Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review



Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, Szent Gellért tér 4, 1111 Budapest, Hungary
*
Authors to whom correspondence should be addressed.
Chemistry 2020, 2(3), 742-758; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030048
Submission received: 22 July 2020
/
Revised: 24 August 2020
/
Accepted: 25 August 2020
/
Published: 27 August 2020
(This article belongs to the Special Issue Organic Chemistry Research in Hungary)
Abstract
:As catalysis plays a significant role in the development of economical and sustainable chemical processes, increased attention is paid to the recovery and reuse of high-value catalysts. Although homogeneous catalysts are usually more active and selective than the heterogeneous ones, both catalyst recycling and product separation pose a challenge for developing industrially feasible methods. In this respect, membrane-supported recovery of organocatalysts represents a particularly useful tool and a valid option for organocatalytic asymmetric synthesis. However, catalyst leaching/degradation and a subsequent decrease in selectivity/conversion are significant drawbacks. As the effectivity of the membrane separation depends mainly on the size of the catalyst in contrast to the other solutes, molecular weight enlargement of small organocatalysts is usually necessary. In the last few years, several synthetic methodologies have been developed to facilitate their recovery by nanofiltration. With the aim of extending the possibilities for the membrane-supported recovery of organocatalysts further, this contribution presents a review of the existing synthetic approaches for the molecular weight enlargement of organocatalysts.
1. Introduction
Doubtless, catalysis has significantly affected the chemical industry as more than 90% of chemical processes utilize catalysts, allowing more economical and often highly selective production. Due to catalysis, a substantial amount of energy and resources are saved, while considerably less waste is generated. The growing demand from the end-user industries leads the market growth, and by the year 2025, the global catalyst market is expected to reach USD 35.63 billion [1]. Consequently, additional development of the field and answering ongoing challenges are both crucial because catalysis plays a significant role in the development of economical and sustainable processes. By definition, the catalyst is not consumed in the reaction, however, deactivation or degradation can take place. Also, the loss of catalyst during work-up is generally experienced. Therefore, catalyst loading and leaching of the catalyst should be minimized [2].
Organocatalysts are generally small, metal-free, organic molecules capable of accelerating chemical transformations. The real “kick-off” of the field began at the turn of the millennium (see Figure 1), when the seminal works of List et al. and MacMillan et al. were published [3,4]. While the former demonstrated that small organic molecules can mimic the enzyme-like catalytic activity and mechanism, the latter conceptualized the field as organocatalysis and revealed a general activation pattern, which is compatible with several organic transformations. These milestones were quickly followed by other important contributions [5,6,7,8].
Preparative chemists quickly recognized the advantages, that organocatalysis could offer for laboratory-scale research. Low cost and toxicity, ease of access, secure handling without the need for special equipment or conditions, and the countless new possibilities for modification have all attracted a multitude of research groups. Although after the exploration of “the low-hanging fruits”, the difficulties of organocatalysis have been slowly unveiled, resulting in new research directions to overcome these drawbacks. Still, high catalyst loading and long reaction time are generally regarded as disadvantages. In the pursuit of improved organocatalyzed chemical reactions, more attention is paid to the recovery and reuse of organocatalysts [9,10].
Looking at the chemical industry, application of organocatalysis in industrial processes is still not significant. However, considering the enormous advances achieved by academic researchers and the potential advantages of organocatalysts, it is easy to understand how they could bring added value to the manufacturing of high value products [11]. To facilitate the adaptation of organocatalysis in industrial settings, academic research should offer more comprehensive studies to tackle the drawbacks of this field, especially in the areas of catalytic activity and catalyst recovery. The inherent contradiction between catalyst loading and the cost of catalyst leaves us with no other choice, but recycling, which is strongly supported by the current approach ruling the chemical industry: sustainable engineering and green chemistry [12,13,14]. Being primarily small organic molecules, the first representatives of organocatalysts were homogeneous, but heterogeneous alternatives quickly followed them. Today, both types offer viable recycling options. However, further improvement of the field is still essential.
Organocatalysts have classically been used as homogeneous ones, and their recovery by chromatography is straightforward. Obviously, this method is normally applicable only on laboratory scale and does not fulfill the expectations of sustainable manufacturing. Nowadays, most industrial catalytic processes are performed in biphasic systems, where the catalyst is heterogeneous [15]. Heterogeneous catalysts provide significant engineering advantages, like the ease of separation from the reaction mixture enabling excellent recycling. Even though homogeneous catalysts are commonly more effective regarding activity and selectivity, both catalyst recycling and product separation pose a challenge to develop industrially feasible processes [16].
Heterogenization of homogeneous organocatalysts is a commonly used method for their recovery from the reaction mixture, either by precipitating the homogeneous catalyst or using an initially heterogeneous ones [17]. The latter method has the advantage that no additive is needed for the catalyst recovery, which is usually performed by microfiltration, centrifugation or magnetic force. Immobilization of organocatalysts on solid supports is usually straightforward, though the anchoring method has a huge impact on the activity: both the ratio of the catalytic unit to the extent of backbone and the linker between them need to be considered [18,19,20,21]. Beside the solid–liquid phase separations, liquid–liquid partition is also a commonly used convenient method [22,23].
Membrane-based separations are known to be sustainable with low energy needs [24,25]. Considering the recent progress made for greener organocatalytic methods [26,27] and more eco-friendly membrane processes [28,29,30,31], the application and membrane-assisted recovery of organocatalysts have been further studied at the Department of Organic Chemistry and Technology, Budapest University of Technology and Economics. We believe that extending the possibilities of the membrane-supported recovery of organocatalysts further could provide a particularly useful tool in the hands of organic chemists not only in the academia, but possibly also in the industry. In this paper, the membrane-assisted recovery of homogeneous organocatalysts is briefly summarized focusing on the results of the Kupai Research Group.
2. Homogeneous Organocatalyst Recovery Using Organic Solvent Nanofiltration
As separation processes account for up to 40–70% of both capital and operating costs, plus they consume 15% of the energy produced in the world, they play a major role in the fine chemical, pharmaceutical, petrochemical, food, agricultural, and related industries [32,33]. Compared to traditional separation techniques (distillation, extraction, evaporation, adsorption, and chromatography), membrane technologies can be advantageous due to their low carbon footprint, ease of scalability, and small spatial requirements. Relative to thermal processes, they are less energy demanding because, in most cases, they do not require a phase change and operate at relatively mild conditions, therefore, membrane separation of sensitive compounds is feasible [30]. Owing to the several attractive features of this field, membrane separations are both well-developed and widely used in the industry [31]. Membrane processes can be classified based on the pore size of the membrane (see Table 1).
Nanofiltration was introduced in the 1980s, and it is located between ultrafiltration (used for the separation of colloidal material, proteins, etc.) and reversed osmosis (typically used in water purification). At the beginning, nanofiltration was mostly applied in water treatment by using water-resistant nanofiltration membranes, particularly for the removal of natural and synthetic organic matters [35,36], salts [37], and dyes [38]. Following the appearance of solvent-resistant membranes around the turn of the millennium, nanofiltration became feasible for organic solutions [39,40].
Organic solvent nanofiltration (OSN), also called as solvent-resistant nanofiltration (SRNF), is capable of distinguishing molecules in the range of 50–2000 Da by applying only a pressure gradient. OSN processes can be categorized as one of the three conceptually simple operating types: purification, solvent exchange, and concentration. These can be arranged in different ways or combined with classical separation techniques to create a broad range of applications [41]. The sustainability evaluation of OSN processes has been carefully performed, and from greener membrane fabrication through more efficient process development to scale-up, significant progress for environmentally friendly solvent-resistant separations has already been made [24,28]. Its scale-up and implementation in continuous and hybrid processes are relatively simple, therefore feasible for industrial utilization [42,43]. Thus, OSN is a sustainable recycling method for homogeneous catalysts [25,44]. However, catalyst leaching and subsequent decrease in selectivity or conversion are considerable obstacles. Therefore, for suitable industrial procedures, practically 99.99% retention of the catalyst is needed [2]. This essential requirement is supported by our observations on the membrane-assisted recovery of crown ethers and camphorsulfonamides [45,46]. In Figure 2, a schematic representation of an ideal OSN-assisted catalyst recycling is presented, where the molecular weight (MW) of the catalyst is manyfold higher than that of the product. The catalyst accumulates in the retentate during the filtration, while the product passes through the membrane and consequently can be retrieved from the permeate.
3. Molecular Weight Enlargement of Homogeneous Organocatalysts for Membrane Filtration
Kragl et al. accomplished first [47] the membrane-based recovery of homogeneous catalyst α,α-diphenyl-l-prolinol, a well-known member of the proline organocatalyst family, which was followed by quick development in the field [25]. As the effectiveness of the membrane separation depends primarily on (i) the MW gap between the catalyst and the other solutes, and (ii) the absolute catalyst retention on the membrane, molecular weight enlargement (MWE) of small catalysts is usually necessary [2,44]. So far, MWE of homogeneous organocatalysts was performed using different synthetic approaches (see Figure 3a): soluble polymers, dendrimers, polyalkylation of multifunctional cores, attachment to benzoyl subunit, or anchoring to cyclodextrin (CD); and these size-enlarged catalysts were utilized in various process configurations (Figure 3b) [47,48,49,50,51,52,53,54,55,56,57,58]. Other than these covalent modifications, salt formation using organic acid/base having high critical area also proved effective to facilitate the separation of the organocatalysts using membrane filtration [59]. Recently, we showed that a two-stage diafiltration cascade is a suitable system, without the application of MWE, for the recovery of a hydroquinine derivative by OSN in the aza-Markovnikov reaction [60]. Furthermore, Großeheilmann et al. also reported the application of OSN for the purification of some cinchona derivatives [61], and a catalytical system where the size of the bifunctional phosphonium catalyst, used for the conversion of butylene oxide and CO2 into cyclic carbonate, was already sufficient for its recovery by membrane filtration and no additional size-enlargement was necessary [62].
3.1. Soluble Polymers
Similarly to enzyme membrane reactors applied on a scale of 100 ton per year for the synthesis of fine chemicals, soluble polymer-bound organocatalysts—also called chemzymes—can be efficiently retained in membrane reactors. As for polymer support, polymethacrylate, polystyrene, polyethylene glycol, and polysiloxane were explored. The retention depends significantly on the MW and the three-dimensional structure of the polymer. Low MW and linear polymers pass faster through the membrane than large-size molecules and branched polymers. However, the synthesis of linear polymers is less cumbersome, and increasing the MW can raise the viscosity of the polymer solution causing low permeance. These homogeneous polymer-supported catalysts demonstrated good to excellent catalytic activities, and selectivities in several asymmetric organic transformations like ketone reduction, meso-anhydride opening, olefin epoxidation, and diethyl-zinc addition to aldehydes (as an example see Scheme 1). As the examined membranes showed excellent rejections for the polymer-bound catalysts, soluble polymer-anchored catalyst (Figure 4) recycling is highly efficient. However, catalyst deactivation (instead of leaching) was found to be a drawback in several cases [47,48,49,50,51,52].
3.2. Dendrimers
