- Luster, A.D.; Alon, R.; Von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012, 24, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.J.; Martin, P. Wound repair: A showcase for cell plasticity and migration. Curr. Opin. Cell Biol. 2016, 42, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Lauffenburger, D.A.; Horwitz, A.F. Cell Migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho GTPases and the Actin Cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Small, J.; Stradal, T.E.; Vignal, E.; Rottner, K. The lamellipodium: Where motility begins. Trends Cell Biol. 2002, 12, 112–120. [Google Scholar] [CrossRef]
- Mattila, P.K.; Lappalainen, P. Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Tsai, F.-C.; Meyer, T. Ca2+ Pulses Control Local Cycles of Lamellipodia Retraction and Adhesion along the Front of Migrating Cells. Curr. Biol. 2012, 22, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, S.J.; Rodgers, M.A.; Perrin, B.J.; Han, J.; Bennin, D.A.; Critchley, D.R.; Huttenlocher, A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 2004, 6, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Bennin, D.A.; Huttenlocher, A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 2010, 285, 11418–11426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glading, A.; Lauffenburger, D.A.; Wells, A. Cutting to the chase: Calpain proteases in cell motility. Trends Cell Biol. 2002, 12, 46–54. [Google Scholar] [CrossRef]
- Giannone, G.; Rondé, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium Rises Locally Trigger Focal Adhesion Disassembly and Enhance Residency of Focal Adhesion Kinase at Focal Adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, M.A.; Maxfield, F.R. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nat. Cell Biol. 1995, 377, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.H.; Falke, J.J. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 16176–16181. [Google Scholar] [CrossRef] [Green Version]
- Larsson, C. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 2006, 18, 276–284. [Google Scholar] [CrossRef]
- Zhao, J.-W.; Gao, Z.-L.; Zhang, H.-Y.; Ji, Q.-Y.; Wang, H.; Yang, Y.-D.; Xing, F.-J.; Meng, L.-J.; Wang, Y. Regulation of Cofilin Activity by CaMKII and Calcineurin. Am. J. Med. Sci. 2012, 344, 462–472. [Google Scholar] [CrossRef]
- Price, L.S.; Langeslag, M.; Hordijk, P.L.; Jalink, K.; Collard, J.G.; Klooster, J.P.T. Calcium Signaling Regulates Translocation and Activation of Rac. J. Biol. Chem. 2003, 278, 39413–39421. [Google Scholar] [CrossRef] [Green Version]
- Dovas, A.; Yoneda, A.; Couchman, J.R. PKCbeta-dependent activation of RhoA by syndecan-4 during focal adhesion formation. J. Cell Sci. 2006, 119, 2837–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Xiang, Y.; Sun, G. The KCTD family of proteins: Structure, function, disease relevance. Cell Biosci. 2013, 3, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayón, Y.; Trinidad, A.G.; De La Puerta, M.L.; Rodriguez, M.D.C.; Bogetz, J.; Rojas, A.M.; De Pereda, J.M.; Rahmouni, S.; Williams, S.; Matsuzawa, S.-I.; et al. KCTD5, a putative substrate adaptor for cullin3 ubiquitin ligases. FEBS J. 2008, 275, 3900–3910. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin Mediates Degradation of RhoA through Evolutionarily Conserved BTB Adaptors to Control Actin Cytoskeleton Structure and Cell Movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Di Marcotullio, L.; Moretti, M.; Pelloni, M.; Occhione, M.A.; Infante, P.; Cucchi, D.; Greco, A.; Pietrosanti, L.; Todorovic, J.; et al. Identification and Characterization of KCASH2 and KCASH3, 2 Novel Cullin3 Adaptors Suppressing Histone Deacetylase and Hedgehog Activity in Medulloblastoma. Neoplasia 2011, 13, 374–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.; Perera, S.; Teh, P.; Chen, J. Obscurin and KCTD6 regulate cullin-dependent small ankyrin-1 (sAnk1.5) protein turnover. Mol. Biol. Cell 2012, 23, 2490–2504. [Google Scholar] [CrossRef]
