
Stability of Rhodamine Lactone Cycle in Solutions: Chain–Ring Tautomerism, Acid–Base Equilibria, Interaction with Lewis Acids, and Fluorescence

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
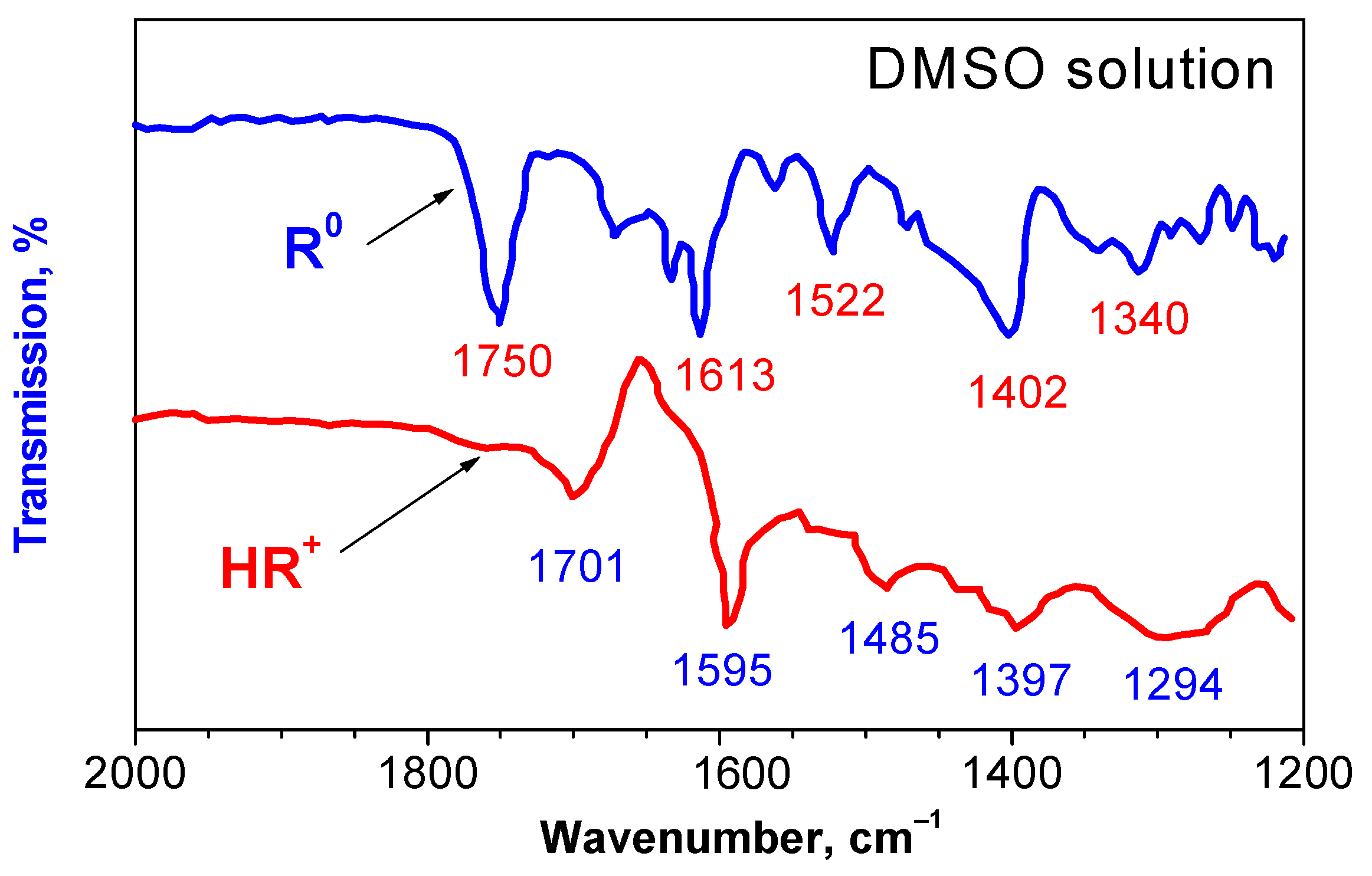
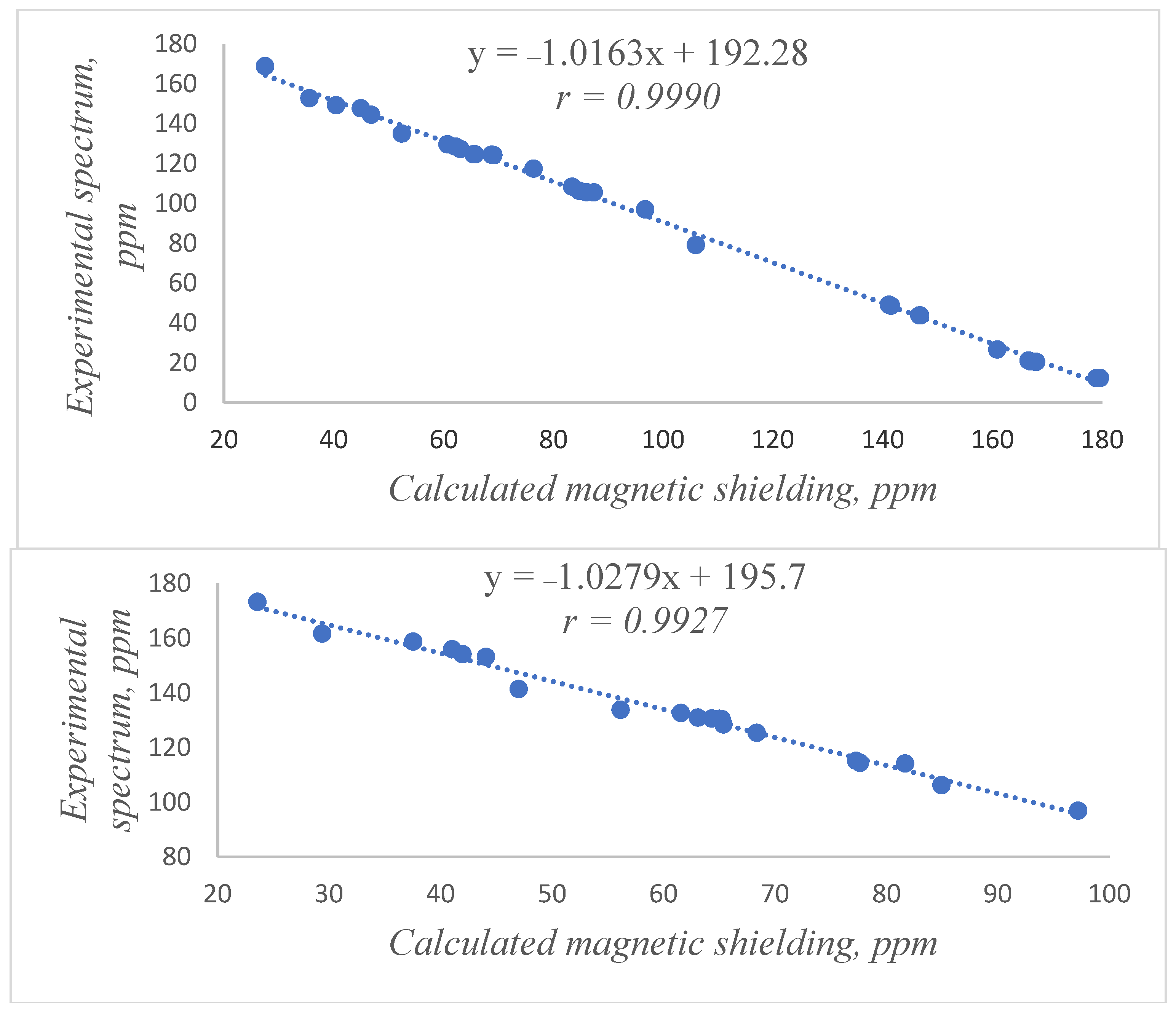
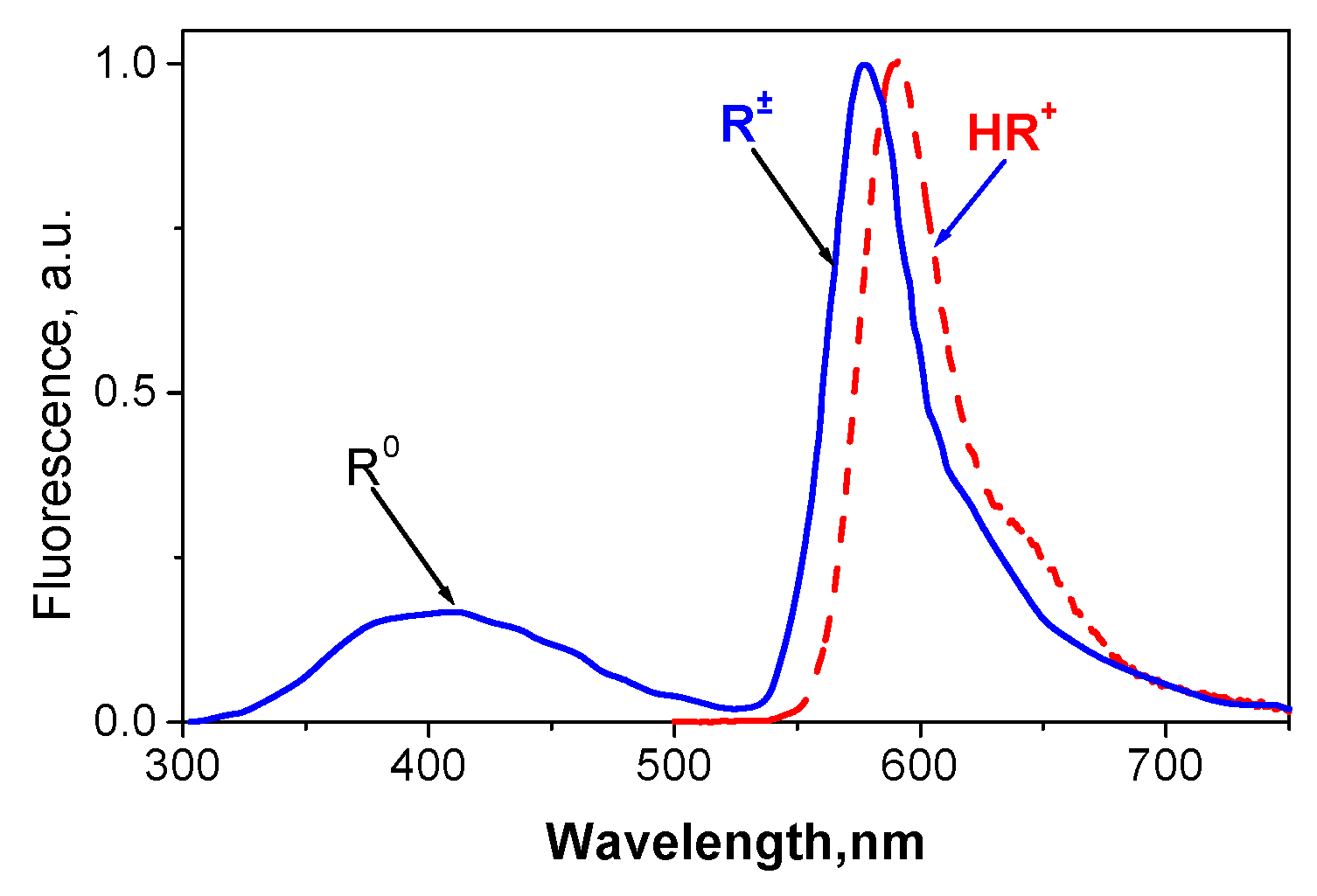
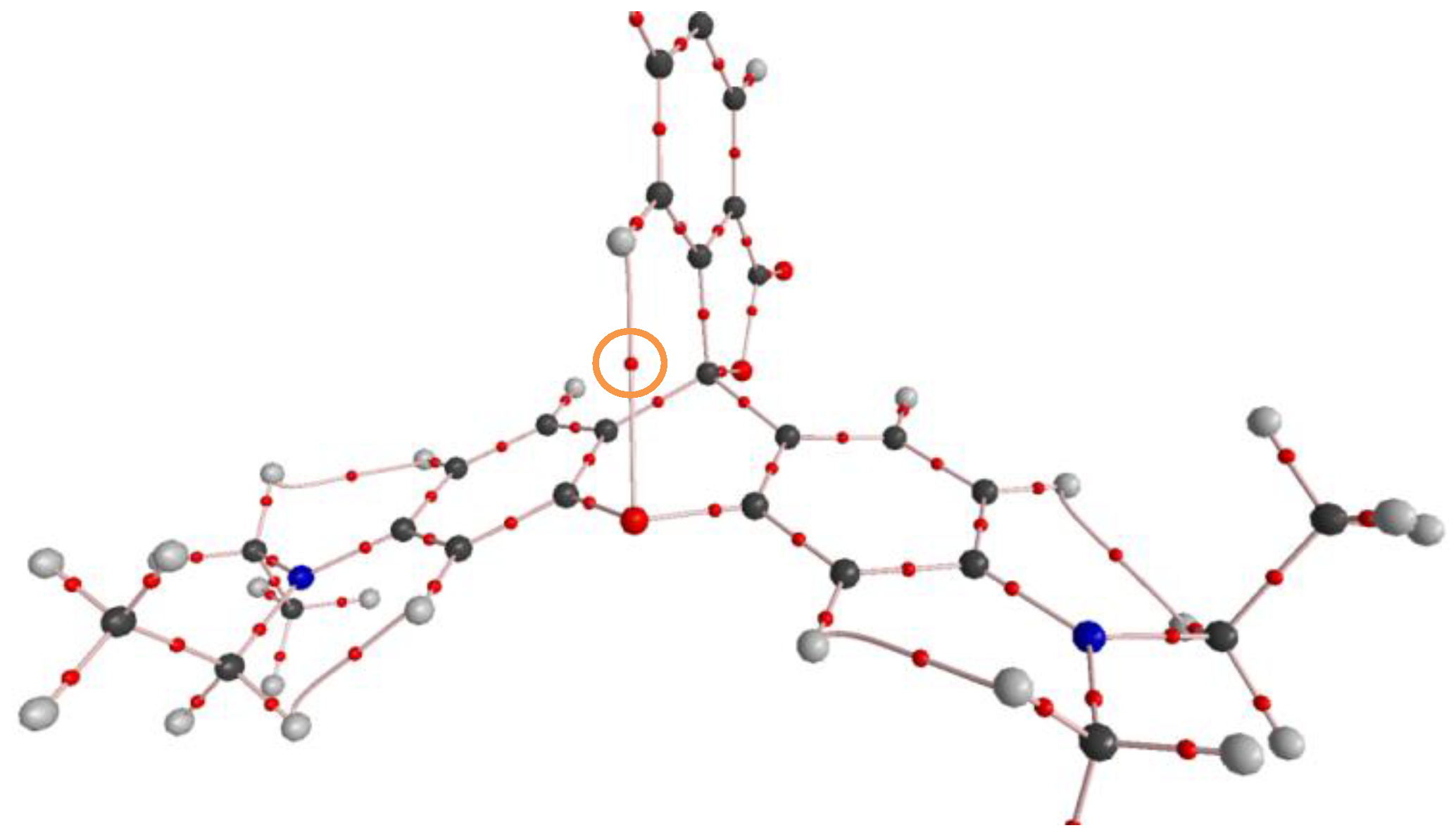
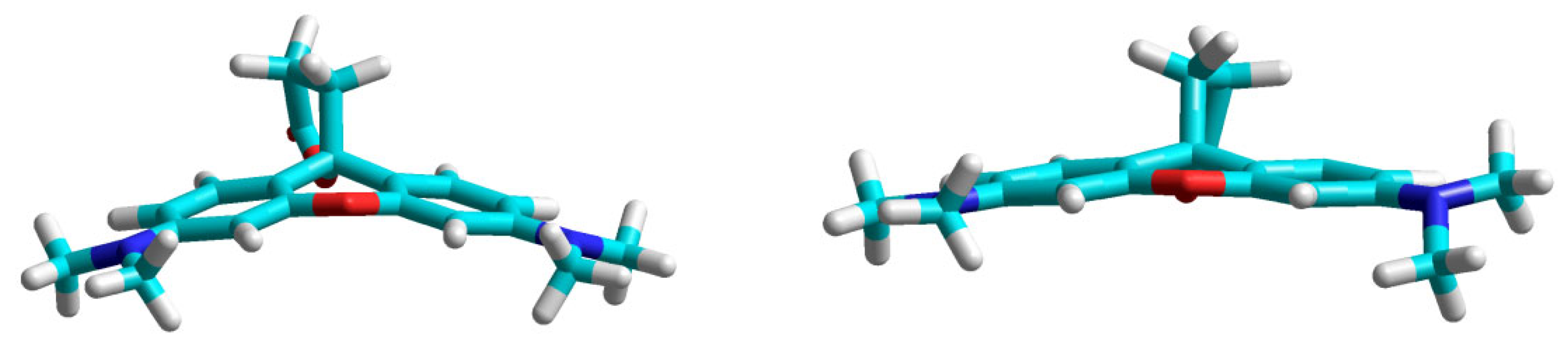
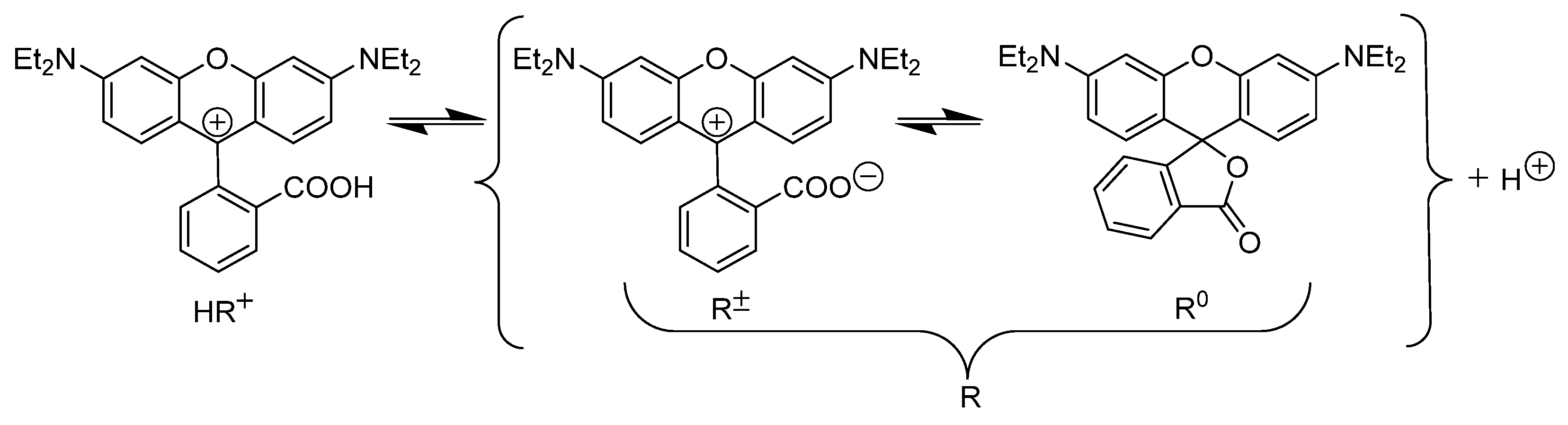
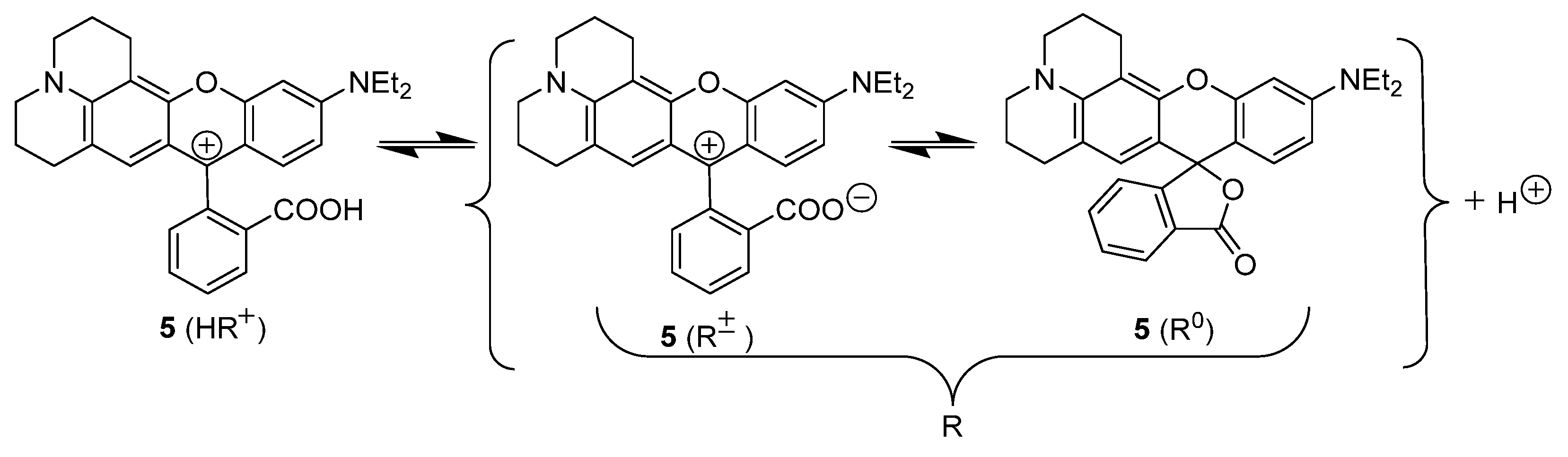
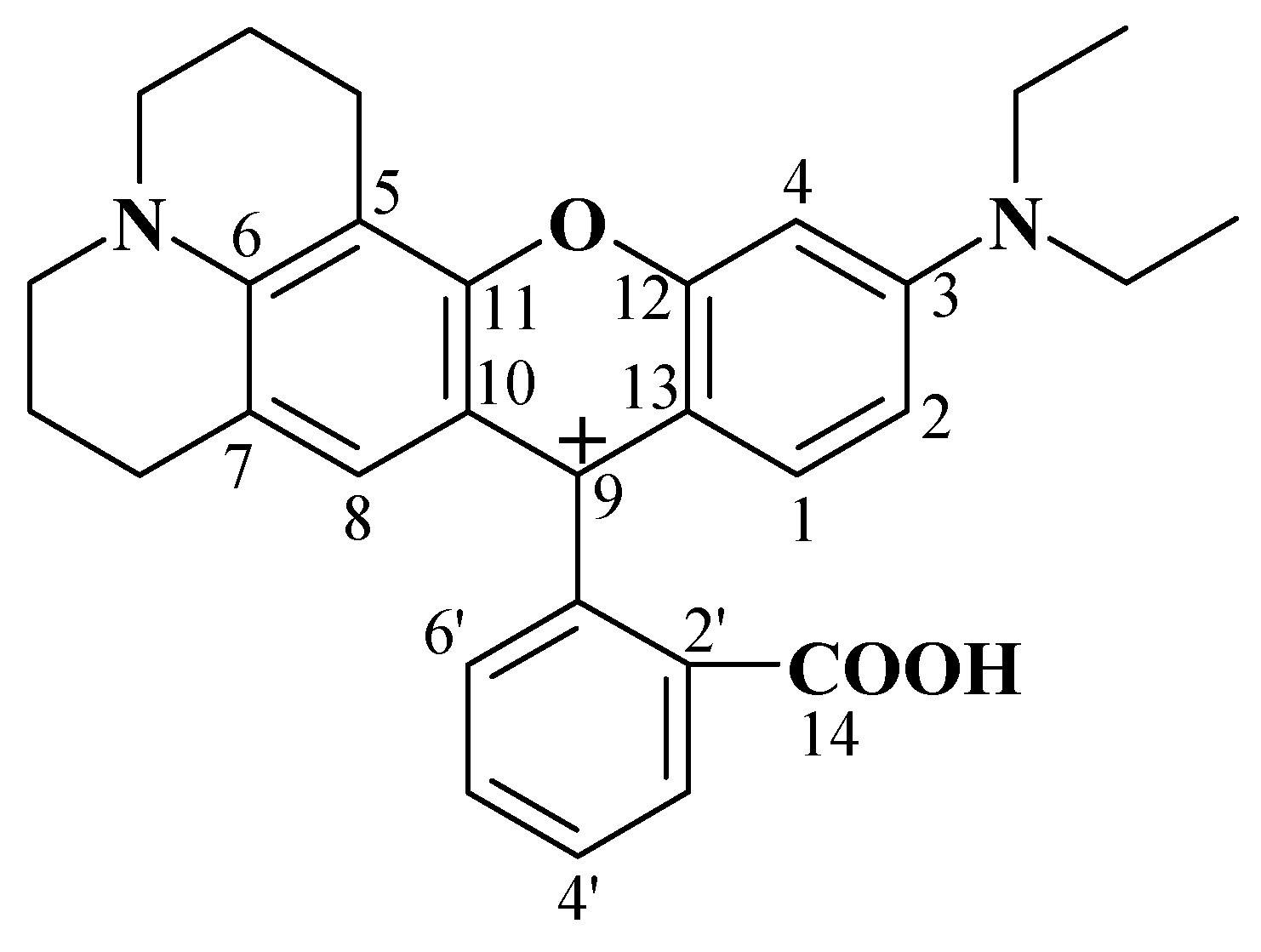
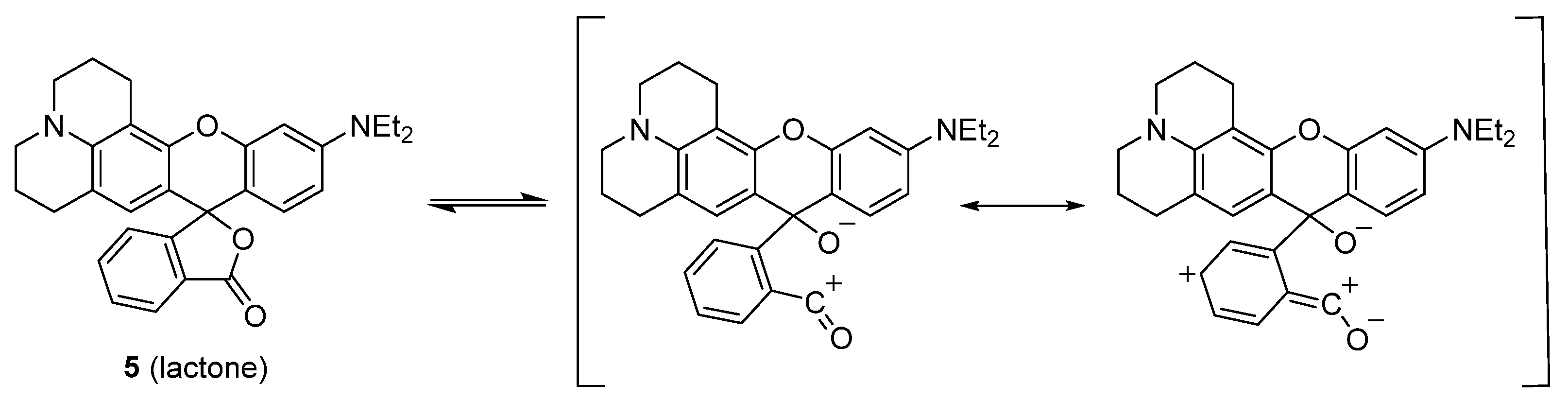
2.1. Identification of the Equilibrium Structures in Solution
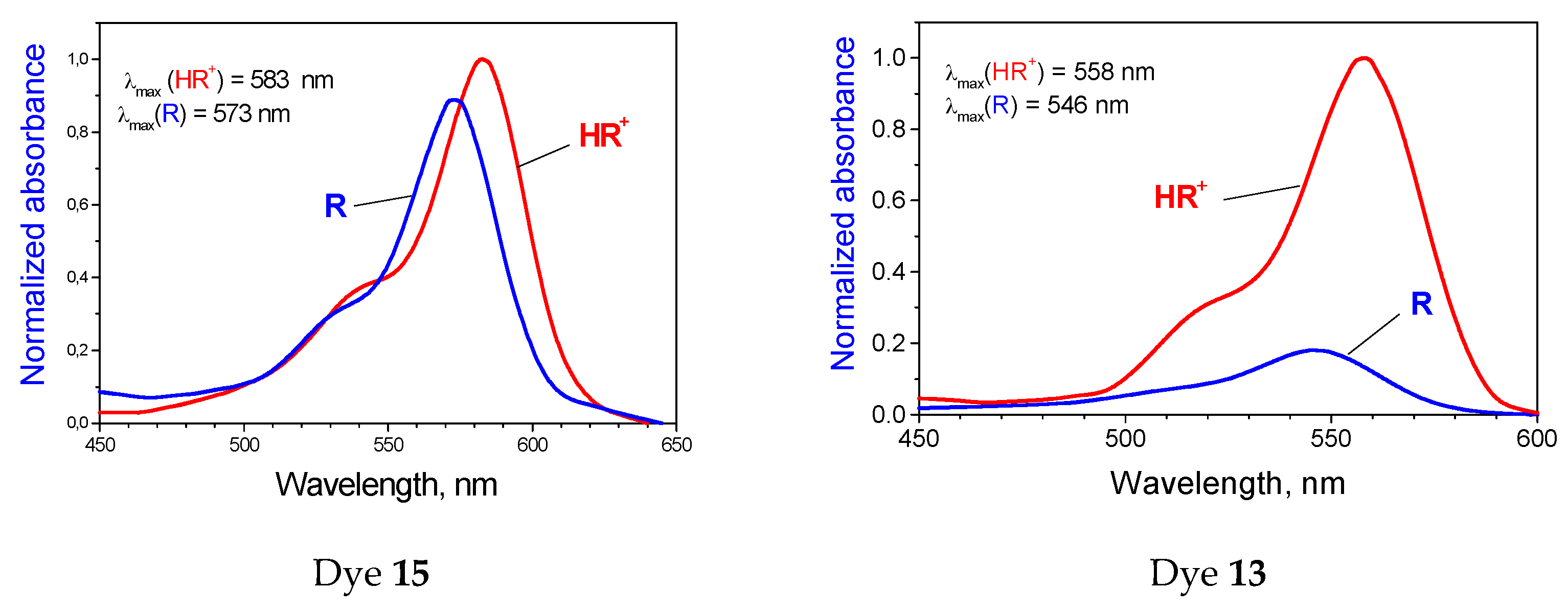
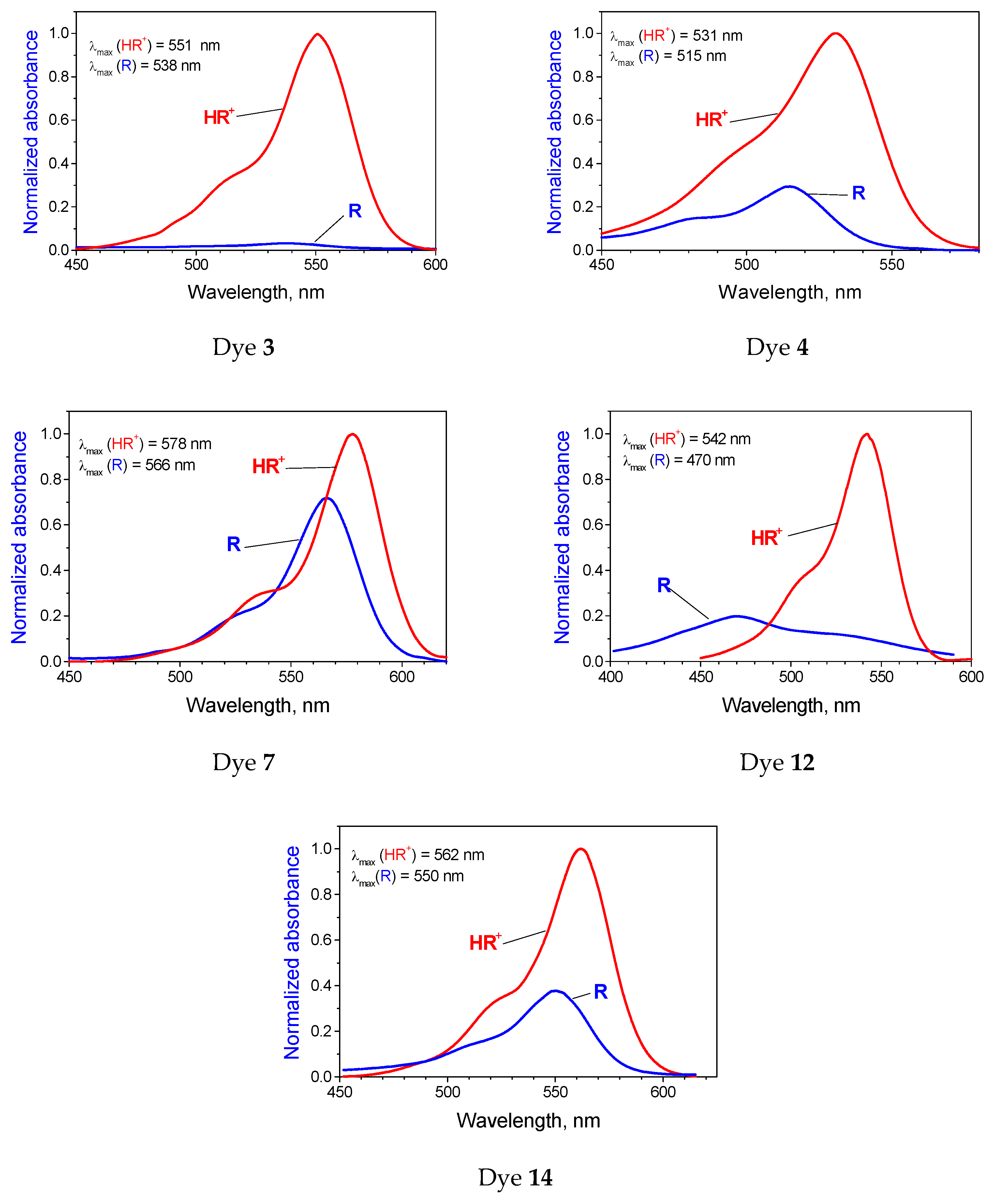
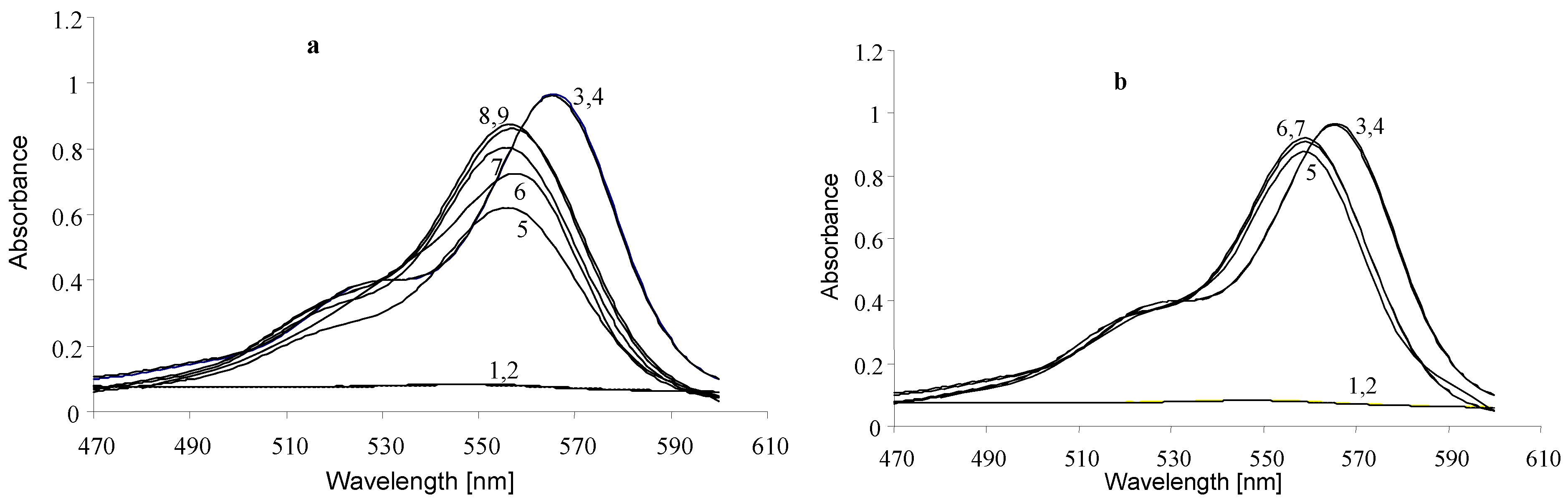
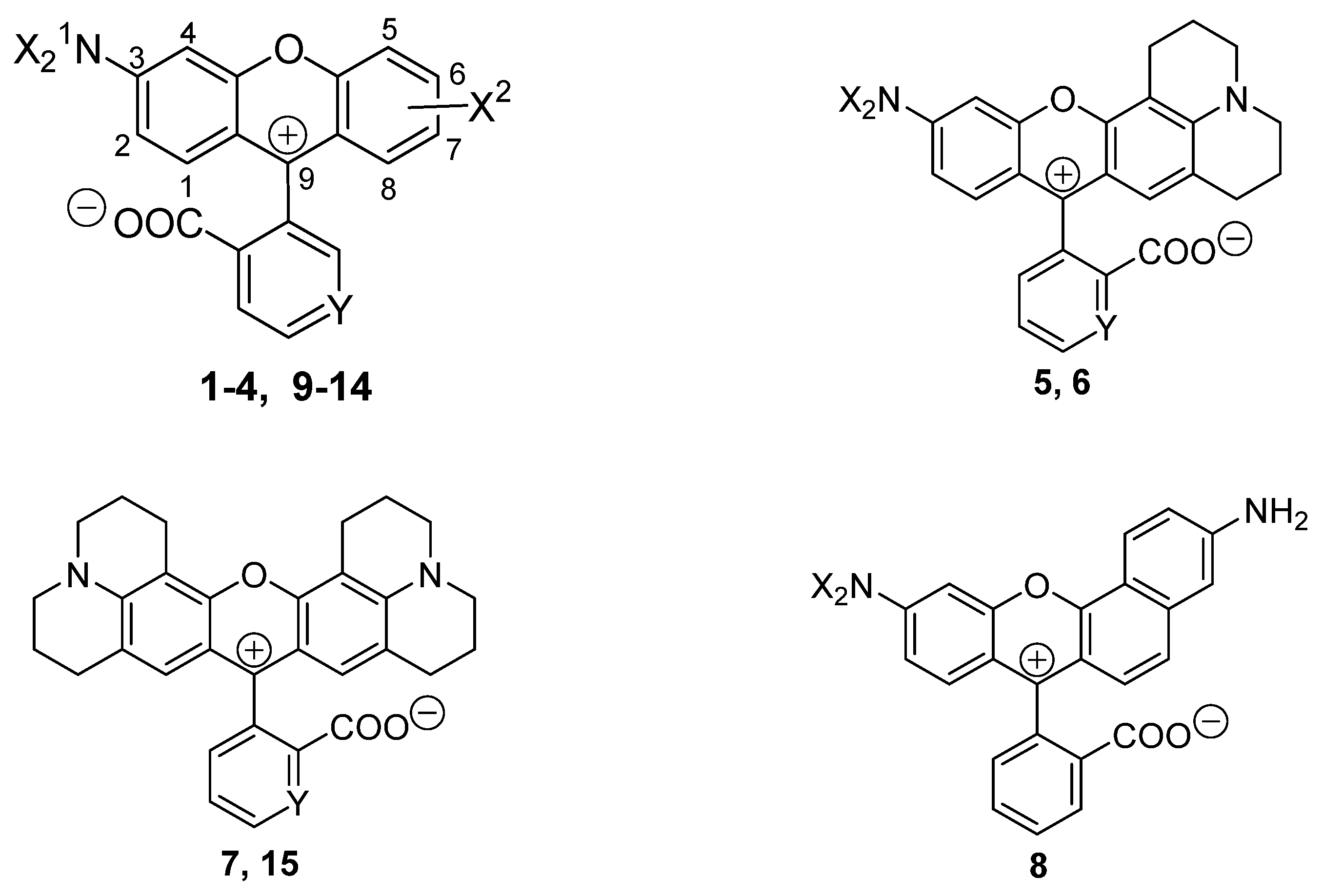
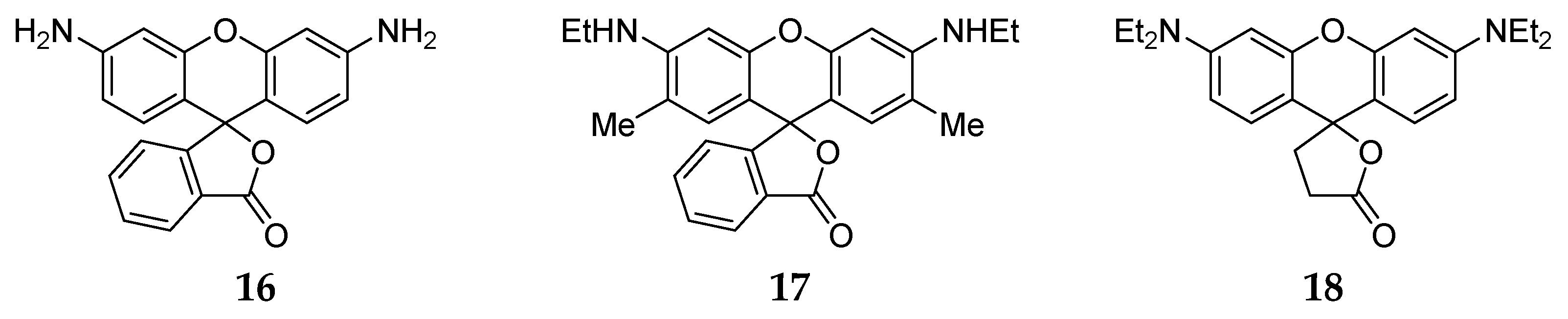
2.2. The Influence of Molecular Structure of Rhodamine Dyes on the Chain–Ring Tautomerism
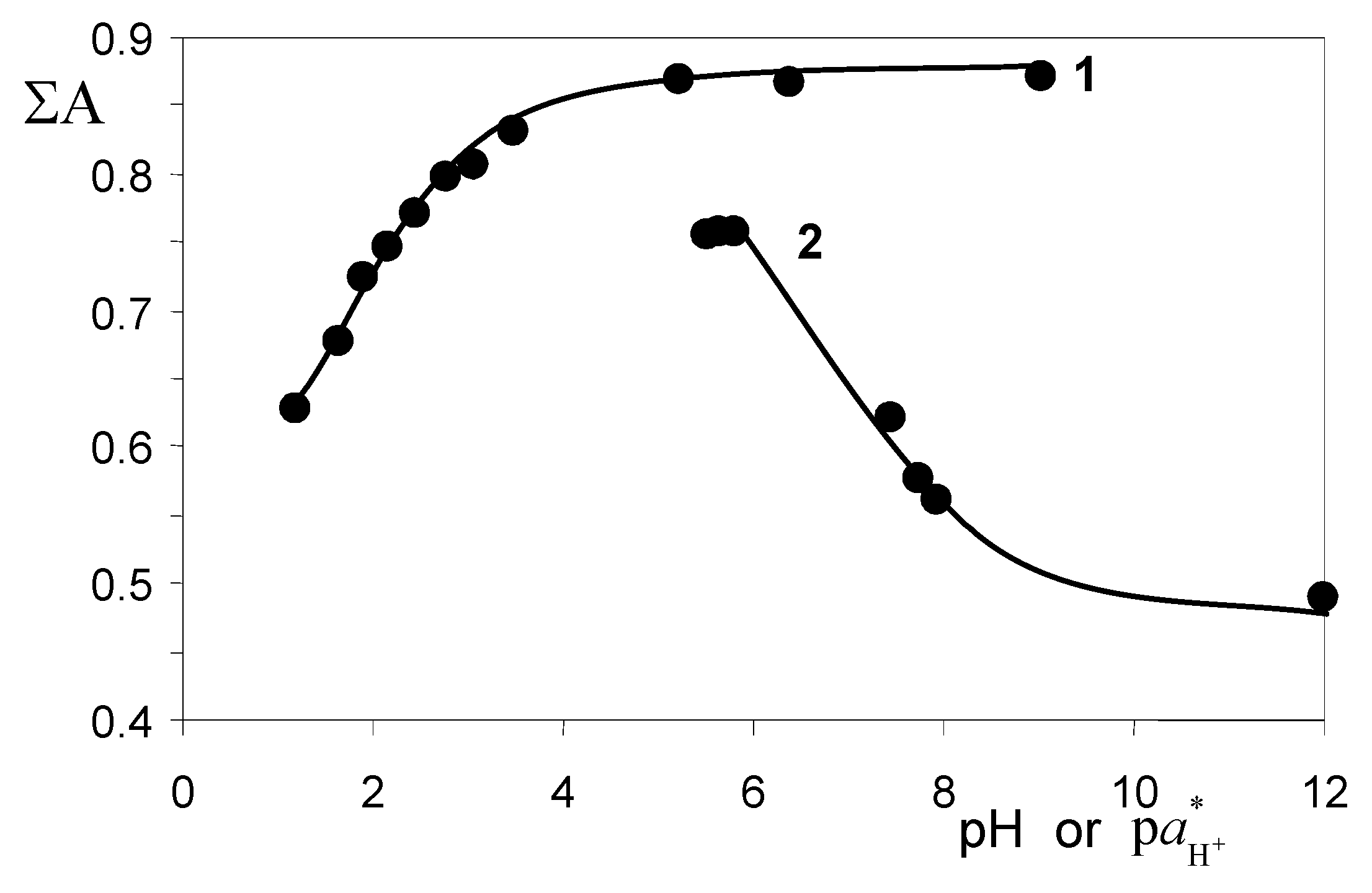
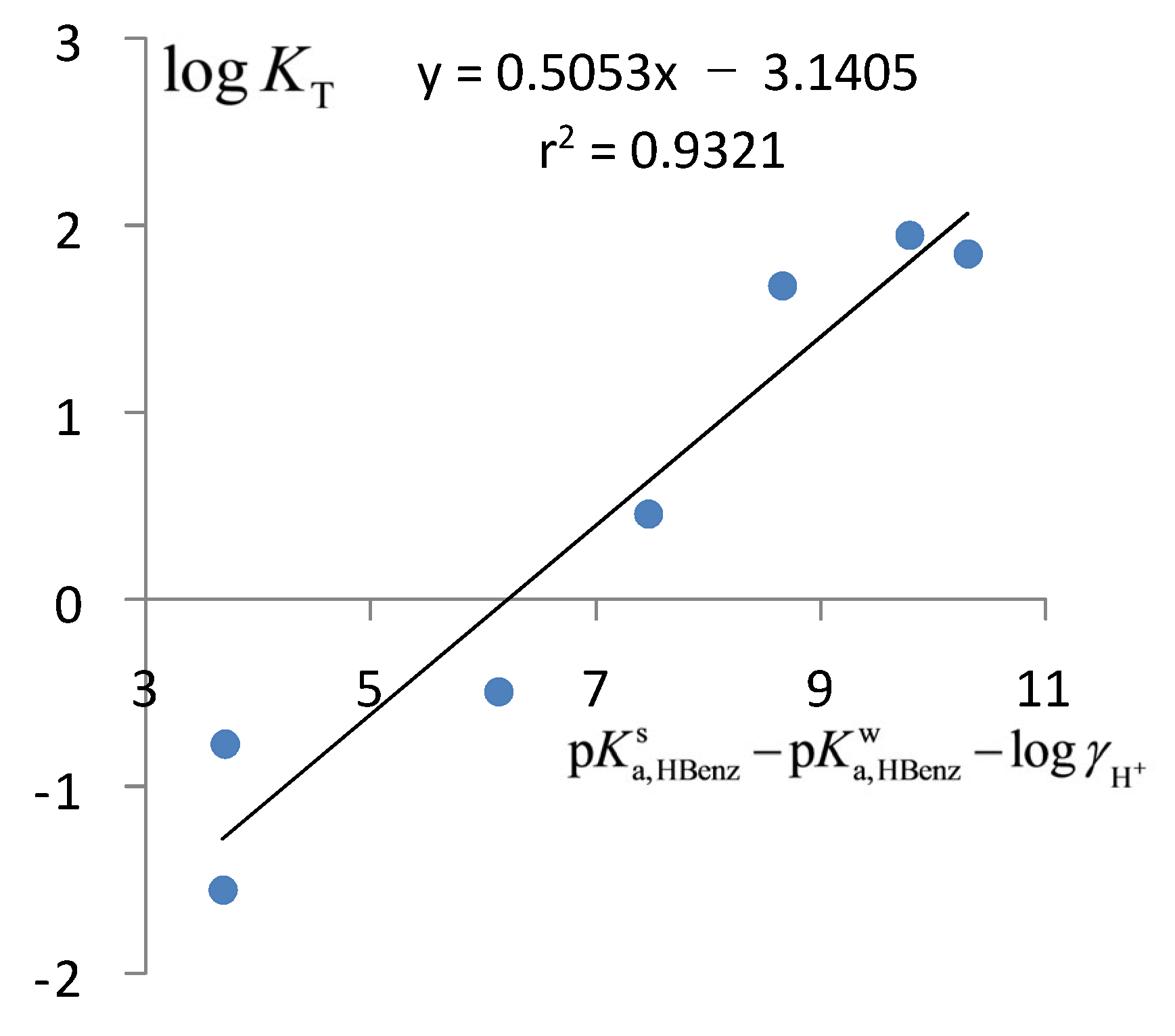
2.3. The Acidic Dissociation of the HR+ Acid and the Charge Type of the Acid/Base Couple

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
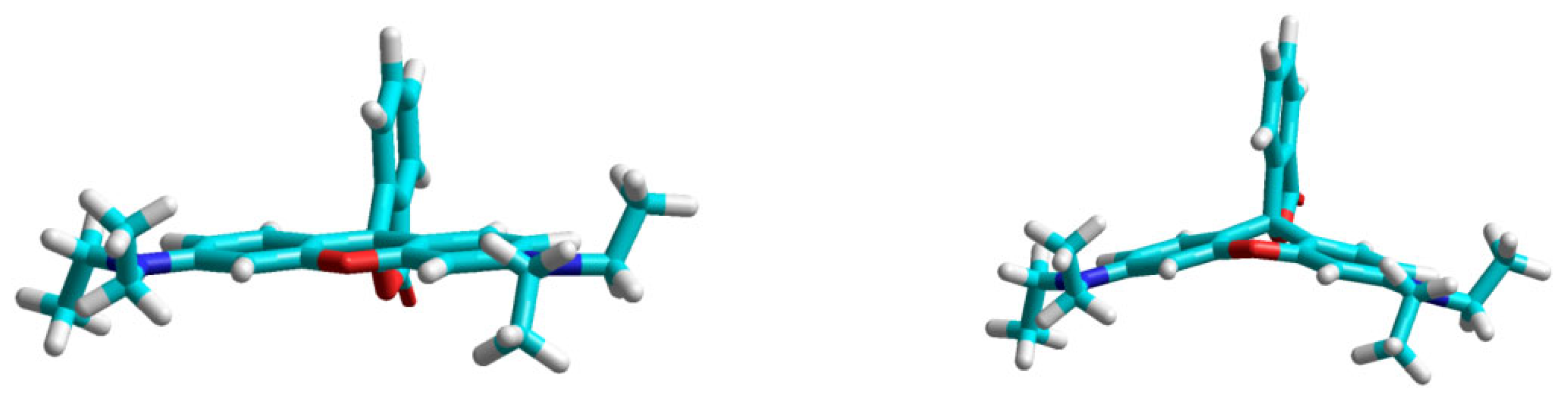{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Rhodamine B (1) | Rhodamine 19 (N,N′-Diethyl-2,7-Dimethylrhodamine) | Rhodamine 5 |
|---|---|---|---|
| Water | 3.2 b; 3.22 c; 3.22 d; 3.41 e; 3.46 f; 3.36 g | 3.15 h; 3.26 i | 3.10 |
| Methanol | 7.6 j; 7.4 k | — | — |
| Ethanol | 8.7 j; 8.7 k | 9.4 h | — |
| N,N-Dimethylformamide | 6.5 k | — | — |
| N-Methyl pyrrolidine-2-one | 5.55 l | — | — |
| 91.3 mass% DMSO | 5.60 k | — | — |
| Acetone + 5 mol% DMSO | 7.50 m | 7.90 m | — |
| 90 mass% Acetone | 6.47 k | — | 7.31 |
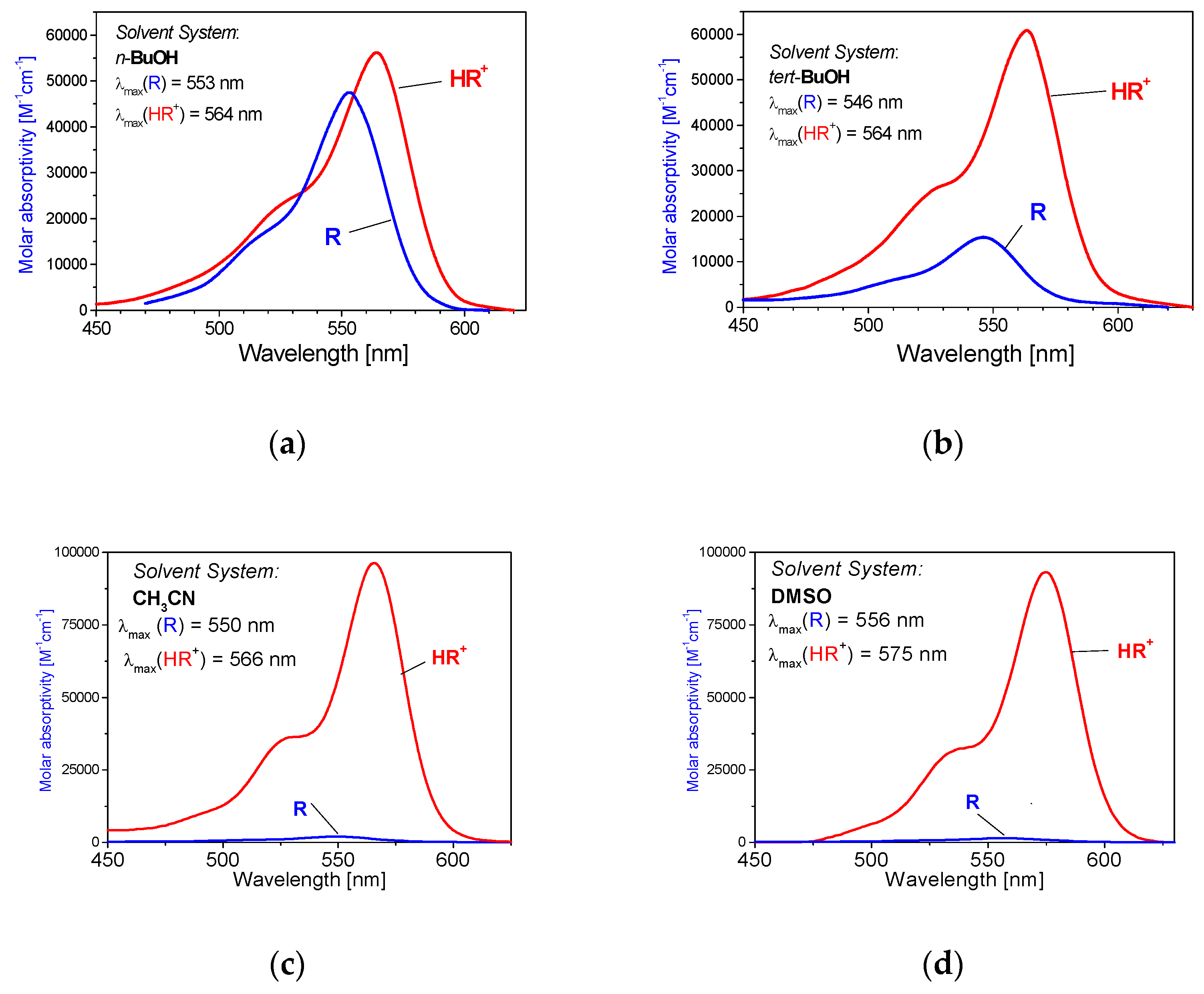
2.4. Tautomerism: Influence of the Solvent Nature
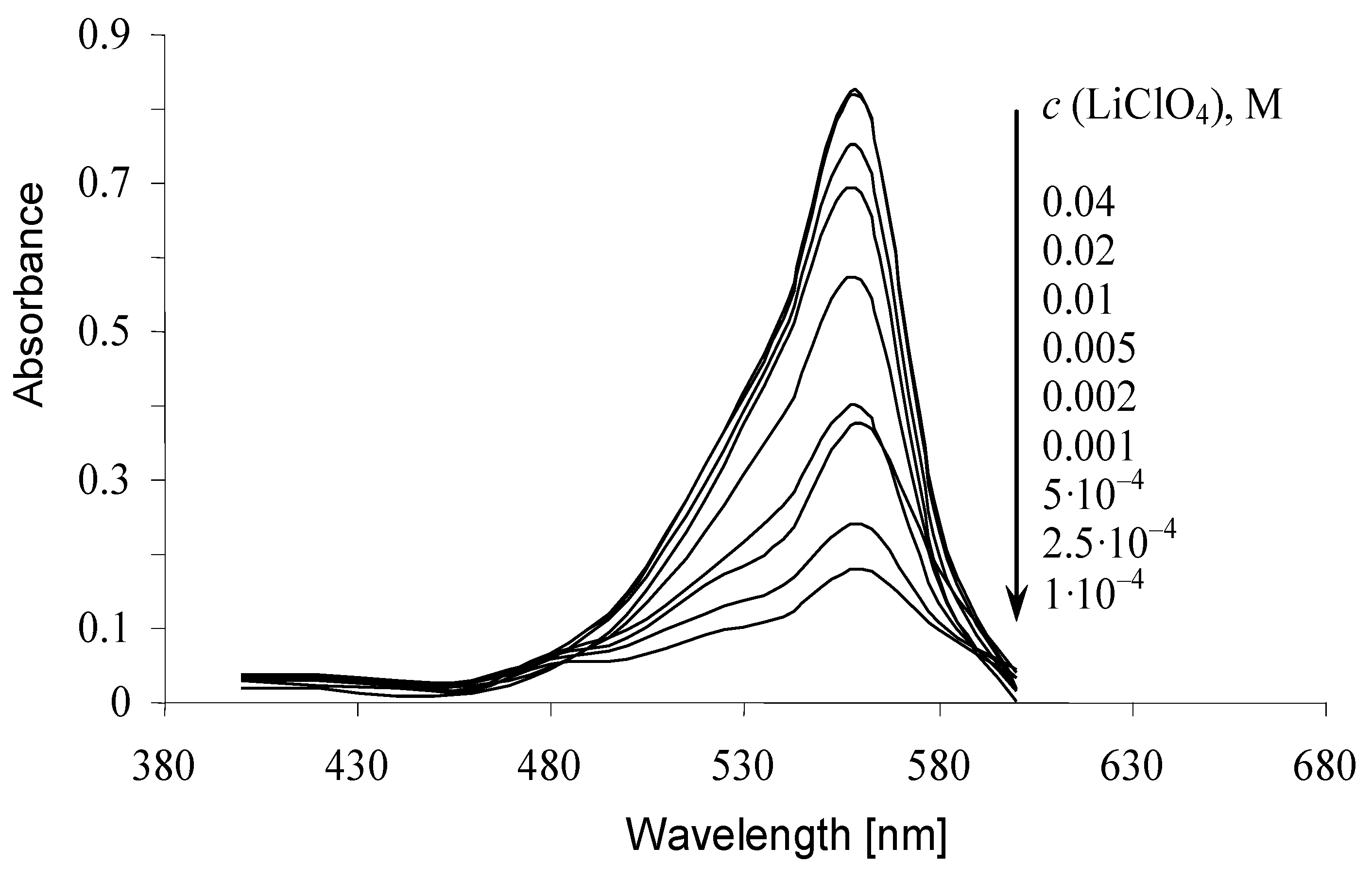
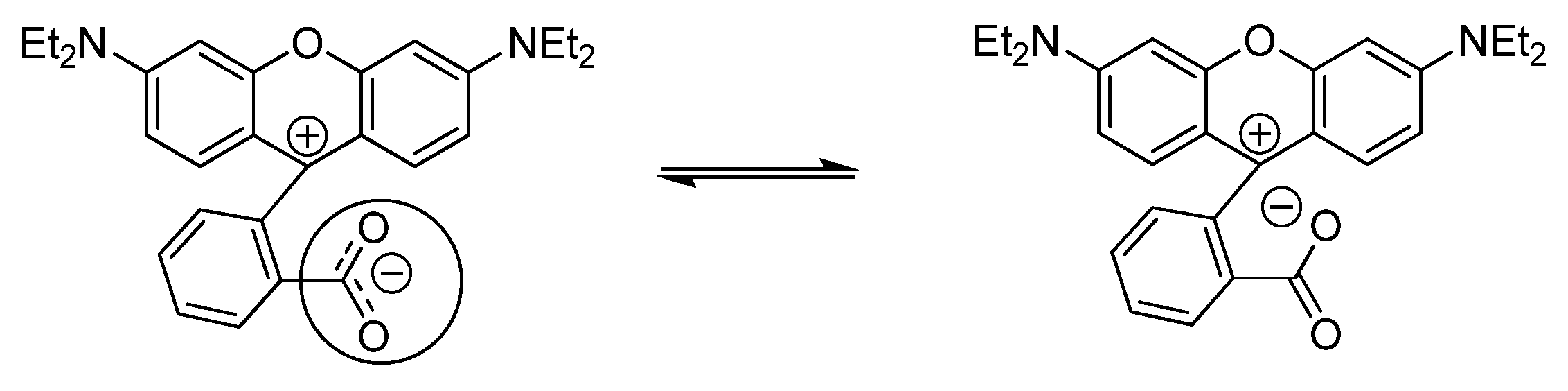
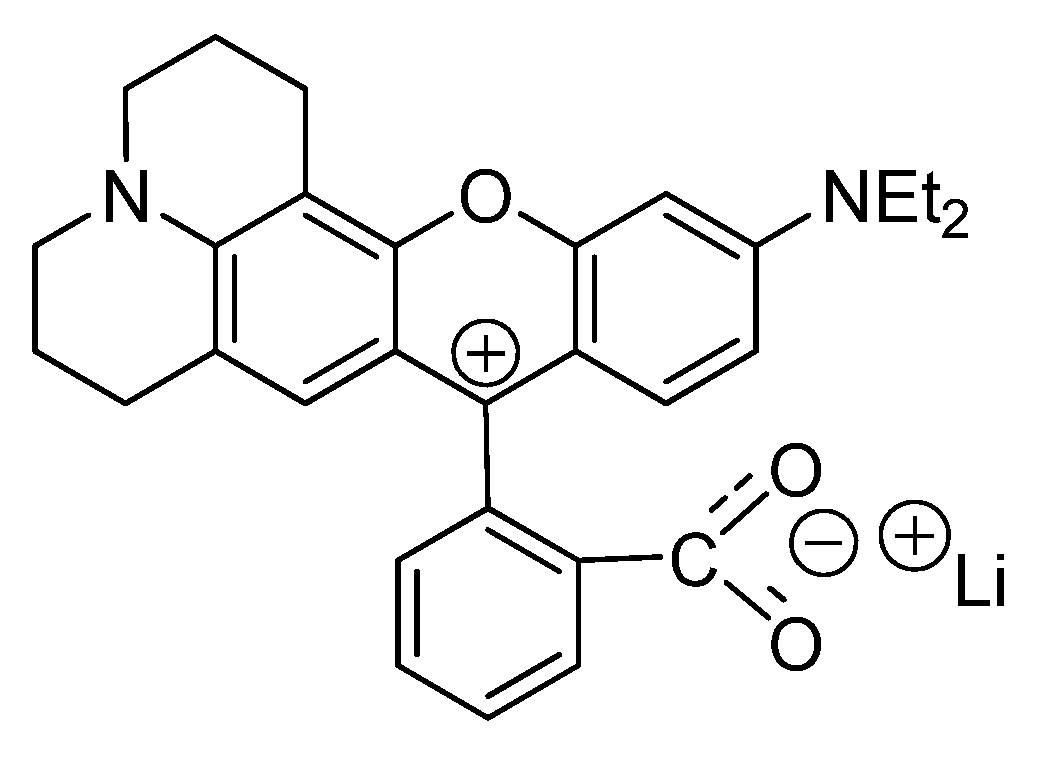
2.5. Interactions with Electrolytes
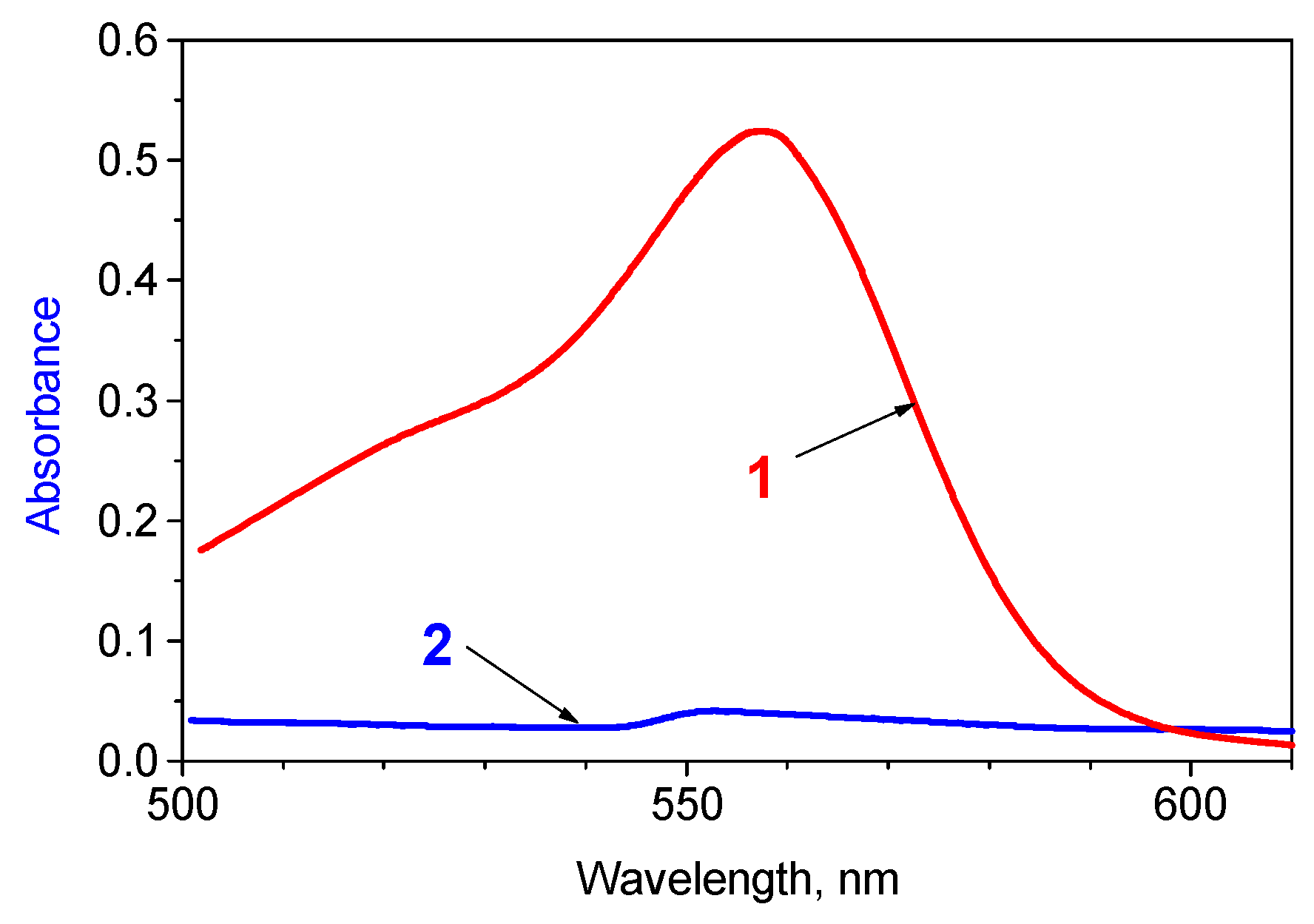
2.6. Fluorescence and Photocleavage of the Lactone Cycle
3. Experimental
3.1. Materials
3.2. Synthesis of Dyes 13–15
3.3. Apparatus
3.4. Procedure
3.5. Quantum-Chemical Calculations
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Beija, M.; Afonso, C.A.M.; Martinho, J.M.G. Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Burgess, K. Fluorescent indicators for intracellular pH. Chem. Rev. 2010, 110, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
- Haugland, R.P. Handbook of Fluorescent Probes and Research Products, 9th ed.; Molecular Probes, Inc.: Eugene, OR, USA, 2002. [Google Scholar]
- Shin, Y.; Kim, K.S.; Kim, B. Loading behavior of pH-responsive P(MMA-co-EGMA) hydrogel microparticles for intelligent drug delivery applications. Polymer 2008, 32, 421–426. [Google Scholar] [CrossRef]
- Liu, W.; Chen, J.; Xu, L.; Wu, J.; Zhang, H.; Wang, P. Reversible “off–on” fluorescence chemosensor for Hg2+ based on rhodamine derivative. Spectrochim. Acta A 2012, 85, 38–42. [Google Scholar] [CrossRef]
- Freddi, S.; Sironi, L.; D’Antuono, R.; Morone, D.; Dona, A.; Cabrini, E.; D’Alfonso, L.; Collini, M.; Pallavicini, P.; Baldi, G.; et al. A Molecular Thermometer for Nanoparticles for Optical Hyperthermia. Nano Lett. 2013, 13, 2004–2010. [Google Scholar] [CrossRef]
- Rittikulsittichai, S.; Singhana, B.; Bryan, W.W.; Sarangi, S.; Jamison, A.C.; Brazdeikis, A.; Lee, T.R. Preparation, characterization, and utilization of multifunctional magnetic-fluorescent composites for bioimaging and magnetic hyperthermia therapy. RSC Adv. 2013, 3, 7838–7849. [Google Scholar] [CrossRef]
- Moreno-Villoslada, I.; Jofre, M.; Miranda, V.; Gonzalez, R.; Sotelo, T.; Hess, S.; Rivas, B.L. pH Dependence of the Interaction between Rhodamine B and the Water-Soluble Poly(sodium 4-styrenesulfonate). J. Phys. Chem. B 2006, 110, 11809–11812. [Google Scholar] [CrossRef]
- Bujdak, J.; Czimerova, A.; Lopez Arbeloa, F. Two-step resonance energy transfer between dyes in layered silicate films. J. Colloid Interface Sci. 2011, 364, 497–504. [Google Scholar] [CrossRef]