Having repetitively branched tree-like structure with a spherical shape, dendrimers are particularly suitable for nanofiltration. In comparison to the less well–defined polymeric systems, catalyst loading of dendrimers can be determined exactly. Therefore, a direct comparison with the unsupported organocatalyst is possible, providing valuable information for catalyst development. Regarding organocatalysis, Chavan et al. used porphyrin-functionalized pyrimidine dendrimers (Figure 5) for the oxidation of different olefins to the desired allylic hydroperoxides with high conversions (>90%) and selectivities (>99%) (as an example see Scheme 2) [53]. Recycling of the dendrimer-enlarged catalysts proved to be efficient using an oxidatively stable membrane. The applied poly(dimethylsiloxane) membrane was modified by incorporating ultra-stable Y zeolite as inorganic filler. During the recycling experiments, they found that the catalytic activity remains high after the first cycle, however, the conversion decreased significantly in the subsequent runs. Photodegradation control experiments showed that the decrease in activity was attributed mostly to the instability of the porphyrin units (oxidative degradation), and only to a lesser extent to the size-enlarged catalyst leaching through the membrane.
Recently, Št’astná et al. demonstrated the application of carbosilane dendrimers as a support to which ammonium and phosphonium units were covalently anchored to form a dendritic ionic liquid. The obtained homogeneous catalysts were used to catalyze the cycloaddition of CO2 to epoxides. Finally, the tested dendritic catalysts of all generations were successfully recovered by nanofiltration and reused up to four times [54].
3.3. Multifunctional Core
Polyalkylation using multifunctional cores, also called as the “hub approach”, is a method for MWE where multiple catalytic motifs are attached to a central unit (hub). In this case, the number of catalytic units in each enlarged catalyst molecule is increased, and the extent of non-functional “spacers” in the enlarged molecule is reduced compared to polymer or dendrimer supports. Additionally, the flexibility of the resulting size-enlarged molecule can be varied by the type and the length of the linker connecting the core and the catalytic units. For example, a short rigid bond between the catalytic subunit and a benzene backbone can decrease the flexibility, thus, maintaining the increased size in all directions and leading to higher rejections [55].
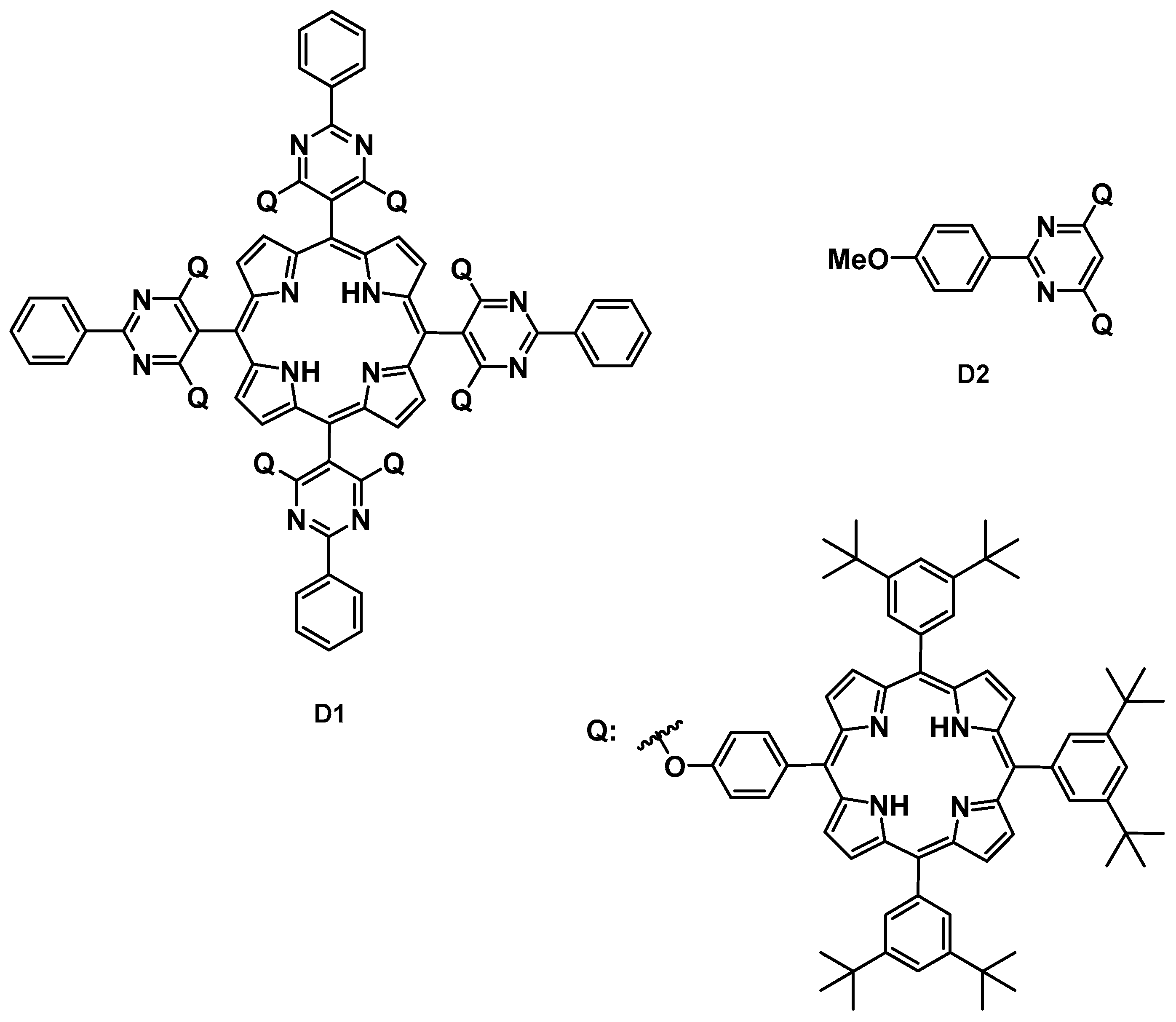
C3-Symmetrical structures gained a special interest in asymmetric catalysis, because they are presumably capable of reducing the number of possible diastereomeric transition states during the catalytic cycle and create a sterically more hindered space, which might lessen disadvantages such as rotation or flexibility [63]. Applying the hub approach, Siew et al. prepared several C3-symmetrical cinchona derivatives (Figure 6) [55]. In comparison to the synthetic precursors, the size-enlarged catalysts showed increased retention during the membrane filtration experiments, while the high catalytic activity was demonstrated in the Michael addition of dimethyl malonate to various nitrostyrenes (92–96% ee, as an example see Scheme 3).
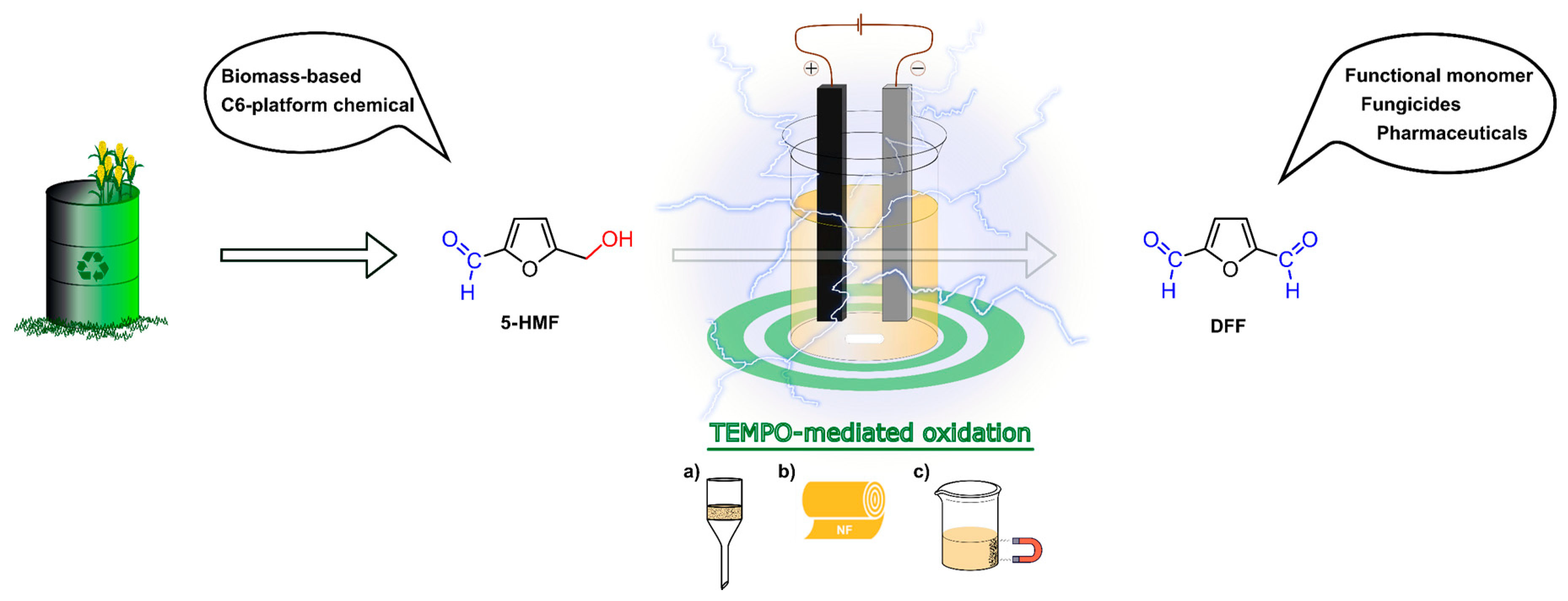
Recently, we successfully applied the hub approach for the development of a C3-symmetrical size-enlarged TEMPO organocatalyst [57]. Biomass-derived 5-hydroxymethylfurfural (HMF) was converted into 2,5-diformylfuran (DFF) in a galvanostatic setup using the compact ElectraSyn reactor in an environmentally friendly organic electrosynthesis (Figure 7). In comparison to the previous methods [64,65,66,67], which used precious metals (like Pt) as the electrode material, graphite (anode) and stainless steel (cathode) were chosen to achieve a cost-effective process. As catalysts, native TEMPO and recyclable homogeneous and heterogeneous derivatives were applied.
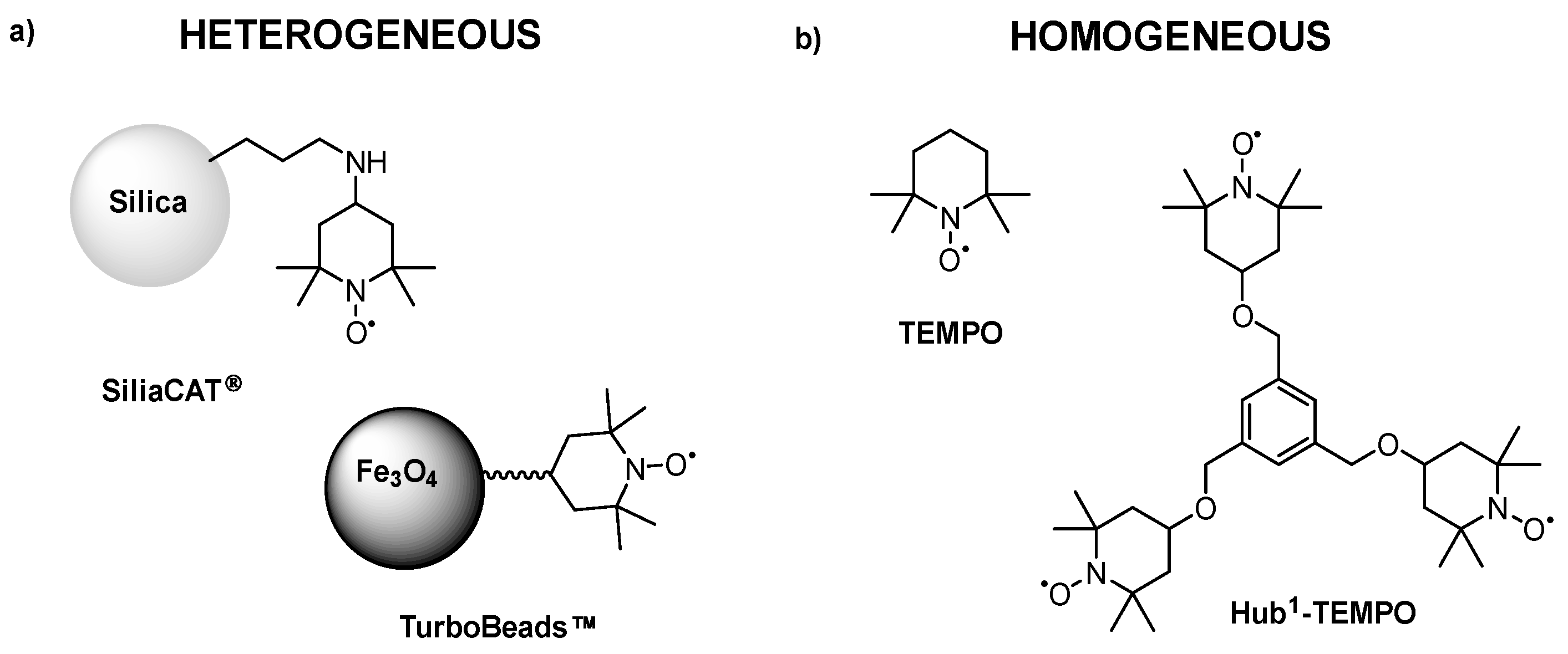
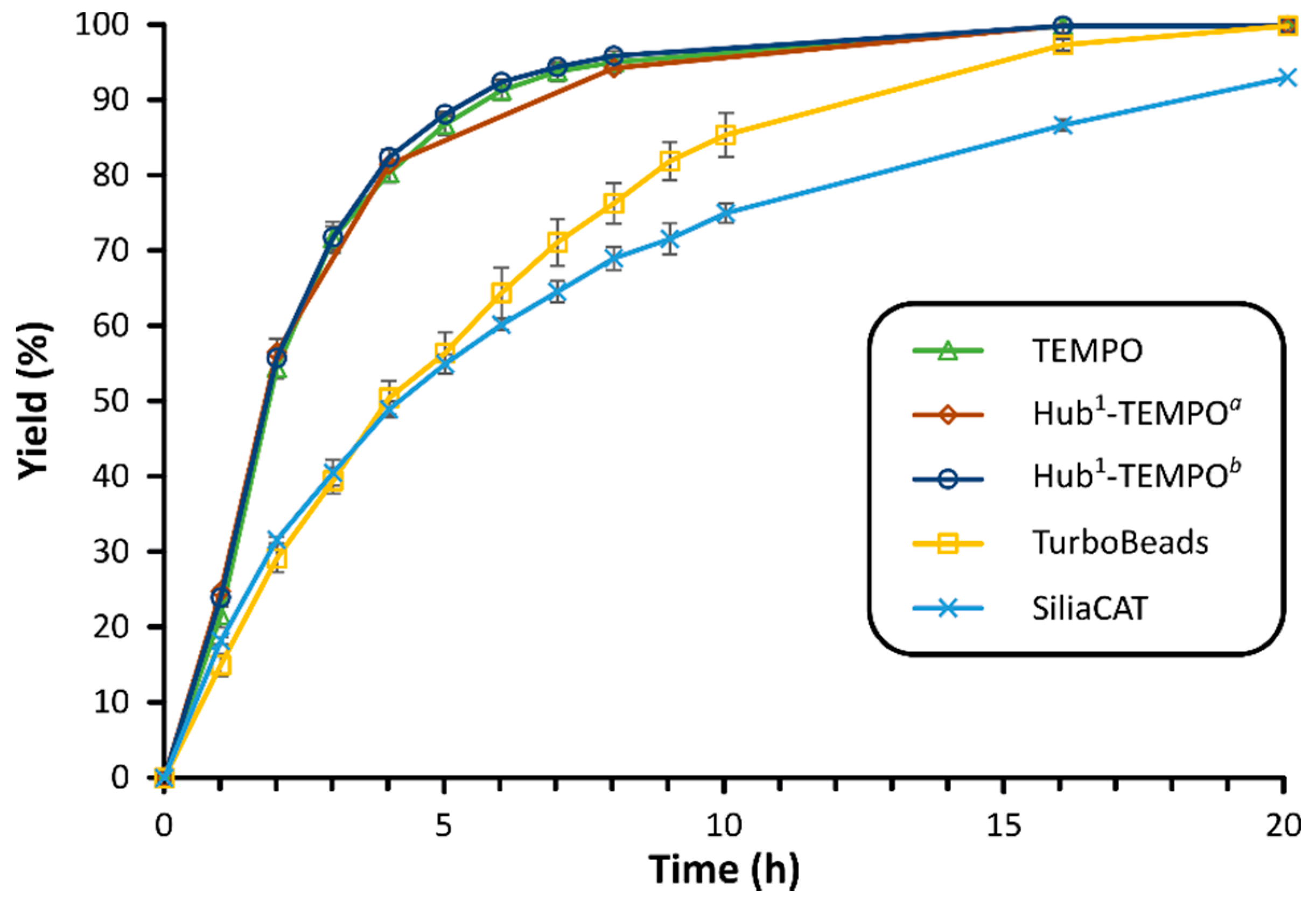
TEMPO and the lutidine (base) showed a synergistic influence on the electrooxidation. During the parameter optimization, the effects of current strength, solvent, stirring rate, temperature, catalyst molar ratio, and electrode surface area were investigated. Following the reaction optimization, two commercially available heterogeneous TEMPO derivatives (SiliaCAT®, TurboBeads™, see Figure 8a) were used which were recovered by using microfiltration or magnetic force (Figure 7a,c), respectively. Additionally, a C3-symmetrical molecular size-enlarged homogeneous catalyst (Hub1-TEMPO) was also designed using the hub approach (Figure 8b) that can be recycled by membrane filtration (Figure 7b). The size-enlarged catalyst design and structure optimization were supported by quantum mechanical modeling. Scheme 4 shows the preparative synthesis of the size-enlarged Hub1-TEMPO catalyst.
The catalytic activities of the different TEMPO derivatives were compared in the electrocatalytic oxidation of HMF (Figure 9). The heterogeneous catalysts rendered moderately slower reactions than the homogeneous native TEMPO system. In comparison to the native TEMPO, the Hub1-TEMPO showed no significant differences in the yield and the progression of the reaction. When the latter catalyst was used in such a way that an equivalent amount of TEMPO units were present in the reaction mixture (one third the mole percentage compared to the native TEMPO), practically no change was observed in the catalytic activity. Hence, we can conclude that the size-enlargement did not adversely affect the catalytic performance.
The homogeneous C3-symmetrical size-enlarged TEMPO derivative (Hub1-TEMPO) was successfully recovered using OSN. Multiple membranes (GMT-oNF-1, NF030306, and DM300) were screened to find the most suitable one for the catalyst recycling by diafiltration. The MW gap between the native TEMPO and the other components, as well as the absolute rejection of the TEMPO by the membrane (approximately 30–70%) were not sufficient for successful separation. In contrast, the retention of Hub1-TEMPO was found to be between 90% and 100% for all the examined membranes. DM300 fully rejected the Hub1-TEMPO, while other solutes have all been effectively purged, showing rejections around 10–20%. This means, that the size-enlargement method is suitable for membrane-supported catalyst recovery.
3.4. Benzoyl Subunit
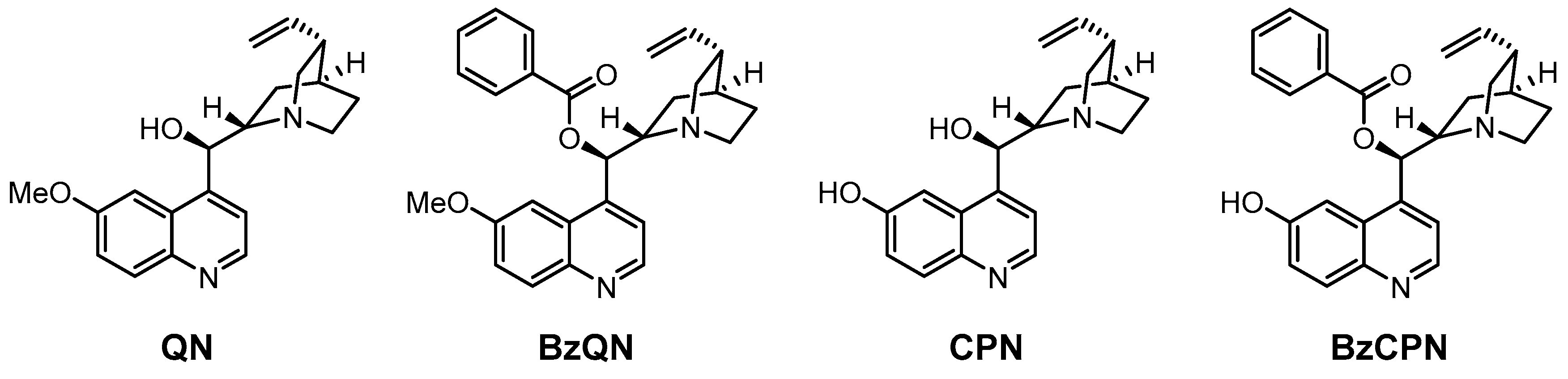
Size-enlargement by attaching a benzoyl subunit was also found to be advantageous for facilitating organocatalyst recycling during membrane filtration. Fahrenwaldt et al. modified the cinchona alkaloid quinine (QN) and its demethylated derivative (cupreine, CPN) by esterification with benzoyl chloride (Figure 10) to facilitate their recovery by OSN [56]. The catalysts containing the benzoyl subunit (BzQN and BzCPN) indeed provided better rejection (about 7% higher than quinine) on the DM300 type membrane. The BzCPN catalyst was applied in the Henry reaction (see Scheme 5) in consecutive batches, and it can be concluded that the catalyst was still fully active and could be easily reused after the nanofiltration steps. However, a decrease in the yield was observed, which could be explained by the loss of catalyst during the diafiltration steps. Another important observation was that a decrease in the rejection of product and catalyst was also found, probably caused by a change in the membrane material/structure.
3.5. Cyclodextrin Anchoring
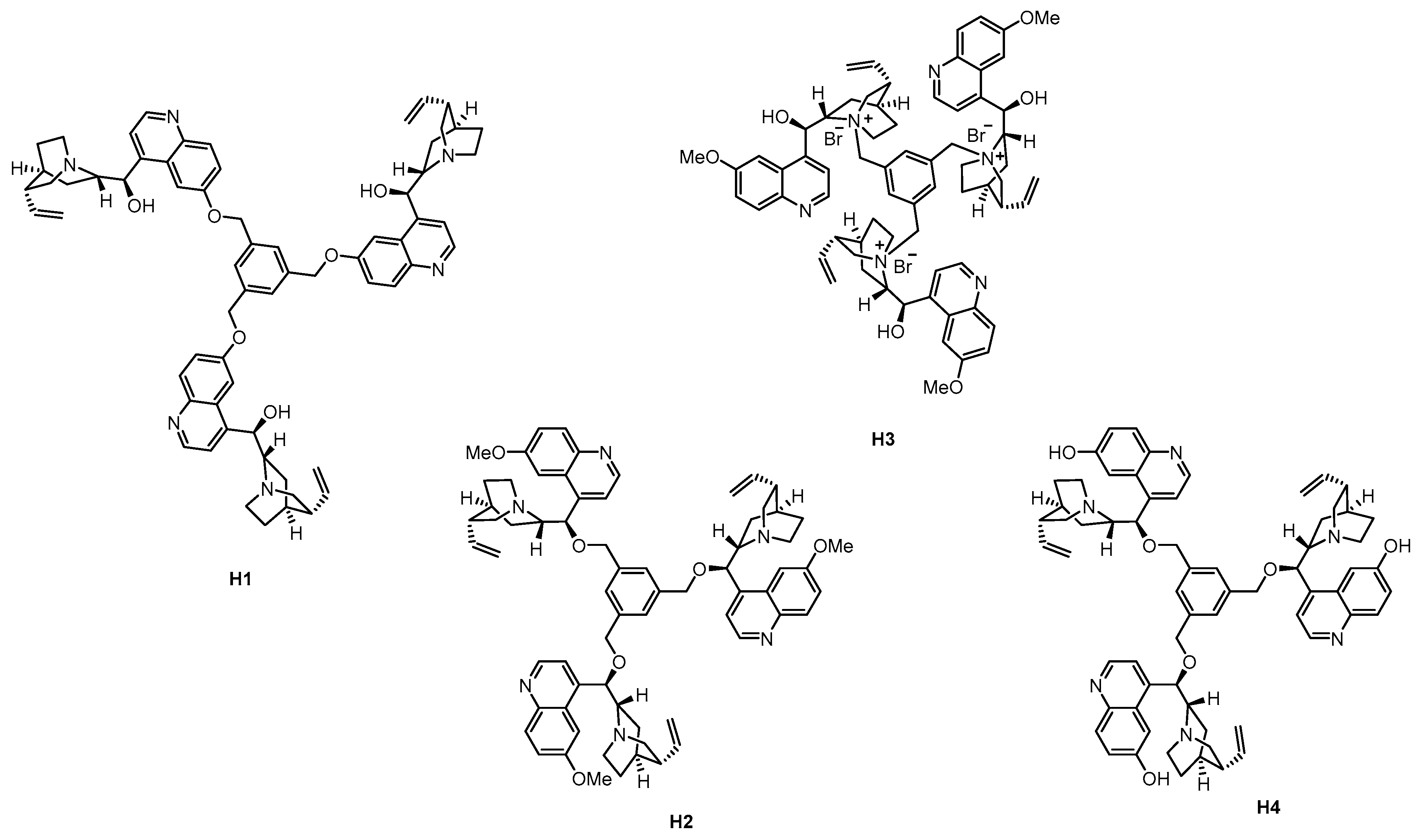
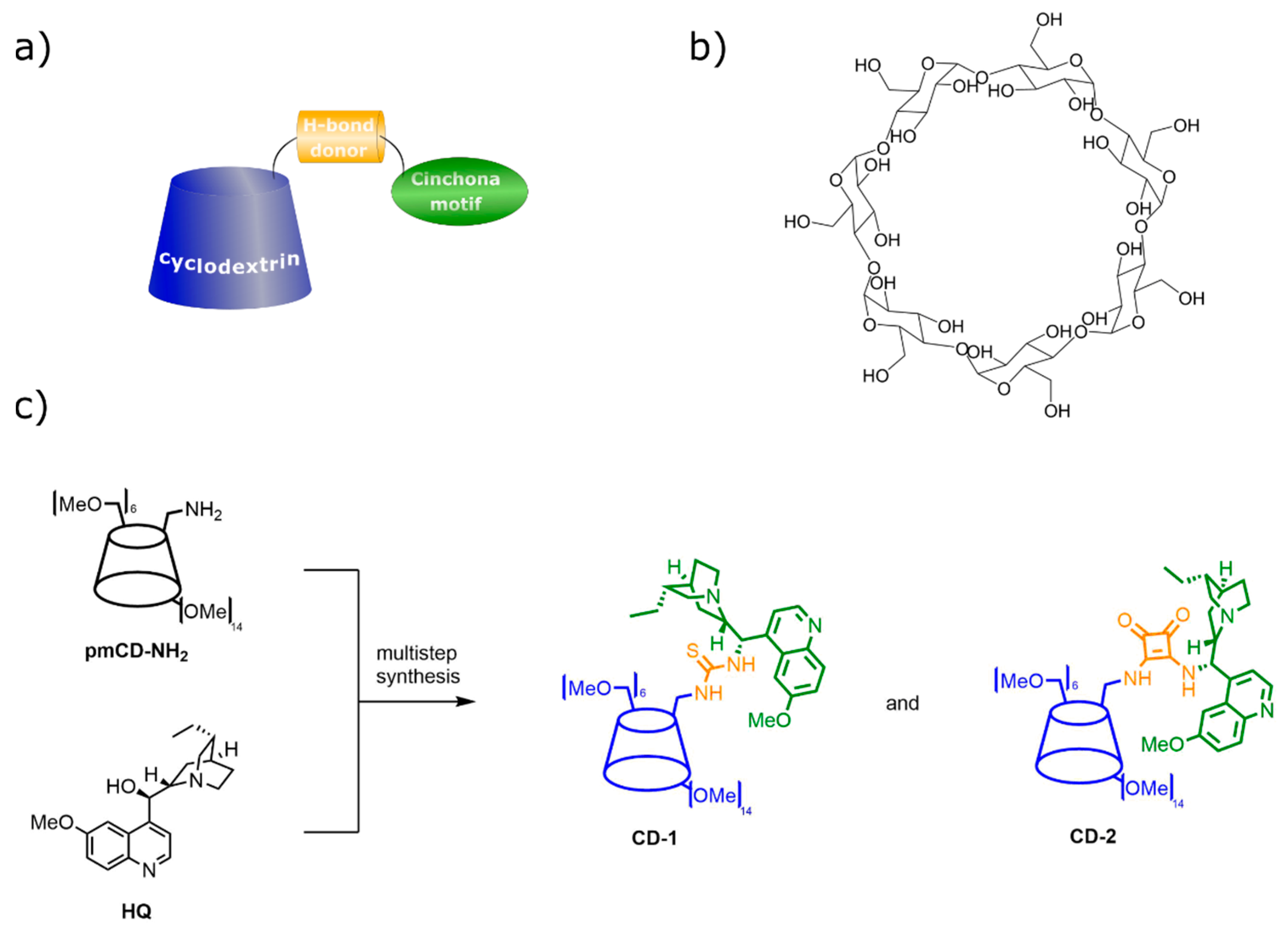
Though the size-enlargement by attaching a benzoyl group to the catalytic unit offered a straightforward and cheap solution compared to other methods, we devised that the application of more bulky subunits can lead to better rejection values. Therefore, a cyclodextrin-enhanced synthetic platform containing cinchona-based organocatalyst for asymmetric synthesis was proposed (Figure 11a) [58]. Cyclodextrins are inherently large, stable compounds that form inclusion complexes with a broad variety of lipophilic molecules (Figure 11b) [68]. The size-enlarged CD-cinchona catalysts (CD-1 and CD-2, see Figure 11c) were prepared from native β-cyclodextrin through a permethylated cyclodextrin amine derivative (pmCD-NH2) and commercially available hydroquinine (HQ). Using the above mentioned β-cyclodextrin derivative, cinchona-thiourea and -squaramide scaffolds were attached to it, forming well-defined and characterized bifunctional hydrogen bonding organocatalysts.
We applied the size-enlarged cinchona organocatalysts in the Michael addition of 1,3-dioxo compounds to trans-β-nitrostyrene (Scheme 6), which gave the adducts with high yields (up to 95%) and excellent enantiomeric excesses (up to 99%). Furthermore, after a solvent screen, including 22 alternative and conventional solvents, a correlation between the enantioselectivity and the hydrogen bond donor Kamlet–Taft solvent parameter (α) was observed: solvents with lower α parameter provided higher enantioselectivities in the Michael addition reaction.
Following the successful batch application of the CD-2 catalyst, a continuous catalysis–separation platform utilizing the biomass-derived 2-methyltetrahydrofuran (2-MeTHF) as solvent was explored. After the experiments in flow mode, several commercial membranes were tested to recycle the CD-2 catalyst from the reaction mixture. DM900 retained practically 100% of the catalyst and showed less than 5% rejection for the other solutes. Finally, coupling of the nanofiltration rig with the continuous-flow reactor was carried out. In the integrated synthesis–separation procedure (Figure 12), the continuous-flow reactor outlet stream (the crude reaction mixture) was diverted to a cross-flow membrane cell. The CD-2 catalyst completely and 50% of the 2-MeTHF solvent were in situ recycled by the membrane with the retentate stream, which were then combined with the inlet flow containing fresh starting materials. The permeate stream containing the highly concentrated solution of the product with a purity of 92% was collected in a vessel kept at room temperature, where the product crystallized allowing the final purity to reach 98% with 99% ee.
The CD anchor had two main roles. First, it advantageously altered the conformation of the catalyst and the reagents, and, consequently, enhanced the catalytic performance. This finding was supported by ab initio calculations revealing improved intermolecular interaction and positively altered distances and angles between the reactants. Second, it made possible the full recovery of the CD-2 catalyst due to the increased size.
3.6. Explored Membrane-Processes
For the membrane recovery of organocatalysts several membrane-types and solvents have been explored so far. In Table 2, a comparison of these separation processes is shown, including the (estimated) MW and the achieved catalyst rejection values.
4. Conclusions and Outlook
Among the broad variety of available types of catalysts, organocatalysts are commonly favored instead of transition metal containing ones. However, high catalyst loading and long reaction time are still drawbacks for organocatalytic transformations. Undoubtedly, catalyst recovery holds a pivotal role in the development of economical organocatalytic processes. Membrane separation is an emerging field with accentuated focus on more sustainable membrane fabrication methods like renewable materials, solvent recycling, and improved selectivity. Considering the recent progress made toward both greener organocatalytic techniques and more eco-friendly membrane processes, the nanofiltration-enabled recovery of organocatalysts have been reviewed in this paper.
We can conclude that molecular weight enlargement of organocatalysts is a reasonable method to facilitate their recovery by OSN. Although their synthesis can sometimes be cumbersome, the efficient recovery of these compounds provides a significant advantage. With proper catalyst design, the most suitable size-enlargement method might be chosen to attain high catalytic activity and excellent selectivity.
The roles of the linker and the anchoring method were both found to be substantial. With the help of QM modeling, the catalyst-substrate interactions might be thoroughly studied and used to design more effective catalysts. Therefore, we propose that, with the development of these computational design methods, a more direct approach can be available to find the suitable catalyst size-enlargement approach, minimizing the need of experimental work.
Finally, nanofiltration-enabled recovery of organocatalysts was also found to be suitable for the development of an integrated synthesis–separation system. This approach could encourage future industrial applications as continuous processing is becoming the preferred production form. Organocatalysis is a suitable method for the construction of optically pure compounds, which is especially crucial for the synthesis of bioactive compounds. At the same time, membrane processes are believed to be viable alternatives to conventional separation techniques (distillation, chromatography, etc.), and they are already widely used in several industrial operations. Therefore, further exploitation of these techniques in the chemical industry is expected. Accordingly, the combination of the two fields suggests a resultful future for industrial organocatalytic processes.
Funding
This work was funded by Janos Bolyai Research Scholarship of the Hungarian Academy of Sciences (JK), the New National Excellence Program of the Ministry of Human Capacities, grant number ÚNKP-19-4-BME-415 (JK), ÚNKP-20-5-BME-322 (JK), ÚNKP-20-4-I-BME-320 (PK) and the Gedeon Richter’s Talentum Foundation (PK). Financial supports of the National Research, Development and Innovation Office (former OTKA, grant number K128473), and the New Széchenyi Development Plan (TÁMOP-4.2.1/B-09/1/KMR-2010-002) are gratefully acknowledged. ![Chemistry 02 00048 i001]()