- Kasahara, K.; Kawakami, Y.; Kiyono, T.; Yonemura, S.; Kawamura, Y.; Era, S.; Matsuzaki, F.; Goshima, N.; Inagaki, M. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat. Commun. 2014, 5, 5081. [Google Scholar] [CrossRef] [Green Version]
- Ji, A.X.; Chu, A.; Nielsen, T.K.; Benlekbir, S.; Rubinstein, J.L.; Privé, G.G. Structural Insights into KCTD Protein Assembly and Cullin3 Recognition. J. Mol. Biol. 2016, 428, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-J.; Kim, S.-H.; Jin, X.; Jin, X.; Kim, H. KCTD2, an adaptor of Cullin3 E3 ubiquitin ligase, suppresses gliomagenesis by destabilizing c-Myc. Cell Death Differ. 2017, 24, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Kovacević, I.; Sakaue, T.; Majolée, J.; Pronk, M.C.; Maekawa, M.; Geerts, D.; Fernandez-Borja, M.; Higashiyama, S.; Hordijk, P. The Cullin-3–Rbx1–KCTD10 complex controls endothelial barrier function via K63 ubiquitination of RhoB. J. Cell Biol. 2018, 217, 1015–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, A.; Maekawa, M.; Kawai, K.; Nakayama, J.; Araki, N.; Semba, K.; Taguchi, T.; Kamei, Y.; Takada, Y.; Higashiyama, S. Cullin-3/KCTD10 E3 complex is essential for Rac1 activation through RhoB degradation in human epidermal growth factor receptor 2-positive breast cancer cells. Cancer Sci. 2019, 110, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Dementieva, I.S.; Tereshko, V.; McCrossan, Z.A.; Solomaha, E.; Araki, D.; Xu, C.; Grigorieff, N.; Goldstein, S.A.N. Pentameric Assembly of Potassium Channel Tetramerization Domain-Containing Protein 5. J. Mol. Biol. 2009, 387, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasco, N.; Pirone, L.; Smaldone, G.; Di Gaetano, S.; Esposito, L.; Pedone, E.M.; Vitagliano, L. Molecular recognition of Cullin3 by KCTDs: Insights from experimental and computational investigations. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1289–1298. [Google Scholar] [CrossRef]
- Rutz, N.; Heilbronn, R.; Weger, S. Interactions of cullin3/KCTD5 complexes with both cytoplasmic and nuclear proteins: Evidence for a role in protein stabilization. Biochem. Biophys. Res. Commun. 2015, 464, 922–928. [Google Scholar] [CrossRef]
- Brockmann, M.; Blomen, V.A.; Nieuwenhuis, J.; Stickel, E.; Raaben, M.; Bleijerveld, O.B.; Altelaar, A.F.M.; Jae, L.T.; Brummelkamp, T.R. Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling. Nat. Cell Biol. 2017, 546, 307–311. [Google Scholar] [CrossRef]
- He, H.; Peng, Y.; Fan, S.; Chen, Y.; Zheng, X.; Li, C. Cullin3/KCTD5 induces monoubiquitination of DeltaNp63alpha and impairs its activity. FEBS Lett. 2018, 592, 2334–2340. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, A.; Uribe, F.; Canales, J.; Romero, C.; Soza, A.; Peña, M.A.; Antonelli, M.; Almarza, O.; Cerda, O.; Toledo, H. KCTD5 and Ubiquitin Proteasome Signaling Are Required for Helicobacter pylori Adherence. Front. Cell. Infect. Microbiol. 2017, 7, 450. [Google Scholar] [CrossRef]
- Stavropoulos, N.; Young, M.W. insomniac and Cullin-3 Regulate Sleep and Wakefulness in Drosophila. Neuron 2011, 72, 964–976. [Google Scholar] [CrossRef] [Green Version]