- Nikishiori, H.; Fujii, T. Molecular forms of rhodamine B in dip-coated thin films. J. Phys. Chem. B 1997, 101, 3680–3687. [Google Scholar] [CrossRef]
- Merka, O.; Yarovyi, V.; Bahnemann, D.W.; Wark, M. pH-Control of the photocatalytic degradation mechanism of rhodamine B over Pb3Nb4O13. J. Phys. Chem. C 2011, 115, 8014–8023. [Google Scholar] [CrossRef]
- Watarai, H.; Funaki, F. Total Internal Reflection Fluorescence Measurements of Protonation Equilibria of Rhodamine B and Octadecylrhodamine B at a Toluene/Water Interface. Langmuir 1996, 12, 6717–6720. [Google Scholar] [CrossRef]
- Tsukahara, S.; Yamada, Y.; Watarai, H. Effect of surfactants on in-plane and out-of-plane rotational dynamics of octadecylrhodamine B at toluene–water interface. Langmuir 2000, 16, 6787–6794. [Google Scholar] [CrossRef]
- Tokimoto, T.; Tsukahara, S.; Watarai, H. Lactone-cleavage reaction kinetics of rhodamine dye at liquid/liquid interfaces studied by micro-two-phase sheath flow/two-photon excitation fluorescence microscope. Langmuir 2005, 21, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Tsukahara, S.; Watarai, H. Single-molecule lactonization of octadecylrhodamine B at a liquid-liquid interface. Langmuir 2012, 28, 15428–15432. [Google Scholar] [CrossRef]
- Zhen, X.-Y.; Harata, A. Confocal fluorescence microscope studies of the adsorptive behavior of dioctadecyl-rhodamine B molecules at a cyclohexane–water interface. Anal. Sci. 2001, 17, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snare, M.J.; Treloar, F.E.; Ghiggino, K.P.; Thistlethwaite, P.J. The photophysics of rhodamine B. J. Photochem. 1982, 18, 335–346. [Google Scholar] [CrossRef]
- Chang, T.-L.; Cheung, H.C. Solvent effect on photoisomerisation rates of the zwitterionic and the cationic forms of rhodamine B in protic solvents. J. Phys. Chem. 1992, 96, 4874–4878. [Google Scholar] [CrossRef]
- Magde, D.; Rogas, G.E.; Seybold, P.G. Solvent dependence of the fluorescence lifetime of xanthene dyes. Photochem. Photobiol. 1999, 70, 737–744. [Google Scholar] [CrossRef]
- Karpiuk, J.; Grabowski, Z.R.; De Schryver, F.C. Photophysics of the Lactone Form of Rhodamine 101. J. Phys. Chem. 1994, 98, 3247–3256. [Google Scholar] [CrossRef]
- El-Rayyes, A.A.; Al-Arfai, A.A.; Klein, U.K.A.; Barri, S.A.I. Acidity of all-slica MCM-41–studied by laser spectroscopy of adsorbed fluorescent probe compounds. Catal. Lett. 2004, 97, 83–90. [Google Scholar] [CrossRef]
- Sinelnikov, A.N.; Artyukhov, V.Y. Fluorescence of the lactone form of rhodamine B. Russ. J. Phys. Chem. A 2013, 87, 1409–1416. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Zhang, Y.; Liu, L. Fluorescence lifetimes and quantum yields of ten rhodamine derivatives: Structural effect on emission mechanism in different solvents. J. Lumin. 2014, 145, 448–453. [Google Scholar] [CrossRef]
- Sagoo, S.K.; Jockusch, R.A. The fluorescence properties of cationic rhodamine B in the gas phase. J. Photochem. Photobiol. A Chem. 2011, 220, 173–178. [Google Scholar] [CrossRef]
- Nagy, A.M.; Talbot, F.; Czar, M.F.; Jockusch, R.A. Fluorescence lifetimes of rhodamine dyes in vacuo. J. Photochem. Photobiol. A Chem. 2012, 244, 47–53. [Google Scholar] [CrossRef]
- Caruana, M.V.; Fava, M.C.; Magri, D.C. A colorimetric and fluorimetric three-input inverted enabled OR logic array by self-assembly of a rhodamine probe in micelles. Asian J. Org. Chem. 2015, 4, 239–243. [Google Scholar] [CrossRef]
- Tahara, K.; Mikuriya, K.; Masuko, T.; Kikuchi, J.-I.; Hisaeda, Y. Dechlorination of DDT catalyzed by visible-light-driven system composed of vitamin B12 derivative and Rhodamine B. J. Porphyr. Phthalocyanines 2013, 17, 135–141. [Google Scholar] [CrossRef]
- Kim, Y.-R.; Kim, H.J.; Kim, J.S.; Kim, H. Rhodamine-based “turn-on” fluorescent chemodosimeter for Cu(II) on ultrathin platinum films as molecular switches. Adv. Mater. 2008, 20, 4428–4432. [Google Scholar] [CrossRef]
