
Is There a Quadruple Fe-C Bond in FeC(CO)3?


Dipartimento di Chimica e Chimica Industriale, Università di Pisa, Via G. Moruzzi 13, I-56124 Pisa, Italy
*
Author to whom correspondence should be addressed.
Computation 2021, 9(9), 95; https://0-doi-org.brum.beds.ac.uk/10.3390/computation9090095
Submission received: 2 August 2021
/
Revised: 25 August 2021
/
Accepted: 26 August 2021
/
Published: 30 August 2021
(This article belongs to the Special Issue Electronic Correlation)
Abstract
:A recent computational paper (Kalita et al., Phys. Chem. Chem. Phys. 2020, 22, 24178–24180) reports the existence of a quadruple bond between a carbon and an iron atom in the FeC(CO) molecule. In this communication, we perform several computations on the same system, using both density functional theory and post-Hartree–Fock methods and find that the results, and in particular the Fe-C bond length and stretching frequency depend strongly on the method used. We ascribe this behavior to a strong multireference character of the FeC(CO) ground state, which explains the non-conclusive results obtained with single-reference methods. We therefore conclude that, while the existence of a Fe-C quadruple bond is not disproved, further investigation is required before a conclusion can be drawn.
In a recent paper [1], a computational investigation on FeC(CO) revealed the unprecedented result that in such a molecule, a quadruple Fe-C bond may be present. Unfortunately, FeC(CO) has never been synthetized, and therefore no experimental characterization exists to confirm such a result: a computational investigation is therefore, at the moment, the only tool available to investigate such a system. In their work, Kalita et al. use the Fe-C bond length and the associated stretching force constant to assess the existence of a quadruple bond. They use Density Functional Theory (DFT), and in particular the M062X exchange correlation functional together with the Def2-TZVP basis set to perform such an analysis. As DFT calculations may be unreliable for multireference systems, they perform CASSCF and CCSD(T) single point calculations on the DFT geometry to confirm the single-reference character of the wavefunction, and thus the reliability of the DFT results.
Given the exceptionality of the results found by Kalita et al., we decided to perform further calculations on FeC(CO) to confirm their results. We performed three different analyses. First, we compared the Fe-C bond distance and harmonic stretching frequency computed with different DFT exchange-correlation functionals to assess the robustness of the results with respect to the latter choice. In particular, we compare results obtained using five popular hybrid functionals: M062X [2], B3LYP [3], MN15 [4], PBE0 [5], and B97X-D [6]. All the DFT calculations were run using the Gaussian 16 [7] suite of programs, employing Dunning’s cc-pVTZ basis set [8,9], using analytical first and second derivatives and a pruned (99,590) grid for the integration of the exchange-correlation potential. The DFT results are reported in Table 1.
The DFT results show a large variability, with bond distances ranging from 1.481 Å to 1.526 Å and harmonic stretching frequencies ranging from 1113 to 1211 wavenumbers, with the M062X functional predicting the shortest and strongest bond and the B3LYP one the longest and weakest. We therefore decided to extend our computational investigation by performing, to the best of our ability, high-level post-HF calculations, increasing the accuracy of the methods systematically. We performed calculations at the Hartree–Fock (HF), second-order Møller–Plesset perturbation theory [10] (MP2), coupled cluster theory with single and double excitations [11] (CCSD), and with single, doubles, and perturbative triples [12] (CCSD(T)), using Dunning’s cc-pVTZ basis set and the frozen-core approximation. Analytical gradients [13,14,15] were used for all the geometry optimizations, while harmonic frequencies were computed by numerically differentiating analytical gradients. All the post-HF calculations were performed using the CFOUR [16,17] suite of programs. The results are reported in Table 2.
The harmonic frequencies obtained at every level of theory are all real, confirming thus that the structure is a minimum. Unfortunately, it is apparent from the results that the treatment of electronic correlation that we could achieve is not sufficient to obtain converging results. The MP2 method fails to give a correct description of the system, with a very long bond distance and an unphysically very low stretching frequency. Going from MP2 to CCSD and CCSD(T), the bond distance seems to approach convergence, but the same cannot be said for the harmonic frequency. It is interesting to note that the CC bond length is significantly longer than the DFT one. Furthermore, while the fluctuations in the computed harmonic frequencies leave us unable to give a quantitative estimate, the converged value is likely to be in between the CCSD and CCSD(T) result, and in any case lower than all the DFT predictions. Unfortunately, the CCSD(T) calculation is already stretching the limits of computational feasibility: a more accurate treatment of correlation, including full triples excitations or even quadruples ones, seems at the moment out of reach.