- Rivas, J.; Díaz, N.; Silva, I.; Morales, D.; Lavanderos, B.; Álvarez, A.; Saldías, M.P.; Pulgar, E.; Cruz, P.; Maureira, D.; et al. KCTD5, a novel TRPM4-regulatory protein required for cell migration as a new predictor for breast cancer prognosis. FASEB J. 2020, 34, 7847–7865. [Google Scholar] [CrossRef] [Green Version]
- Cáceres, M.; Ortiz, L.; Recabarren, T.; Romero, A.; Colombo, A.; Leiva-Salcedo, E.; Varela, D.; Rivas, J.; Silva, I.; Morales, D.; et al. TRPM4 Is a Novel Component of the Adhesome Required for Focal Adhesion Disassembly, Migration and Contractility. PLoS ONE 2015, 10, e0130540. [Google Scholar] [CrossRef] [PubMed]
- Ishida-Takagishi, M.; Enomoto, A.; Asai, N.; Ushida, K.; Watanabe, T.; Hashimoto, T.; Kato, T.; Weng, L.; Matsumoto, S.; Asai, M.; et al. The Dishevelled-associating protein Daple controls the non-canonical Wnt/Rac pathway and cell motility. Nat. Commun. 2012, 3, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, C.; Morales, D.; Mogollones, I.; Vergara-Jaque, A.; Vargas, C.; Álvarez, A.; Riquelme, D.; Leiva-Salcedo, E.; González, W.; Morales, D.; et al. EB1- and EB2-dependent anterograde trafficking of TRPM4 regulates focal adhesion turnover and cell invasion. FASEB J. 2019, 33, 9434–9452. [Google Scholar] [CrossRef] [Green Version]
- Berginski, M.E.; Gomez, S.M. The Focal Adhesion Analysis Server: A web tool for analyzing focal adhesion dynamics. F1000Research 2013, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK–Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef]
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eprevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef]
- Wei, C.; Wang, X.; Zheng, M.; Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 2012, 24, 254–261. [Google Scholar] [CrossRef]
- Launay, P.; Cheng, H.; Srivatsan, S.; Penner, R.; Fleig, A.; Kinet, J.-P. TRPM4 Regulates Calcium Oscillations After T Cell Activation. Science 2004, 306, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.-C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, A.; Nethe, M.; Hordijk, P.L. Ubiquitin links to cytoskeletal dynamics, cell adhesion and migration. Biochem. J. 2012, 442, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, W.; Liu, Y.; Liu, X.; Cai, L.; Kang, J.; Zhang, Y.; Chen, W.; Dong, C.; Zhang, Y.; et al. The CRL3BTBD9 E3 ubiquitin ligase complex targets TNFAIP1 for degradation to suppress cancer cell migration. Signal Transduct. Target. Ther. 2020, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Genau, H.M.; Huber, J.; Baschieri, F.; Akutsu, M.; Dotsch, V.; Farhan, H.; Rogov, V.; Behrends, C. CUL3-KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1-RAC1 signaling. Mol. Cell. 2015, 57, 995–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.F.; Rahmouni, K. The Bardet-Biedl syndrome protein complex regulates cell migration and tissue repair through a Cullin-3/RhoA pathway. Am. J. Physiol. Cell. Physiol. 2019, 317, C457–C465. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-C.; Lee, Y.-R.; Huang, S.-F.; Lin, Y.-M.; Chen, T.-Y.; Chung, H.-C.; Tsai, C.-H.; Chen, H.-Y.; Chiang, C.-T.; Lai, C.-K.; et al. A Cullin3-KLHL20 Ubiquitin Ligase-Dependent Pathway Targets PML to Potentiate HIF-1 Signaling and Prostate Cancer Progression. Cancer Cell 2011, 20, 214–228. [Google Scholar] [CrossRef] [Green Version]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 Ubiquitin-Ligase HACE1 Catalyzes the Ubiquitylation of Active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Mialki, R.K.; Wei, J.; Coon, T.A.; Zou, C.; Chen, B.B.; Mallampalli, R.K.; Zhao, Y. SCF E3 ligase F-box protein complex SCF FBXL19 regulates cell migration by mediating Rac1 ubiquitination and degradation. FASEB J. 2013, 27, 2611–2619. [Google Scholar] [CrossRef] [Green Version]