Conflicts of Interest
The authors declare no conflict of interest.
References
- Catalyst Market by Material, Type, Application, Regions, Industry Analysis, Size, Share, Growth, Trends, and Forecast 2018 to 2025. Available online: https://www.fiormarkets.com (accessed on 18 March 2020).
- Kamer, P.; Vogt, D.; Thybaut, J.W. Contemporary Catalysis: Science, Technology and Applications; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen—Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Akiyama, T. Stronger Brønsted Acids. Chem. Rev. 2007, 107, 5744–5758. [Google Scholar] [CrossRef]
- Enders, D.; Grondal, C.; Hüttl, M.R.M. Asymmetric Organocatalytic Domino Reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The Advent and Development of Organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Oliveira, V.; Cardoso, M.; Forezi, L. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Carlone, A.; Bernardi, L. Enantioselective Organocatalytic Approaches to Active Pharmaceutical Ingredients – Selected Industrial Examples. Phys. Sci. Rev. 2019, 4, 20180097. [Google Scholar] [CrossRef]
- Transforming Our World: The 2030 Agenda for Sustainable Development: Sustainable Development Knowledge Platform. Available online: https://sustainabledevelopment.un.org/post2015/transformingourworld/publication (accessed on 18 March 2020).
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Livingston, A.G.; Trout, B.L.; Horvath, I.T.; Johnson, M.D.; Vaccaro, L.; Coronas, J.; Babbitt, C.W.; Zhang, X.; Pradeep, T.; Drioli, E.; et al. Challenges and Directions for Green Chemical Engineering—Role of Nanoscale Materials. In Sustainable Nanoscale Engineering; Szekely, G., Livingston, A.G., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–18. [Google Scholar]
- Joshi, S.S.; Ranade, V.V. Industrial Catalytic Processes for Fine and Specialty Chemicals; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Mika, L.T.; Horváth, I.T. Fluorous Catalysis. In Green Techniques for Organic Synthesis and Medicinal Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2018; Volume 79, pp. 219–268. [Google Scholar]
- Benaglia, M. Recoverable Organic Catalysts. In Recoverable and Recyclable Catalysts; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 301–340. [Google Scholar]
- Kasaplar, P.; Riente, P.; Hartmann, C.; Pericàs, M.A. A Polystyrene-Supported, Highly Recyclable Squaramide Organocatalyst for the Enantioselective Michael Addition of 1,3-Dicarbonyl Compounds to β-Nitrostyrenes. Adv. Synth. Catal. 2012, 354, 2905–2910. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Y.; Yu, P.; Han, X.; He, J. Exploration of Dependence of Organo-Catalyzed Enantioselective Michael Addition on the Pore Size of Mesoporous Host. ACS Catal. 2012, 2, 1118–1126. [Google Scholar] [CrossRef]
- Rase, H.F. Handbook of Commercial Catalysts; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Nagy, S.; Fehér, Z.; Kárpáti, L.; Bagi, P.; Kisszékelyi, P.; Koczka, B.; Huszthy, P.; Pukánszky, B.; Kupai, J. Synthesis and applications of cinchona squaramide-modified poly(glycidyl methacrylate) microspheres as recyclable polymer-grafted enantioselective organocatalysts. Chem. Eur. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Ghorai, S. Organocatalysis in Water at Room Temperature with In-Flask Catalyst Recycling. Org. Lett. 2012, 14, 422–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Großeheilmann, J.; Vanderveen, J.R.; Jessop, P.G.; Kragl, U. Switchable-Hydrophilicity Solvents for Product Isolation and Catalyst Recycling in Organocatalysis. ChemSusChem 2016, 9, 696–702. [Google Scholar] [CrossRef] [Green Version]
- Szekely, G.; Jimenez-Solomon, M.F.; Marchetti, P.; Kim, J.F.; Livingston, A.G. Sustainability Assessment of Organic Solvent Nanofiltration: From Fabrication to Application. Green Chem. 2014, 16, 4440–4473. [Google Scholar] [CrossRef] [Green Version]
- Cseri, L.; Fodi, T.; Kupai, J.; Balogh, G.T.; Garforth, A.; Szekely, G. Membrane-Assisted Catalysis in Organic Media. Adv. Mater. Lett. 2017, 8, 1094–1124. [Google Scholar] [CrossRef] [Green Version]
- Branco, L.C.; Phillips, F.A.M.; Marques, M.M.; Gago, S.; Branco, P.S. Recent Advances in Sustainable Organocatalysis. In Recent Advances in Organocatalysis; Karamé, I., Srour, H., Eds.; InTech: London, UK, 2016. [Google Scholar]
- Krištofíková, D.; Modrocká, V.; Mečiarová, M.; Šebesta, R. Green Asymmetric Organocatalysis. ChemSusChem 2020, 13, 2828–2858. [Google Scholar] [CrossRef]
- Kim, J.F.; Székely, G.; Valtcheva, I.B.; Livingston, A.G. Increasing the Sustainability of Membrane Processes through Cascade Approach and Solvent Recovery—Pharmaceutical Purification Case Study. Green Chem. 2014, 16, 133–145. [Google Scholar] [CrossRef]
- Phuong, H.A.L.; Ayob, N.A.I.; Blanford, C.F.; Rawi, N.F.M.; Szekely, G. Nonwoven Membrane Supports from Renewable Resources: Bamboo Fiber Reinforced Poly(Lactic Acid) Composites. ACS Sustain. Chem. Eng. 2019, 7, 11885–11893. [Google Scholar] [CrossRef]
- Cheng, X.Q.; Wang, Z.X.; Jiang, X.; Li, T.; Lau, C.; Guo, Z.; Ma, J.; Shao, L. Towards Sustainable Ultrafast Molecular-Separation Membranes: From Conventional Polymers to Emerging Materials. Prog. Mater. Sci. 2018, 92, 258–283. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.P.; Culfaz-Emecen, P.Z.; Ramon, G.Z.; Visser, T.; Koops, G.H.; Jin, W.; Ulbricht, M. Thinking the Future of Membranes: Perspectives for Advanced and New Membrane Materials and Manufacturing Processes. J. Membr. Sci. 2020, 598, 117761. [Google Scholar] [CrossRef]
- Marchetti, P.; Peeva, L.; Livingston, A.G. The Selectivity Challenge in Organic Solvent Nanofiltration: Membrane and Process Solutions. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Galizia, M.; Bye, K.P. Advances in Organic Solvent Nanofiltration Rely on Physical Chemistry and Polymer Chemistry. Front. Chem. 2018, 6, 511. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Lyu, Z.; Gu, Q.; Zhang, L.; Wang, J. Ceramic-Based Membranes for Water and Wastewater Treatment. Colloids Surf. A Physicochem. Eng. Asp. 2019, 578, 123513. [Google Scholar] [CrossRef]
- Schäfer, A.I.; Fane, A.G.; Waite, T.D. Nanofiltration of Natural Organic Matter: Removal, Fouling and the Influence of Multivalent Ions. Desalination 1998, 118, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Košutić, K.; Kaštelan-Kunst, L.; Kunst, B. Porosity of Some Commercial Reverse Osmosis and Nanofiltration Polyamide Thin-Film Composite Membranes. J. Membr. Sci. 2000, 168, 101–108. [Google Scholar] [CrossRef]
- Xu, Y.; Lebrun, R.E. Investigation of the Solute Separation by Charged Nanofiltration Membrane: Effect of pH, Ionic Strength and Solute Type. J. Membr. Sci. 1999, 158, 93–104. [Google Scholar] [CrossRef]
- Van Der Bruggen, B.; Daems, B.; Wilms, D.; Vandecasteele, C. Mechanisms of Retention and Flux Decline for the Nanofiltration of Dye Baths from the Textile Industry. Sep. Purif. Technol. 2001, 22, 519–528. [Google Scholar] [CrossRef]
- Machado, D.R.; Hasson, D.; Semiat, R. Effect of Solvent Properties on Permeate Flow through Nanofiltration Membranes. Part I: Investigation of Parameters Affecting Solvent Flux. J. Membr. Sci. 1999, 163, 93–102. [Google Scholar] [CrossRef]
- Machado, D.R.; Hasson, D.; Semiat, R. Effect of Solvent Properties on Permeate Flow through Nanofiltration Membranes. Part II. Transport Model. J. Membr. Sci. 2000, 166, 63–69. [Google Scholar] [CrossRef]
- Marchetti, P.; Jimenez-Solomon, M.F.; Szekely, G.; Livingston, A.G. Molecular Separation with Organic Solvent Nanofiltration: A Critical Review. Chem. Rev. 2014, 114, 10735–10806. [Google Scholar] [CrossRef] [PubMed]
- Dreimann, J.; Lutze, P.; Zagajewski, M.; Behr, A.; Górak, A.; Vorholt, A.J. Highly Integrated Reactor-Separator Systems for the Recycling of Homogeneous Catalysts. Chem. Eng. Process. Process Intensif. 2016, 99, 124–131. [Google Scholar] [CrossRef]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis-Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Gürsel, V.I.; Noël, T.; Wang, Q.; Hessel, V. Separation/Recycling Methods for Homogeneous Transition Metal Catalysts in Continuous Flow. Green Chem. 2015, 17, 2012–2026. [Google Scholar] [CrossRef]
- Kisszékelyi, P.; Nagy, S.; Tóth, B.; Zeller, B.; Hegedűs, L.; Mátravölgyi, B.; Grün, A.; Németh, T.; Huszthy, P.; Kupai, J. Synthesis and recovery of pyridine- and piperidine-based camphorsulfonamide organocatalysts used for Michael addition reaction. Period. Polytech. Chem. Eng. 2018, 62, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Kupai, J.; Kisszékelyi, P.; Rojik, E.; Dargó, G.; Hegedűs, L.; Bezzegh, D.; Maszler, P.; Szabó, L.; Németh, T.; Balogh, G.T.; et al. Synthesis and determination of pKa values of new enantiopure pyridino- and piperidino-18-crown-6 ethers. Arkivoc 2016, IV, 130–151. [Google Scholar] [CrossRef] [Green Version]
- Kragl, U.; Dreisbach, C. Continuous Asymmetric Synthesis in a Membrane Reactor. Angew. Chem. Int. Ed. 1996, 35, 642–644. [Google Scholar] [CrossRef]
- Giffels, G.; Beliczey, J.; Felder, M.; Kragl, U. Polymer Enlarged Oxazaborolidines in a Membrane Reactor: Enhancing Effectivity by Retention of the Homogeneous Catalyst. Tetrahedron Asymmetry 1998, 9, 691–696. [Google Scholar] [CrossRef]
- Rissom, S.; Beliczey, J.; Giffels, G.; Kragl, U.; Wandrey, C. Asymmetric Reduction of Acetophenone in Membrane Reactors: Comparison of Oxazaborolidine and Alcohol Dehydrogenase Catalysed Processes. Tetrahedron Asymmetry 1999, 10, 923–928. [Google Scholar] [CrossRef]
- Wöltinger, J.; Bommarius, A.S.; Drauz, K.; Wandrey, C. The Chemzyme Membrane Reactor in the Fine Chemicals Industry. Org. Process Res. Dev. 2001, 5, 241–248. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Wöltinger, J.; Jost, C.; Reichert, D.; Kühnle, A.; Krimmer, H.P.; Drauz, K. Juliá-Colonna Asymmetric Epoxidation in a Continuously Operated Chemzyme Membrane Reactor. Synlett 2002, 2002, 707–710. [Google Scholar] [CrossRef]
- Wöltinger, J.; Krimmer, H.P.; Drauz, K. The Potential of Membrane Reactors in the Asymmetric Opening of Meso-Anhydrides. Tetrahedron Lett. 2002, 43, 8531–8533. [Google Scholar] [CrossRef]
- Chavan, S.A.; Maes, W.; Gevers, L.E.M.; Wahlen, J.; Vankelecom, I.F.J.; Jacobs, P.A.; Dehaen, W.; De Vos, D.E. Porphyrin-Functionalized Dendrimers: Synthesis and Application as Recyclable Photocatalysts in a Nanofiltration Membrane Reactor. Chem. Eur. J. 2005, 11, 6754–6762. [Google Scholar] [CrossRef] [PubMed]
- Št’astná, L.C.; Krupková, A.; Petrickovic, R.; Müllerová, M.; Matoušek, J.; Koštejn, M.; Cuřínová, P.; Jandová, V.; Šabata, S.; Strašák, T. Multivalent Bifunctional Carbosilane Dendrimer-Supported Ammonium and Phosphonium Organocatalysts for the Coupling of CO2 and Epoxides. ACS Sustain. Chem. Eng. 2020, 8, 11692–11703. [Google Scholar] [CrossRef]
- Siew, W.E.; Ates, C.; Merschaert, A.; Livingston, A.G. Efficient and Productive Asymmetric Michael Addition: Development of a Highly Enantioselective Quinidine-Based Organocatalyst for Homogeneous Recycling via Nanofiltration. Green Chem. 2013, 15, 663–674. [Google Scholar] [CrossRef]
- Fahrenwaldt, T.; Großeheilmann, J.; Erben, F.; Kragl, U. Organic Solvent Nanofiltration as a Tool for Separation of Quinine-Based Organocatalysts. Org. Process Res. Dev. 2013, 17, 1131–1136. [Google Scholar] [CrossRef]
- Kisszekelyi, P.; Hardian, R.; Vovusha, H.; Chen, B.; Zeng, X.; Schwingenschlogl, U.; Kupai, J.; Szekely, G. Selective Electrocatalytic Oxidation of Biomass-derived 5-Hydroxymethylfurfural to 2,5-Diformylfuran: From Mechanistic Investigations to Catalyst Recovery. ChemSusChem 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. Asymmetric synthesis with cinchona-decorated cyclodextrin in a continuous-flow membrane reactor. J. Catal. 2019, 371, 255–261. [Google Scholar] [CrossRef]
- Gupta, A. Membrane Mediated Organocatalyst Separation Methodology. J. Appl. Chem. 2016, 5, 255–265. [Google Scholar]
- Nagy, S.; Fehér, Z.; Kisszékelyi, P.; Huszthy, P.; Kupai, J. Cinchona derivatives as sustainable and recyclable homogeneous organocatalysts for aza-Markovnikov addition. New J. Chem. 2018, 42, 8596–8602. [Google Scholar] [CrossRef] [Green Version]
- Großeheilmann, J.; Fahrenwaldt, T.; Kragl, U. Organic Solvent Nanofiltration-Supported Purification of Organocatalysts. ChemCatChem 2016, 8, 322–325. [Google Scholar] [CrossRef]
- Großeheilmann, J.; Büttner, H.; Kohrt, C.; Kragl, U.; Werner, T. Recycling of Phosphorus-Based Organocatalysts by Organic Solvent Nanofiltration. ACS Sustain. Chem. Eng. 2015, 3, 2817–2822. [Google Scholar] [CrossRef]
- Min, C.; Han, X.; Liao, Z.; Wu, X.; Zhou, H.-B.; Dong, C. C3-Symmetrical Cinchonine-Squaramide as New Highly Efficient, and Recyclable Organocatalyst for Enantioselective Michael Addition. Adv. Synth. Catal. 2011, 353, 2715–2720. [Google Scholar] [CrossRef]
- Skowroński, R.; Cottier, L.; Descotes, G.; Lewkowski, J. Selective Anodic Oxidation of 5-Hydroxymethylfurfural. Synthesis 1996, 1996, 1291–1292. [Google Scholar] [CrossRef]
- Cao, T.; Wu, M.; Ordomsky, V.V.; Xin, X.; Wang, H.; Métivier, P.; Pera-Titus, M. Selective Electrogenerative Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandialdehyde. ChemSusChem 2017, 10, 4851–4854. [Google Scholar] [CrossRef]
- Delorme, A.E.; Sans, V.; Licence, P.; Walsh, D.A. Tuning the Reactivity of TEMPO during Electrocatalytic Alcohol Oxidations in Room-Temperature Ionic Liquids. ACS Sustain. Chem. Eng. 2019, 7, 11691–11699. [Google Scholar] [CrossRef]
- Rafiee, M.; Konz, Z.M.; Graaf, M.D.; Koolman, H.F.; Stahl, S.S. Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids Catalyzed by 4-Acetamido-TEMPO: An Alternative to “Anelli” and “Pinnick” Oxidations. ACS Catal. 2018, 8, 6738–6744. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
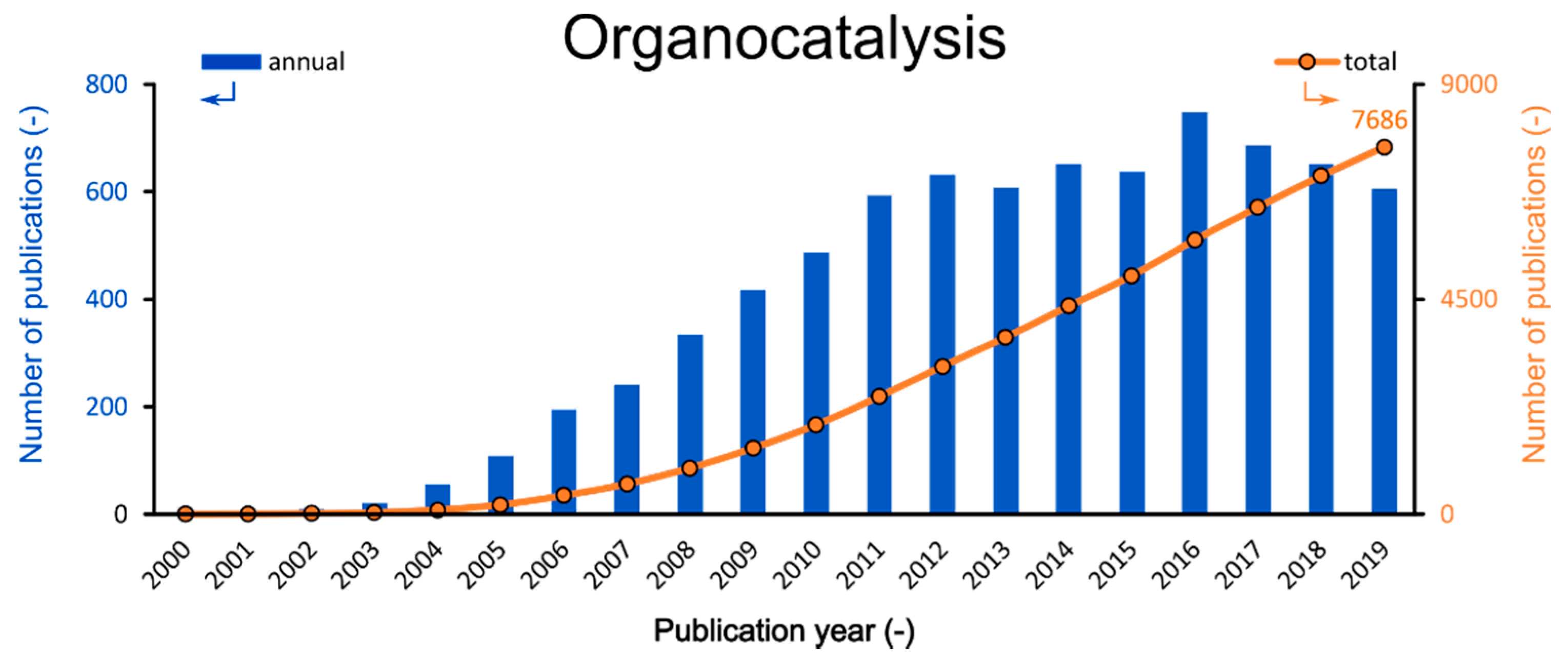
Figure 1.
Annual (blue) and total (orange) numbers of publications related to organocatalysis (Search engine: Web of Science; keyword: organocatalysis; 1 March 2020).
Figure 1.
Annual (blue) and total (orange) numbers of publications related to organocatalysis (Search engine: Web of Science; keyword: organocatalysis; 1 March 2020).