- Doumani, N.; Bou-Maroun, E.; Maalouly, J.; Tueni, M.; Dubois, A.; Bernhard Denat, F.; Cayot, P.; Sok, N. A new pH-dependent macrocyclic rhodamine B-based fluorescence probe for copper detection in white wine. Sensors 2019, 19, 4514. [Google Scholar] [CrossRef] [Green Version]
- Kamino, S.; Horio, Y.; Komeda, S.; Minoura, K.; Ichikawa, H.; Horigome, J.; Tatsumi, A.; Kaji, S.; Yamaguchi, T.; Usami, Y.; et al. A new class of rhodamine luminophores: Design, syntheses and aggregation-induced emission enhancement. Chem. Commun. 2010, 46, 9013–9015. [Google Scholar] [CrossRef]
- Kamino, S.; Muranaka, A.; Murakami, M.; Tatsumi, A.; Nagaoka, N.; Shirasaki, Y.; Watanabe, K.; Yoshida, K.; Horigome, J.; Komeda, S.; et al. A red-emissive aminobenzopyrano-xamthene dye: Elucidation of fluorescence emission mechanisms in solution and in the aggregate state. Phys. Chem. Chem. Phys. 2013, 15, 2131–2140. [Google Scholar] [CrossRef]
- Kamino, S.; Murakami, M.; Tanioka, M.; Shirasaki, Y.; Watanabe, K.; Horigone, J.; Ooyama, Y.; Enomoto, S. Design and syntheses of highly emissive aminobenzopyrano-xanthene dyes in the visible and far-red regions. Org. Lett. 2014, 16, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, M.; Kamino, S.; Muranaka, A.; Ooyama, Y.; Ota, H.; Shirasaki, Y.; Horigome, J.; Ueda, M.; Uchiyama, M.; Sawada, D.; et al. Reversible near-infrared/blue mechanofluorochromism of aminobenzopyranoxanthene. J. Am. Chem. Soc. 2015, 137, 6436–6439. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, Y.; Okamoto, Y.; Muranaka, A.; Kamino, S.; Sawada, D.; Hashizume, D.; Uchiyama, M. Fused-fluoran leuco dyes with large coloe-change derived from two-step equilibrium: Iso-aminobenzopyranoxanthenes. J. Org. Chem. 2016, 81, 12046–12051. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, M.; Kamino, S.; Muranaka, A.; Shirasaki, Y.; Ooyama, Y.; Ueda, M.; Uchiyama, M.; Ueda, M.; Enomoto, S.; Sawada, D. Water-tunable solvatochromic and nanoaggregate fluorescence: Dual color visualization and quantification of trace water in tetrahydrofuran. Phys. Chem. Chem. Phys. 2017, 19, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Obukhova, E.N.; Mchedlov-Petrossyan, N.O.; Vodolazkaya, N.A.; Patsenker, L.D.; Doroshenko, A.O.; Marynin, A.I.; Krasovitskii, B.M. Absorption, fluorescence, and acid-base equilibria of rhodamines in micellar media of sodium dodecyl sulfate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 170, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Ramette, R.W.; Sandell, E.B. Rhodamine B Equilibria. J. Am. Chem. Soc. 1956, 78, 4872–4878. [Google Scholar] [CrossRef]
- Ferguson, J.; Mau, A.W.H. Absorption studies of acidbase equilibria of dye solutions. Chem. Phys. Lett. 1972, 17, 543–546. [Google Scholar] [CrossRef]
- Sadkowski, P.J.; Fleming, G.R. Photophysics of the acid and base forms of rhodamine B. Chem. Phys. Lett. 1978, 57, 526–529. [Google Scholar] [CrossRef]
- Rosenthal, I.; Peretz, P.; Muszkat, K.L. Thermochromic and hyperchromic effects in rhodamine B solutions. J. Phys. Chem. 1979, 83, 350–353. [Google Scholar] [CrossRef]
- Faraggi, M.; Peretz, P.; Rosenthal, I.; Weinraub, D. Solution properties of dye lasers. Rhodamine B in alcohols. Chem. Phys. Lett. 1984, 103, 310–314. [Google Scholar] [CrossRef]
- Hinckley, D.A.; Seybold, P.G.; Borris, D.P. Solvatochromism and thermochromism of rhodamine solutions. Spectrochim. Acta 1986, 42A, 747–754. [Google Scholar] [CrossRef]
- Lopez Arbeloa, I.; Rohatgi-Mukerjee, K.K. Solvent effects on the photophysics of the molecular forms of rhodamine B internal conversion mechanism. Chem. Phys. Lett. 1986, 129, 607–614. [Google Scholar] [CrossRef]
- Lopez, F.; Ruiz Ojeda, P.; Lopez Arbeloa, I. Fluorescence self-quenching of the molecular forms of rhodamine B in aqueous and ethanolic solutions. J. Lumin. 1989, 44, 105–112. [Google Scholar] [CrossRef]
- López Arbeloa, F.; López Arbeloa, T.; Tapia Estévez, M.J.; López Arbeloa, I. Photophysics of rhodamines: Molecular structure and solvent effects. J. Phys. Chem. 1991, 95, 2203–2208. [Google Scholar] [CrossRef]
- Kanasaki, E. Thermochromism and photochromism of rhodamine B. Kagaku Kogyo Chem. Chem. Ind. 1987, 40, 870–871. (In Japanese) [Google Scholar]