- Oberoi, T.K.; Dogan, T.; Hocking, J.C.; Scholz, R.-P.; Mooz, J.; Anderson, C.L.; Karreman, C.; Zu Heringdorf, D.M.; Schmidt, G.; Ruonala, M.; et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2011, 31, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Lluva, S.; Tan, C.T.; Daugaard, M.; Sorensen, P.H.B.; Malliri, A. The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation. Oncogene 2012, 32, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.K.; Kholodenko, B.N.; Von Kriegsheim, A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases 2016, 9, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, J.; Metz, M.; Zolles, G.; Turecek, R.; Fritzius, T.; Bildl, W.; Tarusawa, E.; Kulik, A.; Unger, A.; Ivankova, K.; et al. Native GABAB receptors are heteromultimers with a family of auxiliary subunits. Nat. Cell Biol. 2010, 465, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Cao-Ehlker, X.; Zong, X.; Hammelmann, V.; Gruner, C.; Fenske, S.; Michalakis, S.; Wahl-Schott, C.; Biel, M. Up-regulation of Hyperpolarization-activated Cyclic Nucleotide-gated Channel 3 (HCN3) by Specific Interaction with K+ Channel Tetramerization Domain-containing Protein 3 (KCTD3). J. Biol. Chem. 2013, 288, 7580–7589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbet, G.; Demion, M.; Moura, I.C.; Serafini, N.; Léger, T.; Vrtovsnik, F.; Monteiro, R.C.; Guinamard, R.; Kinet, J.-P.; Launay, P. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat. Immunol. 2008, 9, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzmann, C.; Kappel, S.; Kilch, T.; Jochum, M.M.; Urban, S.K.; Jung, V.; Stöckle, M.; Rother, K.; Greiner, M.; Peinelt, C. Transient receptor potential melastatin 4 channel contributes to migration of androgen-insensitive prostate cancer cells. Oncotarget 2015, 6, 41783–41793. [Google Scholar] [CrossRef] [PubMed]
- Kappel, S.; Stokłosa, P.; Hauert, B.; Ross-Kaschitza, D.; Borgström, A.; Baur, R.; Galván, J.A.; Zlobec, I.; Peinelt, C. TRPM4 is highly expressed in human colorectal tumor buds and contributes to proliferation, cell cycle, and invasion of colorectal cancer cells. Mol. Oncol. 2019, 13, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Wirtz, D. Focal Adhesion Size Uniquely Predicts Cell Migration. Biophys. J. 2013, 104, 319a. [Google Scholar] [CrossRef]
- Li, L.; Duan, T.; Wang, X.; Zhang, R.-H.; Zhang, M.; Wang, S.; Wang, F.; Wu, Y.; Huang, H.; Kang, T. KCTD12 Regulates Colorectal Cancer Cell Stemness through the ERK Pathway. Sci. Rep. 2016, 6, 20460. [Google Scholar] [CrossRef] [Green Version]
- Greif, P.A.; Eck, S.H.; Konstandin, N.P.; Benet-Pagè, A.; Ksienzyk, B.; Dufour, A.; Vetter, A.T.; Popp, H.D.; Lorenz-Depiereux, B.; Meitinger, T.; et al. Identification of recurring tumor-specific somatic mutations in acute myeloid leukemia by transcriptome sequencing. Leukemia 2011, 25, 821–827. [Google Scholar] [CrossRef]
- Darmanis, S.; Cui, T.; Drobin, K.; Li, S.-C.; Öberg, K.; Nilsson, P.; Schwenk, J.M.; Giandomenico, V. Identification of Candidate Serum Proteins for Classifying Well-Differentiated Small Intestinal Neuroendocrine Tumors. PLoS ONE 2013, 8, e81712. [Google Scholar] [CrossRef]