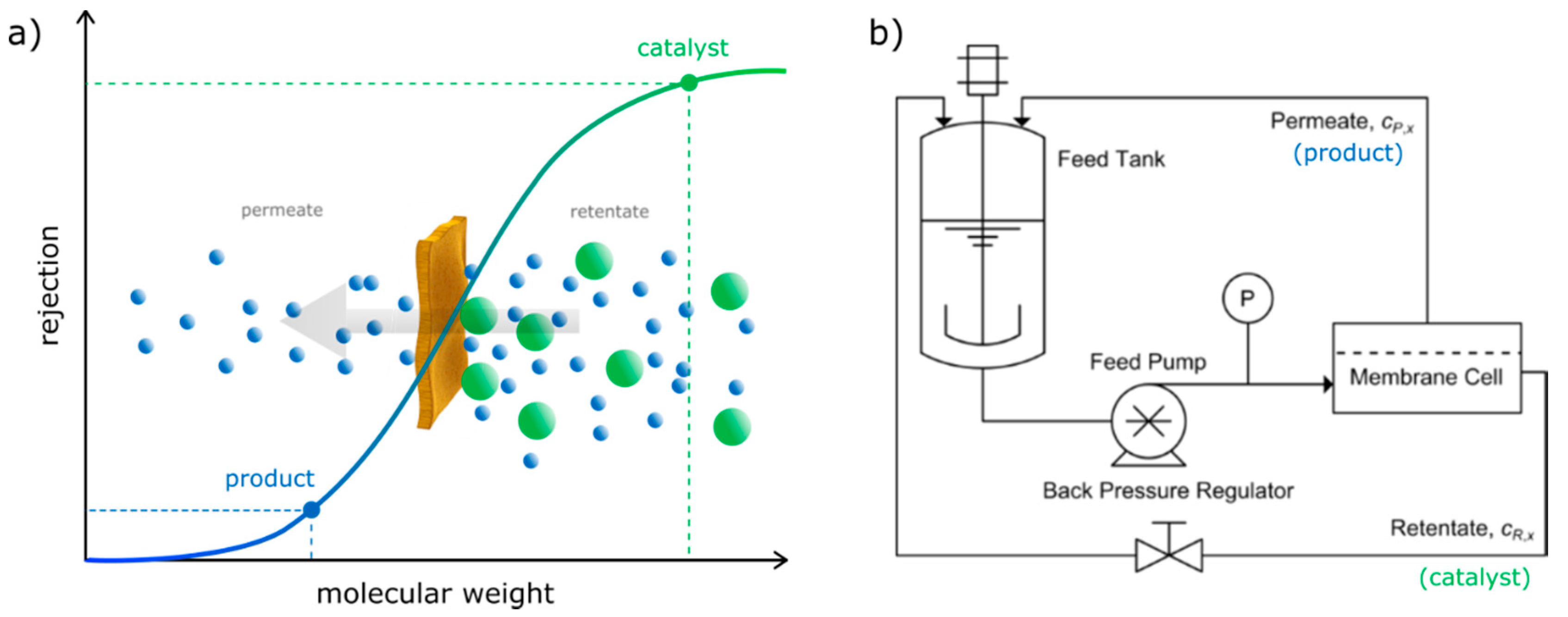
Figure 2.
(a) Simplified representation of an optimal catalyst recovery by nanofiltration: having manyfold higher MW the catalyst stays in the retentate, while the product can easily pass through the membrane; (b) Schematic diagram of a nanofiltration set-up: after the crude mixture is pumped through the membrane cell, the permeate contains the smaller components (product), while the retentate contains the components with high molecular size (catalyst).
Figure 2.
(a) Simplified representation of an optimal catalyst recovery by nanofiltration: having manyfold higher MW the catalyst stays in the retentate, while the product can easily pass through the membrane; (b) Schematic diagram of a nanofiltration set-up: after the crude mixture is pumped through the membrane cell, the permeate contains the smaller components (product), while the retentate contains the components with high molecular size (catalyst).