- Adamovich, L.P.; Melnik, V.V.; Mchedlov-Petrossyan, N.O. Rhodamine B equilibria in aqueous salt solutions. Zhurnal Fiz. Khimii 1979, 53, 356–359. [Google Scholar]
- Kurtaliev, E.N.; Khonkeldieva, M.T.; Talipov, S.A.; Nizomov, N.; Ibragimov, B.T. X-ray study of structure of chloride rhodamine B disolvate with chloroform. Uzb. J. Phys. 2015, 17, 261–264. (In Russian) [Google Scholar]
- Kvick, A.; Vaughan, G.B.M.; Wang, X.; Sun, Y.; Long, Y. A synchrotron-radiation study of the lactone form of rhodamine B at 120 K. Acta Crystallogr. C Cryst. Struct. Commun. 2000, C56, 1232–1233. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Song, M.; Lomg, Y. Synthesis, characterization, and crystal structure of the lactone form of rhodamine B. J. Solid State Chem. 2001, 156, 325–330. [Google Scholar] [CrossRef]
- Lopez Arbeloa, F.; Urrecha Aquirresacona, I.; Lopez Arbeloa, I. Influence of the molecular structure and the nature of the solvent on the absorption and fluorescence characteristics of rhodamines. Chem. Phys. 1989, 130, 371–378. [Google Scholar] [CrossRef]
- Barigelletti, F. Effect of temperature on the photophysics of rhodamine 101 in a polar solvent. Chem. Phys. Lett. 1987, 140, 603–606. [Google Scholar] [CrossRef]
- Elderfield, R.C. (Ed.) Heterocyclic Compounds; Wiley: New York, NY, USA, 1954; Volume 2. [Google Scholar]
- Sivakumar, M.; Pandit, A.B. Ultrasound enhanced degradation of rhodamine B: Optimization with power density. Ultrason. Sonochem. 2001, 8, 233–240. [Google Scholar] [CrossRef]
- Mallah, H.A.; Naoufal, D.M.; Safa, A.I.; El Jamal, M.M. Study of the Discoloration Rate of Rhodamine B as a Function of the Operating Parameters at Pt and BDD Electrodes. Port. Electrochim. Acta 2013, 31, 185–193. [Google Scholar] [CrossRef]
- Beshir, W.B.; Eid, S.; Gafar, S.M.; Ebraheem, S. Application of solutions of Rhodamine B in dosimetry. Appl. Radiat. Isot. 2014, 89, 13–17. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Kholin, Y.V. Aggregation of Rhodamine B in Water. Russ. J. Appl. Chem. 2004, 77, 414–422. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O. Ionization and tautomeric interconversions of rhodamine dyes. Zhurnal Fiz. Khimii 1985, 59, 3000–3004. (In Russian) [Google Scholar]
- Mchedlov-Petrossyan, N.O.; Salinas Mayorga, R.; Surov, Y.N. Ionization and tautomerism of xanthene dyes in mixtures of water with dimethyl sulfoxide. Zhurnal Obs. Khimii (Russ. J. Gen. Chem.) 1991, 61, 225–233. (In Russian) [Google Scholar]
- Mchedlov-Petrosyan, N.O.; Fedorov, L.A.; Sokolovskii, S.A.; Surov, Y.N.; Salinas Maiorga, R. Structural conversions of rhodamines in solutions. Bull. Russ. Acad. Sci. Divis. Chem. Sci. 1992, 3, 403–409. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Kukhtik, V.I.; Bezugliy, V.D. Dissociation, tautomerism and electroreduction of xanthene and sulfonephthalein dyes in N,N-dimethylformamide and other solvents. J. Phys. Org. Chem. 2003, 16, 380–397. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O. Ionization constant of triple-charged cation of rhodamine B. Zhurnal Fiz. Khimii 1979, 53, 2041–2042. [Google Scholar]
- Ponomarev, O.A.; Doroshenko, A.O.; Pedash, Y.F.; Mchedlov-Petrosyan, N.O. On the anomalous Stokes shift of fluorescence of lactones of rhodamine dyes. Zhurnal Fiz. Khimii 1989, 63, 2219–2222. (In Russian) [Google Scholar]
- Ramos, S.S.; Vilhena, A.F.; Santos, L.; Almeida, P. 1H and 13C NMR spectra of commercial rhodamine ester derivatives. Magn. Reson. Chem. 2000, 38, 475–478. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Mchedlov-Petrossyan, N.O. Chemical Equilibrium of Multifunctional Compounds of Xanthenes and Triphenylmethane Series in Nonaqueous Media. Ph.D. Dissertation, Kharkov State University, Kharkov, Ukraine, 1992. [Google Scholar]
- Mollin, J.; Kucerova, T. Acid-base properties and isomerism of nicotinic acid. Chem. Zvesti 1979, 33, 52–56. [Google Scholar]
