Metabolic Reprogramming and Predominance of Solute Carrier Genes during Acquired Enzalutamide Resistance in Prostate Cancer



Abstract
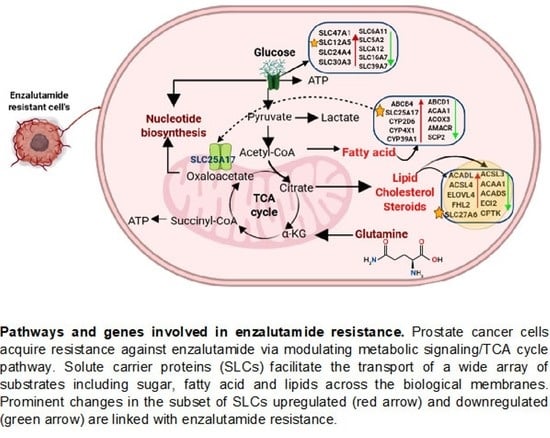
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Development of Enzalutamide Resistance
2.3. RNA Isolation and NGS Sequencing
2.4. RNA Sequencing and Data Analysis
2.5. Quantitative Real-Time PCR
2.6. Pathway and Gene Set Enrichment Analysis
2.7. Gene Network Analysis
2.8. Lactic Acid Assay
2.9. Citric Acid Assay
2.10. Glucose Uptake Assay
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
3.1. Development of Enzalutamide Resistant Prostate Cancer Cell Lines
3.2. Comparison and Validation of Gene Expression Levels between Enzalutamide Resistant and Parental Cells
3.3. Pathway Enrichment Analysis and Mining of Disease Association
3.4. Gene Set Enrichment Analysis (GSEA)
3.5. Identification of Genes Dysregulated in Glucose, Fatty Acid, and Lipid Metabolism
3.6. Altered Expression of Glycolysis End Product and SLC Protein Expression
3.7. Solute Carrier Genes and Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; de Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene 2015, 34, 1745–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019, 22, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Clegg, N.J.; Scher, H.I. Anti-androgens and androgen-depleting therapies in prostate cancer: New agents for an established target. Lancet Oncol. 2009, 10, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Han, R.; Li, J.; Liu, H.; Zheng, L. Molecular mechanism of R-bicalutamide switching from androgen receptor antagonist to agonist induced by amino acid mutations using molecular dynamics simulations and free energy calculation. J. Comput. Aided Mol. Des. 2016, 30, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-generation antiandrogens: From discovery to standard of care in castration resistant prostate cancer. Front Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.-S.; Kimura, G.; et al. Enzalutamide in men with chemotherapy-naive metastatic prostate cancer (mCRPC): Results of phase III PREVAIL study. J. Clin. Oncol. 2014, 32 (Suppl. 4), LBA1. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. Arches: A randomized, phase iii study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, C.; Varchi, G. Non-Steroidal androgen receptor antagonists and prostate cancer: A survey on chemical structures binding this fast-mutating target. Curr. Med. Chem. 2019, 26, 6053–6073. [Google Scholar] [CrossRef]
- Helsen, C.; Van den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen receptor antagonists for prostate cancer therapy. Endocr. Relat. Cancer 2014, 21, T10–T118. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.M.; Pierce, W.; Maher, V.E.; Karuri, S.; Tang, S.; Chiu, H.-J.; Palmby, T.; Zirkelbach, J.F.; Marathe, D.; Mehrotra, N.; et al. Enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer who have previously received docetaxel: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6067–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Gao, A.C. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (Enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.; Antonarakis, E.S.; Luo, J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm. Cancer. 2014, 5, 265–273. [Google Scholar] [CrossRef]
- Van Soest, R.J.; van Royen, M.E.; de Morrée, E.S.; Molla, J.M.; Teubela, W.; Wiemerc, E.A.C.; Mathijssenc, R.H.J.; de Witc, R.; van Weerden, W.M. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur. J. Cancer. 2013, 49, 3821–3830. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinf. Biomath. 2013, 3, 71–85. [Google Scholar]
- Thalmann, G.N.; Anezinis, P.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 5410, 2577–2581. [Google Scholar]
- Saylor, P.J.; Karoly, E.D.; Smith, M.R. Prospective study of changes in the metabolomic profiles of men during their first three months of androgen deprivation therapy for prostate cancer. Clin. Cancer Res. 2012, 18, 3677–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Sadée, W. Membrane transporters and channels in chemoresistance and-sensitivity of tumor cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Zawierucha, P.; Andrzejewska, M.; Ruciński, M.; Zabel, M. Microarray-based detection and expression analysis of ABC and SLC transporters in drug-resistant ovarian cancer cell lines. Biomed. Pharmacother. 2013, 67, 240–245. [Google Scholar] [CrossRef]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug. Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Christianson, J.; Liu, Z.X.; Tian, L.; Choi, C.S.; Neschen, S.; Dong, J.; Wood, P.A.; Shulman, G.I. Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab. 2010, 11, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Liu, Z.-X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [Green Version]
- Rossi Sebastiano, M.; Konstantinidou, G. Targeting long chain acyl-coa synthetases for cancer therapy. Int. J. Mol. Sci. 2019, 20, 3624. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.C.; Chou, S.K.; Kan, J.Y.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. New insight on solute carrier family 27 member 6 (SLC27A6) in tumoral and non-tumoral breast cells. Int. J. Med. Sci. 2019, 16, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Domenichini, A.; Adamska, A.; Falasca, M. ABC transporters as cancer drivers: Potential functions in cancer development. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer. 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Ben-Shabat, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipids and lipid-processing pathways in drug delivery and therapeutics. Int. J. Mol. Sci. 2020, 21, 3248. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Deng, F.; Li, Y.; Daniels, G.; Du, X.; Ren, Q.; Wang, J.; Wang, L.H.; Yang, Y.; Zhang, V.; et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget 2015, 6, 44849–44863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflug. Arch. Eur. J. Physiol. 2004, 447, 689–709. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, S.; Shankar, E.; Chan, E.R.; Gupta, S. Metabolic Reprogramming and Predominance of Solute Carrier Genes during Acquired Enzalutamide Resistance in Prostate Cancer. Cells 2020, 9, 2535. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122535
Verma S, Shankar E, Chan ER, Gupta S. Metabolic Reprogramming and Predominance of Solute Carrier Genes during Acquired Enzalutamide Resistance in Prostate Cancer. Cells. 2020; 9(12):2535. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122535
Chicago/Turabian StyleVerma, Shiv, Eswar Shankar, E. Ricky Chan, and Sanjay Gupta. 2020. "Metabolic Reprogramming and Predominance of Solute Carrier Genes during Acquired Enzalutamide Resistance in Prostate Cancer" Cells 9, no. 12: 2535. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122535