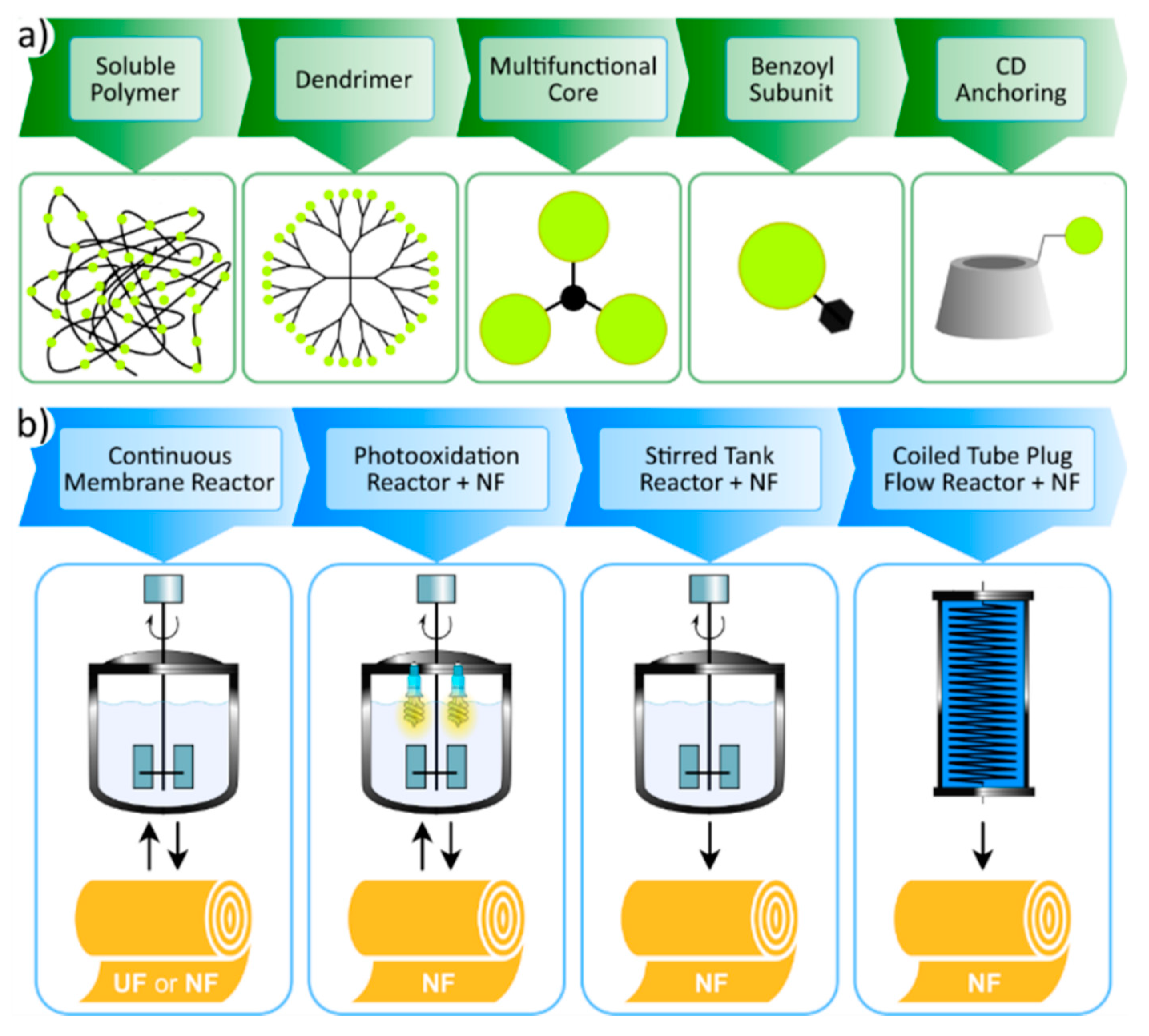
Figure 3.
Membrane assisted recovery of homogeneous organocatalysts: catalyst size-enlargement approaches for efficient retention by the membranes (a), and the applied reactor types in the hybrid processes (b). This figure is reprinted from reference [58] with permission from Elsevier, available by license CC BY-NC-ND 4.0. UF: ultrafiltration, NF: nanofiltration.
Figure 3.
Membrane assisted recovery of homogeneous organocatalysts: catalyst size-enlargement approaches for efficient retention by the membranes (a), and the applied reactor types in the hybrid processes (b). This figure is reprinted from reference [58] with permission from Elsevier, available by license CC BY-NC-ND 4.0. UF: ultrafiltration, NF: nanofiltration.