- Niazi, M.S.K.; Mollin, J. Dissociation constants of some amino acids and pyridinecarboxylic acids in ethanol–water mixtures. Bull. Chem. Soc. Jpn. 1987, 60, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Mchedlov-Petrossyan, N.O.; Kalembet, O.A.; Arias Cordova, E. Tautomerism of rhodamines. I. Kharkov Univ. Bull. 1989, 340, 3–18. [Google Scholar]
- Bates, R. Determination of pH, Theory and Practice, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1973. [Google Scholar]
- Kolthoff, I.M.; Chantooni, M.K.; Bhowmik, S. Dissociation constants of uncharged and monovalent cation acids in dimethyl sulfoxide. J. Am. Chem. Soc. 1968, 90, 23–28. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O. Protolytic equilibrium in lyophilic nano-sized dispersions: Differentiating influence of the pseudophase and salt effects. Pure Appl. Chem. 2008, 80, 1459–1510. [Google Scholar] [CrossRef]
- Breant, M.; Auroux, A.; Lavergne, M. Détermination spectrophotométrique de la partie acide de l’échelle de ph dans la n-méthylpyrrolidone et le diméthylacétamide. Anal. Chim. Acta 1976, 83, 49–57. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Filatov, D.; Goga, S.T.; Lebed, A.V. Ionic equilibrium in mixtures of protophobic and protophilic polar non-hydrogen bond donor solvents: Acids, salts, and indicators in acetone containing 5 mol% DMSO. J. Phys. Org. Chem. 2010, 23, 418–430. [Google Scholar] [CrossRef]
- Ioffe, I.O.; Shapiro, A.L. Investigations in the field of rhodamine dyes and relative compounds. Zhurnal. Org. Khimii 1972, 8, 1726–1729. (In Russian) [Google Scholar]
- Barra, M.; de Rossi, R.H. Erythromicin as a supramolecular receptor. Tetrahedron Lett. 1988, 29, 1119–1122. [Google Scholar] [CrossRef]
- Barra, M.; Cosa, J.J.; de Rossi, R.H. Erythromicin A as a supramolecular catalyst: Effect on rhodamine B lactonization. J. Org. Chem. 1990, 55, 5850–5853. [Google Scholar] [CrossRef]
- Grigoryeva, T.M.; Ivanov, V.L.; Nizamov, N.; Kuzmin, M.G. Adiabatic photodissociation of the C–O in rhodamine C lactone. Doklady AN SSSR (Proc. Acad. Sci. USSR) 1977, 232, 1108–1111. (In Russian) [Google Scholar]
- Pikaev, A.K.; Kartasheva, L.I.; Chulkov, V.N. A new, highly-sensitive dosimetric composition based on a solution of rhodamine B lactone in chloroform. Mendeleev Commun. 1997, 7, 178–180. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Wrona, P.K.; Zielkowska, U.; Reichardt, C. Empirical parameters of Lewis acidity and basicity for aqueous binary solvent mixtures. Tetrahedron 1985, 41, 4519–4527. [Google Scholar] [CrossRef]
- Kalidas, C.; Hefter, G.; Marcus, Y. Gibbs Energies of Transfer of Cations from Water to Mixed Aqueous Organic Solvents. Chem. Rev. 2000, 100, 819–852. [Google Scholar] [CrossRef] [PubMed]
- Chantooni, M.K.; Kolthoff, I.M. Proton solvation in the lower aliphatic alcohols with emphasis on isopropyl and tert-butyl alcohols. J. Phys. Chem. 1978, 82, 994–1000. [Google Scholar] [CrossRef]
- Hojo, M.; Ueda, T.; Yamasaki, M.; Inoue, A.; Tokita, S.; Yanagita, M. 1 H and 13 C NMR Detection of the Carbocations or Zwitterions from Rhodamine B Base, a Fluoran-Based Black Color Former, Trityl Benzoate, and Methoxy-Substituted Trityl Chlorides in the Presence of Alkali Metal or Alkaline Earth Metal Perchlorates in Acetonitrile Solution. Bull. Chem. Soc. Jpn. 2002, 75, 1569–1576. [Google Scholar] [CrossRef]
- Hojo, M.; Ueda, T.; Inoue, A.; Tokita, S. Interaction of a practical fluoran-based black color former with possible color developers, various acids and magnesium ions, in acetonitrile. J. Mol. Liq. 2009, 148, 109–113. [Google Scholar] [CrossRef]
- Shakhverdov, T.A.; Ergashev, R. Photophysics of metal complexes of rhodamine B. Opt. Spectrosc. 1999, 87, 236–242. (In Russian) [Google Scholar]
- Nizamov, N.; Shakhverdov, T.A.; Ergashev, R.; Atakhodjaev, A.K. Spectral-fluorescence studies of rhodamine B with metal ions in liquid solutions. Proc. Acad. Sci. Uzb. Repub. 1986, 5, 72–76. (In Russian) [Google Scholar]
- Ergashev, R. The Study of Complex Formation of Rhodamine Dyes with Metal Ions by Spectral-Luminescent Methods. In Spectroscopy of Intermolecular Interactions and Molecular Motion; Paper Collection; Alisher Navoi Samarkand State University: Samarkand, Uzbekistan, 1985; pp. 58–64. (In Russian) [Google Scholar]