- Zazzeroni, F.; Nicosia, D.; Tessitore, A.; Gallo, R.; Verzella, D.; Fischietti, M.; Vecchiotti, D.; Ventura, L.; Capece, D.; Gulino, A.; et al. KCTD11 Tumor Suppressor Gene Expression Is Reduced in Prostate Adenocarcinoma. BioMed Res. Int. 2014, 2014, 380398. [Google Scholar] [CrossRef]
- Hasegawa, T.; Asanuma, H.; Ogino, J.; Hirohashi, Y.; Shinomura, Y.; Iwaki, H.; Kikuchi, H.; Kondo, T. Use of potassium channel tetramerization domain-containing 12 as a biomarker for diagnosis and prognosis of gastrointestinal stromal tumor. Hum. Pathol. 2013, 44, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Kubota, D.; Yoshida, A.; Tsuda, H.; Suehara, Y.; Okubo, T.; Saito, T.; Orita, H.; Sato, K.; Taguchi, T.; Yao, T.; et al. Gene Expression Network Analysis of ETV1 Reveals KCTD10 as a Novel Prognostic Biomarker in Gastrointestinal Stromal Tumor. PLoS ONE 2013, 8, e73896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; Wang, C.; Yan, W.J.; Gao, R.; Li, Y.H.; Zhou, X.H. Knockdown of TNFAIP1 inhibits growth and induces apoptosis in osteosarcoma cells through inhibition of the nuclear factor-kappaB pathway. Oncol. Rep. 2014, 32, 1149–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Li, Y.; Li, B.; Zheng, L.; Xie, X.; Le, J.; Lu, Y.; Li, T.; Chen, F.; Jia, L. Downregulation of KCTD12 contributes to melanoma stemness by modulating CD271. Cancer Biol. Med. 2019, 16, 498–513. [Google Scholar] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Isotype | Dilution | Final µg/mL | Source | Type | Catalog# | RRID | Purifi- Cation |
|---|---|---|---|---|---|---|---|---|
| Mouse anti-KCTD5 | IgG2b | 1:1000 (IB) | 1 | Origene | mAb | TA501035 | AB_11140321 | Asc |
| Mouse anti-tubulin | IgG1 | 1:5000 (IB) | 1 | Sigma-Aldrich | mAb | T5168 | AB_477579 | Asc |
| Mouse anti-GFP | IgG2a | 1:2000 (IB) | 0.1 | Santa Cruz Biotechnology, Inc. | mAb | sc-9996 | AB_627695 | NA |
| Mouse anti-vinculin | IgG1 | 1:200 (IF) | NA | Sigma-Aldrich | mAb | V4505 | AB_477617 | Asc |
| Mouse anti-c-Myc | IgG1 | 1:200 (IF) | 10 | Sigma-Aldrich | mAb | M4439 | AB_439694 | AP |
| Mouse anti-c-Myc | IgG1 | 1:2000 (IB) | 1 | Sigma-Aldrich | mAb | M4439 | AB_439694 | AP |
| Mouse anti-Rac1 | IgG | 1:500 (IB) | 1 | Cytoskeleton, Inc. | mAb | ARC03 | AB_2721173 | NA |
| Alexa Fluor 555 conjugated goat anti-Mouse IgG1 | IgG | 1:1000 | 2 | Thermo Fisher Scientific | pAb | A-21127 | AB_2535769 | AP |
| Alexa Fluor 647 conjugated goat anti-Mouse IgG1 | IgG | 1:1000 | 2 | Thermo Fisher Scientific | pAb | A-21240 | AB_2535809 | AP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canales, J.; Cruz, P.; Díaz, N.; Riquelme, D.; Leiva-Salcedo, E.; Cerda, O. K+ Channel Tetramerization Domain 5 (KCTD5) Protein Regulates Cell Migration, Focal Adhesion Dynamics and Spreading through Modulation of Ca2+ Signaling and Rac1 Activity. Cells 2020, 9, 2273. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102273
Canales J, Cruz P, Díaz N, Riquelme D, Leiva-Salcedo E, Cerda O. K+ Channel Tetramerization Domain 5 (KCTD5) Protein Regulates Cell Migration, Focal Adhesion Dynamics and Spreading through Modulation of Ca2+ Signaling and Rac1 Activity. Cells. 2020; 9(10):2273. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102273
Chicago/Turabian StyleCanales, Jimena, Pablo Cruz, Nicolás Díaz, Denise Riquelme, Elías Leiva-Salcedo, and Oscar Cerda. 2020. "K+ Channel Tetramerization Domain 5 (KCTD5) Protein Regulates Cell Migration, Focal Adhesion Dynamics and Spreading through Modulation of Ca2+ Signaling and Rac1 Activity" Cells 9, no. 10: 2273. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102273