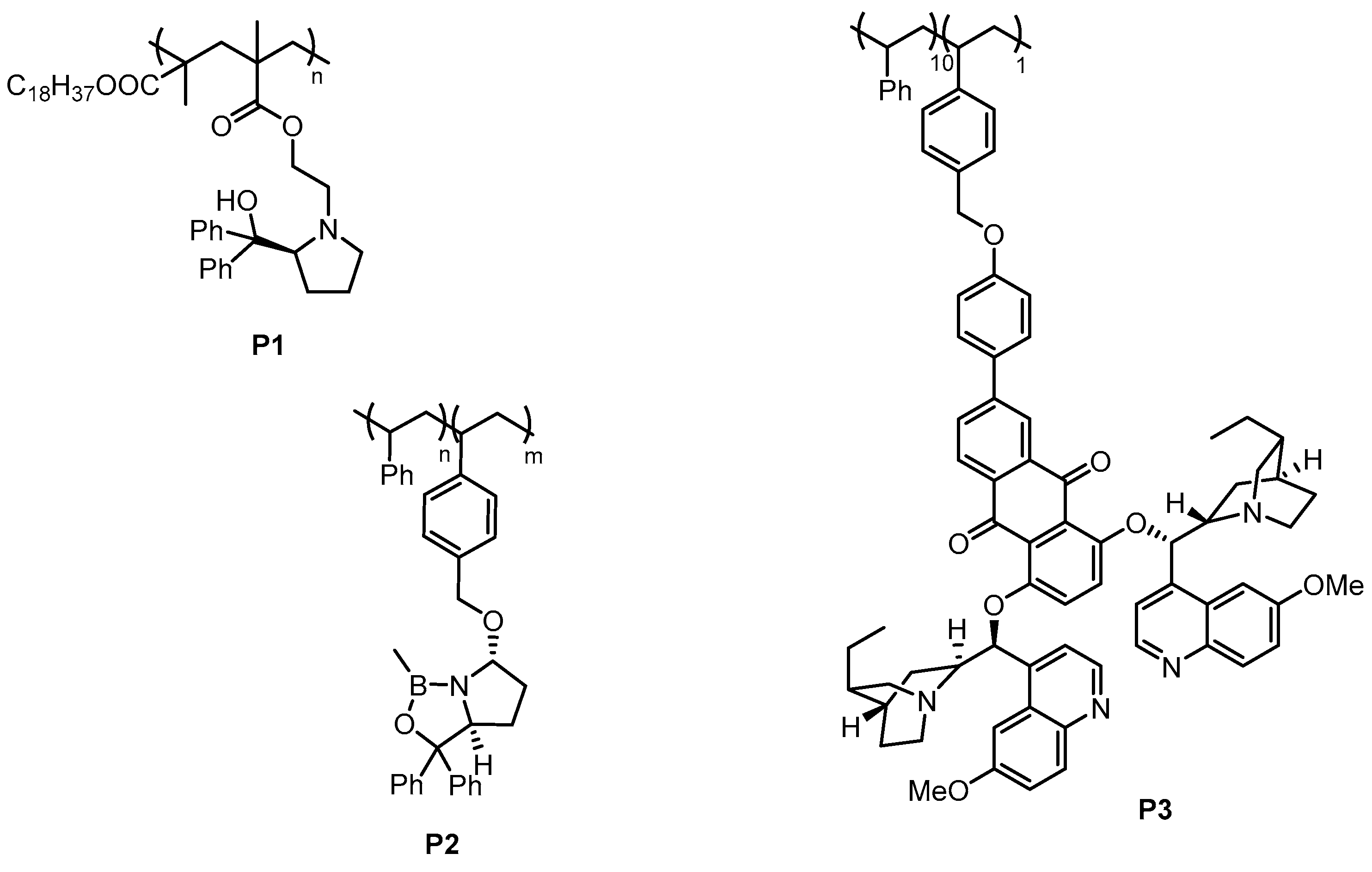
Figure 4.
Examples for polymer-supported homogeneous catalysts, which were recycled by membrane filtration [47,48,49,50,51,52].