- Ergashev, R. Photophysics of Organic Anions and Their Complexes with Metals. Ph.D. Dissertation, Leningrad State University, Leningrad, Russia, 1989. [Google Scholar]
- Refat, M.S.; Killa, H.M.A.; Mansour, A.F.; Ibrahim, M.Y.; Fetooh, H. Cu(II), Co(II), and Mn(II) Complexes of Rhodamine C and Rhodamine 640 Perchlorate: Synthesis, Spectroscopic, Thermal, Fluorescence, and Photostability Studies. J. Chem. Eng. Data 2011, 56, 3493–3503. [Google Scholar] [CrossRef]
- Klein, U.K.A.; Hafner, F.W. A new dual fluorescence with rhodamine lactone. Chem. Phys. Lett. 1976, 43, 141–145. [Google Scholar] [CrossRef]
- Djumadinov, R.K. Spectroscopic Study of Intermolecular Interactions in Solutions of Rhodamine Dyes. Ph.D. Dissertation, State University, Leningrad, Russia, 1977. [Google Scholar]
- Lippert, E. Dipolmoment und Elektronenstruktur von angeregten Molekülen. Z. Nat. A 1955, 10a, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, L.; Moore, M.K.; Corrie, J.E.H.; Fraternali, F. Quantum Mechanics Calculations on Rhodamine Dyes Require Inclusion of Solvent Water for Accurate Representation of the Structure. J. Phys. Chem. A 2004, 108, 7744–7751. [Google Scholar] [CrossRef]
- Karpiuk, J. Photoinduced electron transfer in Malachite Green lactone. Phys. Chem. Chem. Phys. 2003, 5, 1078–1090. [Google Scholar] [CrossRef]
- Karpiuk, J. Dual Fluorescence from Two Polar Excited States in One Molecule. Structurally Additive Photophysics of Crystal Violet Lactone. J. Phys. Chem. A 2004, 108, 11183–11195. [Google Scholar] [CrossRef]
- Bizjak, T.; Karpiuk, J.; Lochbrunner, S.; Riedle, E. 50-fs Photoinduced Intramolecular Charge Separation in Triphenylmethane Lactones. J. Phys. Chem. A 2004, 108, 10763–10769. [Google Scholar] [CrossRef]
- Simmons, H.E.; Fukunaga, T. Spiroconjugation. J. Am. Chem. Soc. 1967, 89, 5208–5215. [Google Scholar] [CrossRef]
- Schweig, A.; Weidner, U.; Hellwinkel, D.; Krapp, W. Spiroconjugation. Angew. Chem. Int. Engl. Ed. 1973, 12, 310–311. [Google Scholar] [CrossRef]
- Schweig, A.; Weidner, U.; Hill, R.K.; Cullison, D.A. Theory and application of photoelectron spectroscopy. 36. Quantitative account of spiroconjugation. J. Am. Chem. Soc. 1973, 95, 5426–5427. [Google Scholar] [CrossRef]
- Dürr, H.; Gleiter, R. Spiroconjugation. Angew. Chem. Int. Engl. Ed. 1978, 17, 559–569. [Google Scholar] [CrossRef]
- Maslak, P.; Chopra, A.; Moylan, C.R.; Wortmann, R.; Lebus, S.; Rheingold, A.L.; Yap, G.P.A. Optical Properties of Spiroconjugated Charge-Transfer Dyes. J. Am. Chem. Soc. 1996, 118, 1471–1481. [Google Scholar] [CrossRef]
- Wu, C.-C.; Liu, W.-G.; Hung, W.-Y.; Liu, T.-L.; Lin, Y.-T.; Lin, H.-W.; Wong, K.-T.; Chien, Y.-Y.; Chen, R.-T.; Hung, T.-H.; et al. Spiroconjugation-enhanced intermolecular charge transport. Appl. Phys. Lett. 2005, 87, 052103. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, C.; Liu, Z.; Yang, C.; Qin, J. New intramolecular through-space charge transfer emission: Tunable dual fluorescence of terfluorenes. Chem. Commun. 2010, 46, 6666–6668. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, C.; Liu, C.; Liu, Z.; Qin, J.; Yang, C. Spiroconjugated Intramolecular Charge-Transfer Emission in Non-Typical Spiroconjugated Molecules: The Effect of Molecular Structure upon the Excited-State Configuration. Chem. Phys. Chem. 2013, 14, 982–989. [Google Scholar] [CrossRef]
- Wössner, J.S.; Grenz, D.C.; Kratzert, D.; Esser, B. Tuning the optical properties of spiro-centered charge-transfer dyes by extending the donor or acceptor part. Org. Chem. Front. 2019, 6, 3649–3656. [Google Scholar] [CrossRef]
- Wössner, J.S.; Esser, B. Spiroconjugated Donor−σ–Acceptor Charge-Transfer Dyes: Effect of the π-Subsystems on the Optoelectronic Properties. J. Org. Chem. 2020, 85, 5048–5057. [Google Scholar] [CrossRef] [PubMed]
- Sinel’nikov, A.N.; Artyukhov, V.Y. Quantum chemical investigation of spectral-luminescent properties of zwitterion and lactone forms of rhodamine B. Russ. Phys. J. 2012, 54, 1057–1066. [Google Scholar] [CrossRef]
- Luzanov, A.V. The Structure of the Electronic Excitation of Molecules in Quantum-chemical Models. Russ. Chem. Rev. 1980, 49, 1033–1048. [Google Scholar] [CrossRef]
- Luzanov, A.V.; Zhikol, O.A. Electron invariants and excited state structural analysis for electronic transitions within CIS, RPA, and TDDFT models. Int. J. Quantum Chem. 2010, 110, 902–924. [Google Scholar] [CrossRef]
- Luzanov, A.V.; Zhikol, O.A. Excited State Structural Analysis: TDDFT and Related Models. In Practical Aspects of Computational Chemistry I; Leszczynski, J., Shukla, M.K., Eds.; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 15–18. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef]
- Lippert, E.; Rettig, W.; Bonacic-Koutecky, V.; Heisel, F.; Miehe, J.A. Photophysics of internal twisting. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1987; pp. 1–174. [Google Scholar]
- Vollmer, F.; Rettig, W.; Birckner, E. Photochemical Mechanisms Producing Large Fluorescence Stokes Shifts. J. Fluoresc. 1994, 4, 65–69. [Google Scholar] [CrossRef]
- Rettig, W.; Klock, A. Intramolecular fluorescence quenching in aminocoumarines. Identification of an excited state with full charge separation. Can. J. Chem. 1985, 63, 1649–1653. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, M.; Phaniraj, P.; Dogra, S.K. Absorptiometric and fluorimetric study of solvent dependence and prototropism of benzimidazole homologues. J. Chem. Soc. Perkin Trans. 2 1986, 1917–1925. [Google Scholar] [CrossRef]
- Fujiki, K.; Iwanaga, C.; Koizumi, M. Some Spectral Studies of the Aqueous Solution of Pyronine G. Bull. Chem. Soc. Jpn. 1962, 35, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Golovina, A.P.; Mitzel, Y.A.; Levshin, L.V.; Bobrovskaya, E.A. Vestnik MGU, Fizyka. Astonomiya (Bull. Mosc. State Univ. Phys. Astron.) 1969, 4, 36–41. (In Russian) [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox, Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Bylaska, E.J.; de Jong, W.A.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Valiev, M.; Wang, D.; Apra, E.; Windus, T.L.; Hammond, J.; et al. A Computational Chemistry Package for Parallel Computers; Version 5.1; Pacific Northwest National Laboratory: Richland, WA, USA, 2007.























| Atom | Lactone R0 | Zwitterion R± |
|---|---|---|
| 1 | 128.41 | 132.62 |
| 2 | 105.50 | 114.16 |
| 3 | 144.43 | 155.95 |
| 4 | 96.95 | 96.58 |
| 5 | 105.59 | 106.23 |
| 6 | 134.97 | 153.19 |
| 7 | 117.41 | 125.35 |
| 8 | 124.63 | 128.46 |
| 9 | 79.16 | 173.33 |
| 10 | 106.32 | 114.36 |
| 11 | 147.75 | 154.17 |
| 12 | 149.23 | 158.74 |
| 13 | 108.37 | 115.17 |
| 14 | 168.78 | 161.69 |
| 1′ | 152.76 | 133.82 |
| 2′ | 127.25 | 141.42 |
| 3′ | 124.26 | 130.93 |
| 4′ | 129.70 | 130.63 |
| 5′ | 124.63 | 130.43 |
| 6′ | 124.26 | 130.54 |
| Atoms | Cation HR+ of 1 | Esters of 1 | R± of 1 | R0 of 1 | |
|---|---|---|---|---|---|
| From Ref. [60] | From Ref. [64] | From Ref. [60] | From Ref. [60] | ||
| CD3OD | DMSO(d6) | CDCl3 | CD3OD + NaOH | DMSO(d6) + NaOH | |
| 1, 8 | 131.7 | 131.9 | 131.1–131.3 | 132.4 | — |
| 2, 7 | 114.7 | 115.2 | 114.2–114.9 | 114.1 | 109.2 |
| 3, 6 | 158.6 | 157.4 | 154.9–155.8 | 158.6 | 150.2 |
| 4, 5 | 97.0 | 97.0 | 96.3–95.4 | 96.3 | 97.9 |
| 11, 12 | 156.4 | 155.8 | — | 156.1 | 153.7 |
| 10, 13 | 114.2 | 113.5 | — | 114.2 | 106.1 |
| 9 | 160.3 | – | 158.7–158.9 | 162.5 | — |
| 14 | 167.2 | 167.4 | 164.9–165.5 | 172.8 | 169.9 |
| 1′ | – | 134.2 | 133.2–133.5 | 133.1 | — |
| 2′ | 133.2 | 135.4 | 129.7–130.3 | 141.0 | — |
| No. | Compound | /nm | KT = [R0]/[R±] | ||
|---|---|---|---|---|---|
| HR+ | R± | ||||
| 1 |  | 550 | 540 | 10 (337) | 16.6 [58] |
| 2 |  | 552 | 540 | 12 (402) | 16.9 |
| 3 |  | 551 | 538 | 13 (438) | 29.8 |
| 4 |  | 531 | 515 | 16 (585) | 2.19 |
| 5 |  | 565 | 555 | 10 (319) | 1.05 |
| 6 |  | 577 | 566 | 11 (337) | 0.32 |
| 7 |  | 578 | 566 | 12 (367) | 0.38 |
| 8 |  | 575 | – a | – | – |
| 9 |  | 522 | – a | – | – |