Scheme 1.
Continuous reduction of tetralone by borane using oxazaborolidine organocatalyst P2 in a membrane reactor [48].
Scheme 1.
Continuous reduction of tetralone by borane using oxazaborolidine organocatalyst P2 in a membrane reactor [48].

Figure 5.
An example for dendrimer-bound homogeneous organocatalyst containing porphyrin motifs [53].
Figure 5.
An example for dendrimer-bound homogeneous organocatalyst containing porphyrin motifs [53].

Scheme 2.
Photooxidation of olefins catalyzed by dendrimer-enlarged organocatalysts [53].
Scheme 2.
Photooxidation of olefins catalyzed by dendrimer-enlarged organocatalysts [53].

Figure 6.
C3-Symmetrical molecular size-enlarged cinchona organocatalysts applied by Siew et al. [55].
Figure 6.
C3-Symmetrical molecular size-enlarged cinchona organocatalysts applied by Siew et al. [55].

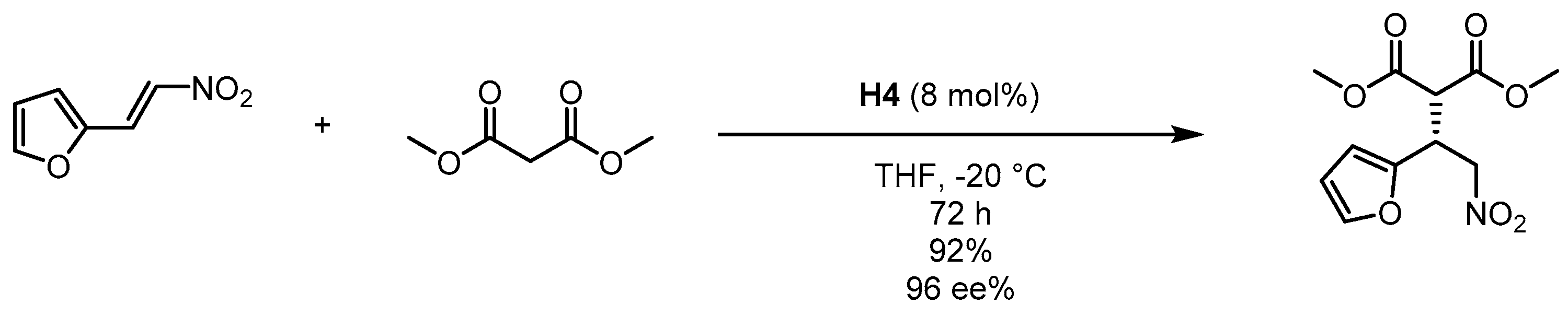
Scheme 3.
Michael addition of 2-(2-nitrovinyl) furan to dimethyl malonate catalyzed by C3-symmetrical molecular size-enlarged cinchona organocatalyst H4 [55].
Scheme 3.
Michael addition of 2-(2-nitrovinyl) furan to dimethyl malonate catalyzed by C3-symmetrical molecular size-enlarged cinchona organocatalyst H4 [55].

Figure 7.
Schematic representation of recyclable TEMPO mediated electrochemical oxidation of biomass-based HMF; mediator recovered by (a) standard filtration, (b) nanofiltration, (c) magnetic separation. NF: nanofiltration [57].
Figure 7.
Schematic representation of recyclable TEMPO mediated electrochemical oxidation of biomass-based HMF; mediator recovered by (a) standard filtration, (b) nanofiltration, (c) magnetic separation. NF: nanofiltration [57].

Figure 8.
Schematic representation of the applied (a) heterogeneous, and (b) homogeneous TEMPOs in the electrocatalytic oxidation of HMF to DFF [57].
Figure 8.
Schematic representation of the applied (a) heterogeneous, and (b) homogeneous TEMPOs in the electrocatalytic oxidation of HMF to DFF [57].

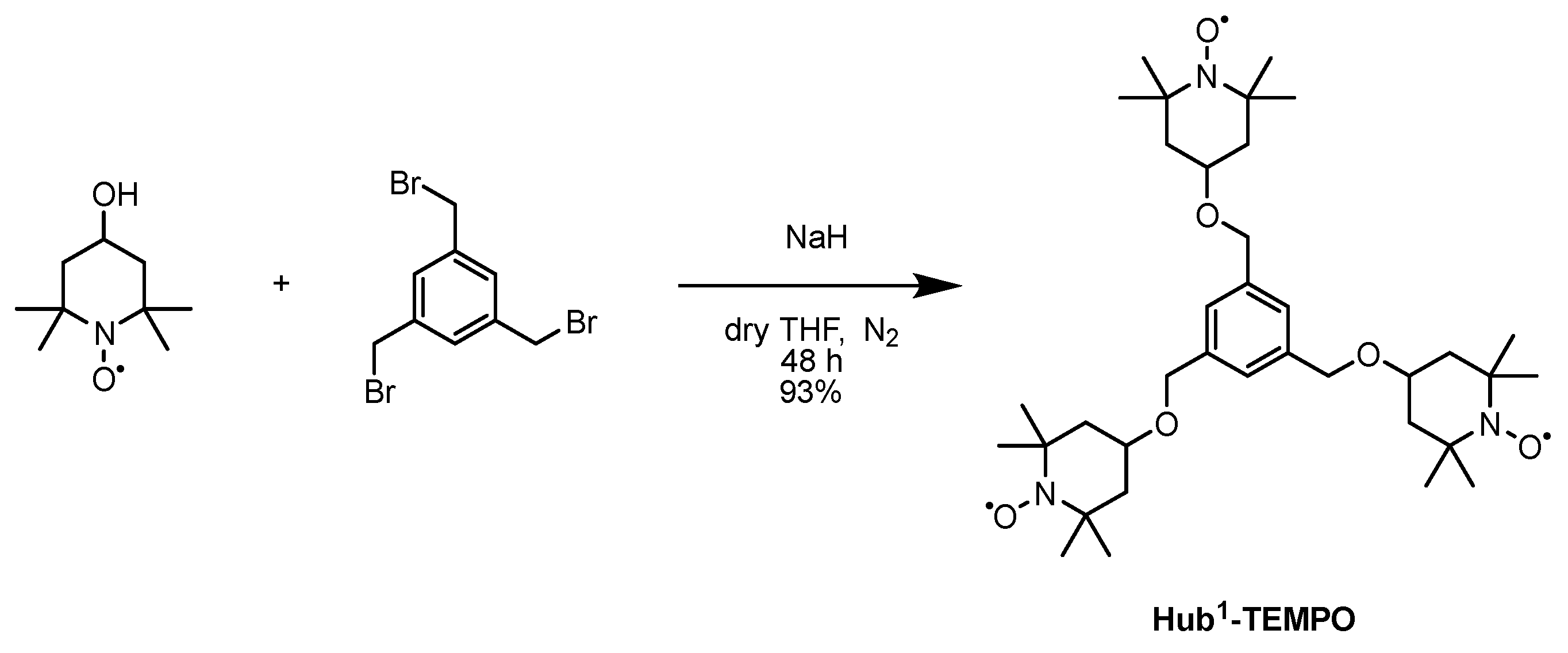
Scheme 4.
Synthesis of the size-enlarged C3-symmetrical Hub1-TEMPO catalyst using Williamson-type etherification [57].
Scheme 4.
Synthesis of the size-enlarged C3-symmetrical Hub1-TEMPO catalyst using Williamson-type etherification [57].