| 10 |  | 576 | – a | – | – |
| 11 |  | 494, 525 | – a | – | – |
| 12 |  | 542 | 470 | 72 (2826) | 4.04 |
| 13 |  | 558 | 546 | 12 (394) | 4.10 |
| 14 |  | 562 | 550 | 12 (483) | 1.65 |
| 15 |  | 583 | 573 | 10 (299) | 0.12 |
| Solvent | KT = [R0]/[R±] |
|---|---|
| Water | 0.005–0.01 a,b |
| Methanol | 0.1 c |
| Ethanol | 0.28 d |
| 90 mass% aqueous acetone | 16.6 |
| 91.3 mass% aqueous DMSO | 59 e |
| CHCl3 saturated with water | 90 f |
| N,N-Dimethylformamide | 100 g |
| 98.3 mass% aqueous acetone | 234 h |
| Acetone | 250 a |
| Solvent a | /nm | KT | |||
|---|---|---|---|---|---|
| HR+ | R b | ||||
| Methanol (32) | 564 | 556 | 8 | 0.028 | 0.762 |
| Benzene—ethanol—water c (12.8) | 567 | 556 | 11 | 0.155 | 0.587 |
| 1-Propanol (20.4) | 566 | 556 | 10 | 0.16 | 0.617 |
| 1-Butanol (17.4) | 564 | 553 | 11 | 0.19 | 0.586 |
| 2-Methyl-propane-1-ol (17.9) | 566 | 554 | 12 | 0.23 | 0.552 |
| 2-Propanol (19.9) | 564 | 550 | 14 | 0.32 | 0.546 |
| 2-Methyl-propane-2-ol (12.5) d | 563 | 546 | 17 | 2.94 | 0.389 |
| 90% Acetone—10% water e (24) | 565 | 555 | 10 | 1.06 | 0.57 |
| 95% Acetone—5% water e (22) | 568 | 560 | 8 | 5.76 | 0.49 |
| 90% CH3CN—10% water e (39.4) | 567 | 558 | 9 | 0.17 | 0.692 |
| CH3CN (35.9) | 566 | 550 | 9 | 48.3 | 0.460 |
| 91.3% DMSO e (56) | 574 | 560 | 14 | 2.89 | 0.50 |
| DMSO (46.4) | 577 | 566 | 11 | 70.7 | 0.444 |
| N,N-Dimethylformamide (36.7) | 570 | 555 | 5 | 89.9 | 0.404 |
| Solvent | Cation HR+ (0.01 M H2SO4) | Neutral Form (R± ⇄ R0) (0.02 M DBU) | ||
|---|---|---|---|---|
| λmax, nm | Emax × 10−3, M−1 cm−1 | λmax, nm | Emax × 10−3, M−1 cm−1 | |
| Methanol | 564 | 98.0 | 556 | 95.3 |
| Acetonitrile | 566 | 98.6 | 550 | 2.00 |
| DMF | 570 | 110.0 | 555 | 1.21 |
| DMSO | 575 | 93.2 | 556 | 1.30 |
| Solvent | ||||
|---|---|---|---|---|
| Methanol | 9.40 | +1.52 | 3.68 | −1.55 |
| 90% acetonitrile (x2 = 0.798) | 9.1 | +1.21 | 3.70 | −0.77 |
| 2-Propanol | 11.75 | +1.42 | 6.13 | −0.49 |
| 91% DMSO (x2 = 0.708) | 8.05 | −3.61 | 7.46 | +0.48 |
| Acetonitrile | 20.7 | +7.85 | 8.65 | +1.68 |
| DMF | 12.3 | –2.52 | 9.78 | +1.95 |
| DMSO | 11.1 | –3.40 | 10.3 | +1.85 |
| Transition | Wavelength, Wavenumber, Oscillator Strength | Electronic Localization Indices | Charge Transfer Indices | ||
|---|---|---|---|---|---|
 |  |  | |||
| Absorption (S0-state optimized geometry, DFT) | |||||
| S0-S1 | 352 nm 28,400 cm−1 0.005 | 24.7 | 24.7 | 50.6 |  |
| S0-S2 | 342 nm 29,200 cm−1 0.0004 | 22.2 | 22.2 | 54.1 |  |
| Fluorescence (S1-state optimized geometry, TDDFT) | |||||
| S0-S1 | 496 nm 20,150 cm−1 0.005 | 4.2 | 44.1 | 51.2 |  |
| Compound | , nm (from Absorption Spectrum) | , nm | Stokes Shift, cm−1 |
|---|---|---|---|
| 2 | 313 | 450 | 9700 |
| 3 | 310 | 454 | 10,300 |
| 4 | 301 | 389 | 7500 |
| 5 | 312 | 478 | 11,150 |
| 6 | 318 | 486 | 10,900 |
| 7 | 319 | 491 | 11,050 |
| 9 | 325 | 456 | 8850 |
| 10 | 315 | 462 | 10,100 |
| 12 | 351 | 485 | 7900 |
| 14 | 341 | 511 | 9750 |
| 15 | 310 | 435 a | 9300 |
| 585 b | 15,200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obukhova, O.M.; Mchedlov-Petrossyan, N.O.; Vodolazkaya, N.A.; Patsenker, L.D.; Doroshenko, A.O. Stability of Rhodamine Lactone Cycle in Solutions: Chain–Ring Tautomerism, Acid–Base Equilibria, Interaction with Lewis Acids, and Fluorescence. Colorants 2022, 1, 58-90. https://0-doi-org.brum.beds.ac.uk/10.3390/colorants1010006
Obukhova OM, Mchedlov-Petrossyan NO, Vodolazkaya NA, Patsenker LD, Doroshenko AO. Stability of Rhodamine Lactone Cycle in Solutions: Chain–Ring Tautomerism, Acid–Base Equilibria, Interaction with Lewis Acids, and Fluorescence. Colorants. 2022; 1(1):58-90. https://0-doi-org.brum.beds.ac.uk/10.3390/colorants1010006
Chicago/Turabian StyleObukhova, Olena M., Nikolay O. Mchedlov-Petrossyan, Natalya A. Vodolazkaya, Leonid D. Patsenker, and Andrey O. Doroshenko. 2022. "Stability of Rhodamine Lactone Cycle in Solutions: Chain–Ring Tautomerism, Acid–Base Equilibria, Interaction with Lewis Acids, and Fluorescence" Colorants 1, no. 1: 58-90. https://0-doi-org.brum.beds.ac.uk/10.3390/colorants1010006