Figure 9.
Comparison of homogeneous and solid-supported TEMPO derivatives in the oxidation of HMF; a 10 mol% catalyst (3 equivalent active units); b 3.3 mol% catalyst (1 equivalent active unit). This figure is reprinted from reference [57] with permission from Wiley-VCH Verlag GmbH & Co. KGaA., available by license CC BY.
Figure 9.
Comparison of homogeneous and solid-supported TEMPO derivatives in the oxidation of HMF; a 10 mol% catalyst (3 equivalent active units); b 3.3 mol% catalyst (1 equivalent active unit). This figure is reprinted from reference [57] with permission from Wiley-VCH Verlag GmbH & Co. KGaA., available by license CC BY.

Figure 10.
Structures of the cinchona derivatives used by Fahrenwaldt et al. during the membrane filtration experiments [56].
Figure 10.
Structures of the cinchona derivatives used by Fahrenwaldt et al. during the membrane filtration experiments [56].

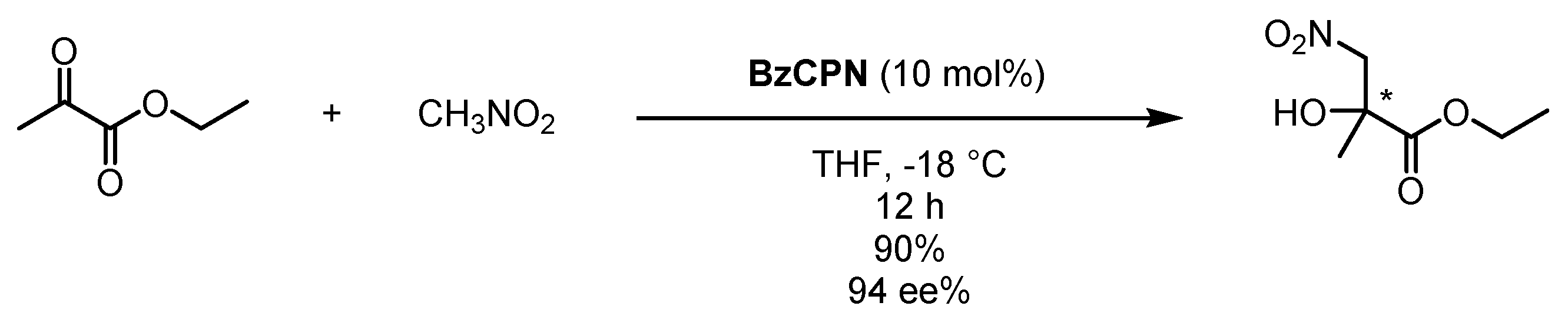
Scheme 5.
Enantioselective Henry reaction of ethyl pyruvate and nitromethane catalyzed by BzCPN [56].
Scheme 5.
Enantioselective Henry reaction of ethyl pyruvate and nitromethane catalyzed by BzCPN [56].

Figure 11.
Cinchona-decorated cyclodextrin derivatives, a new method used for organocatalyst size-enlargement: (a) schematic representation of the CD-anchoring through a H-bond donor unit; (b) the structure of β-cyclodextrin; (c) synthesis of CD-anchored cinchona thiourea (CD-1) and cinchona squaramide (CD-2) [58].
Figure 11.
Cinchona-decorated cyclodextrin derivatives, a new method used for organocatalyst size-enlargement: (a) schematic representation of the CD-anchoring through a H-bond donor unit; (b) the structure of β-cyclodextrin; (c) synthesis of CD-anchored cinchona thiourea (CD-1) and cinchona squaramide (CD-2) [58].

Scheme 6.
Michael addition of 1,3-diphenylpropane-1,3-dione to trans-β-nitrostyrene catalyzed by cyclodextrin-anchored organocatalyst CD-2 [58].
Scheme 6.
Michael addition of 1,3-diphenylpropane-1,3-dione to trans-β-nitrostyrene catalyzed by cyclodextrin-anchored organocatalyst CD-2 [58].

Figure 12.
Schematic process diagram for the continuous catalysis–separation platform. The coiled tube plug flow reactor and the membrane cell were thermostated at 20 °C and 50 °C, respectively. The reactor inlet flow rate was set at 4 mL min−1, the recycle ratio was 50%, and 2-MeTHF was used as solvent. The length and volume of the reactor were 21 m and 9.6 mL, respectively. This figure is reprinted from reference [58] with permission from Elsevier, available by license CC BY-NC-ND 4.0.
Figure 12.
Schematic process diagram for the continuous catalysis–separation platform. The coiled tube plug flow reactor and the membrane cell were thermostated at 20 °C and 50 °C, respectively. The reactor inlet flow rate was set at 4 mL min−1, the recycle ratio was 50%, and 2-MeTHF was used as solvent. The length and volume of the reactor were 21 m and 9.6 mL, respectively. This figure is reprinted from reference [58] with permission from Elsevier, available by license CC BY-NC-ND 4.0.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
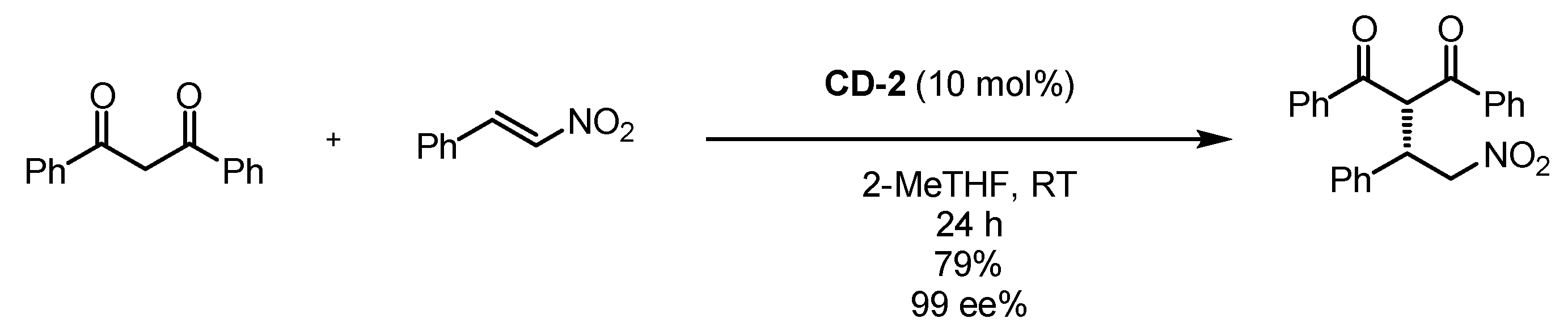{kind=link}
Table 1.
Classification of membrane process types and some examples for application areas [34].
Table 1.
Classification of membrane process types and some examples for application areas [34].
| Process Type | Pore Size of Membrane (nm) | Examples for Application |
|---|---|---|
| microfiltration | 50–500 | yeast, fungus, bacteria, oil emulsion |
| ultrafiltration | 2–50 | colloidal solid, virus, protein, polysaccharide |
| nanofiltration | ≤2 | catalysts, dyes, antibiotics, API impurities |
| reverse osmosis | 0.3–0.6 | water, inorganic ions |
Table 2.
Various membrane-types for organocatalyst recycling/purification by OSN.
| Reference | Type of Molecular Weight Enlargement | Catalyst MW [g mol−1] | Membrane Type a | Solvent b | Catalyst Retention [%] |
|---|---|---|---|---|---|
| Kupai et al. [46] | - | 351–435 | PBI | toluene | 97–100 |
| Kisszekelyi et al. [45] | - | 322–538 | PBI | IPA, THF, toluene | 48–99 |
| Nagy et al. [60] | - | 325 | GMT-oNF-1, -2, -3, PBI | MeCN | 88–99 |
| Großeheilmann et al. [62] | - | 374 | DM150, 200, 300, 500 | EtOH, acetone, butylene carbonate | 84–99 |
| Kragl et al. [47] | soluble polymer | ~96,000 | Nadir UF PA20 | n-hexane | 100 |
| Giffels et al. [48] | soluble polymer | ~13,800 | MPF-50 | THF | n.a. |
| Rissom et al. [49] | soluble polymer | ~14,000 | MPF-50 | THF | n.a. |
| Wöltinger et al. [50] | soluble polymer | ~22,640 | MPF-50 | THF | 99–100 |
| Tsogoeva et al. [51] | soluble polymer | n.a. | MPF-50 | THF | 99 |
| Wöltinger et al. [52] | soluble polymer | n.a. | n.a. | toluene:MeOH | n.a. |
| Chavan et al. [53] | dendrimer | 2117–8650 | MPF-50, PDMS, PDMS-USY-PAN | IPA, CHCl3 | 40–99 |
| Št’astná et al. [54] | dendrimer | 1342–7724 | regenerated cellulose | MeOH | 20–98 c |
| Siew et al. [55] | multifunctional core | 1044–1332 | DM500, 300 | THF | >99 |
| Kisszekelyi et al. [57] | multifunctional core | 631 | GMT-oNF-1, NF030306, DM300 | MeCN | 90–100 |
| Fahrendwaldt et al. [56] | benzoyl subunit | 324–429 | DM150, 200, 300, 500 | THF | 90–100 |
| Kisszekelyi et al. [58] | cyclodextrin-anchoring | 1817 | GMT-oNF-1, NF030306, NF010306, DM300, 500, 900 | 2-MeTHF | 98–100 |
| Großeheilmann et al. [61] | esterification | 409–521 | DM200, 300 | EtOH | 53–100 |
| Gupta [59] | salt formation | 115–548 | PDCPD | DCM, MeOH | 0–99 |
a PBI: polybenzimidazole, DM: Duramem, PDMS: polydimethylsiloxane, PDMS-USY-PAN: ultra-stable Y zeolite modified PDMS, PDCPD: polydicyclopentadiene; b IPA: propane-2-ol, MeCN: acetonitrile, 2-MeTHF: 2-methyltetrahydrofuran; c Calculated based on reference: [54].
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kisszékelyi, P.; Nagy, S.; Fehér, Z.; Huszthy, P.; Kupai, J. Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review. Chemistry 2020, 2, 742-758. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030048
AMA Style
Kisszékelyi P, Nagy S, Fehér Z, Huszthy P, Kupai J. Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review. Chemistry. 2020; 2(3):742-758. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030048
Chicago/Turabian StyleKisszékelyi, Péter, Sándor Nagy, Zsuzsanna Fehér, Péter Huszthy, and József Kupai. 2020. "Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review" Chemistry 2, no. 3: 742-758. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030048