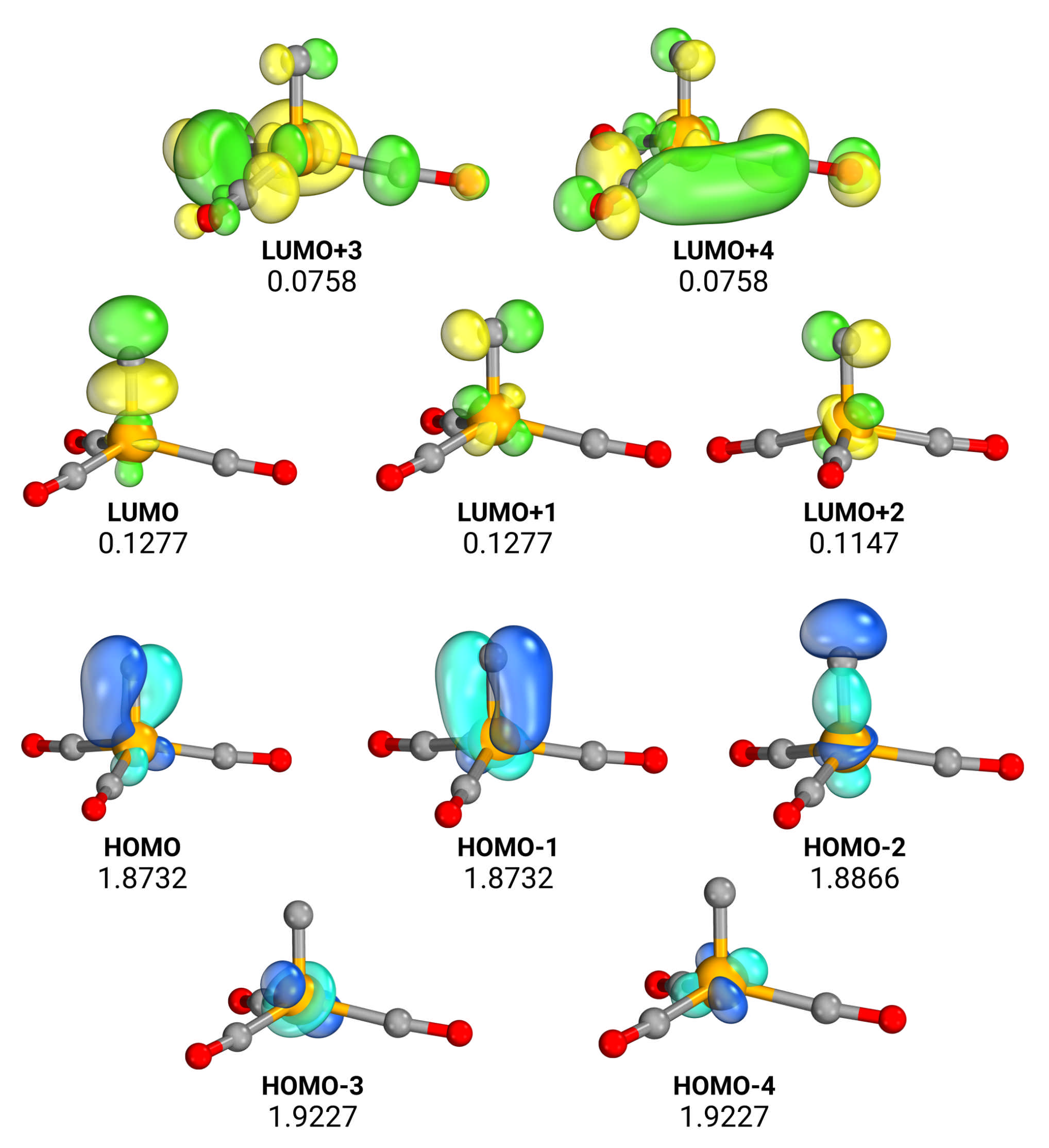
The large oscillations observed in the results motivated us to investigate whether the source of the lack of robustness of both the DFT and post-HF results stems from static correlation. We therefore performed further complete active space self-consistent field (CASSCF) calculations [18,19]. Using Dunning’s cc-pVDZ basis set, which is sufficient for qualitatively correct results, and the CFOUR suite of programs, we optimized the geometry of FeC(CO) using analytical gradients and computed harmonic frequencies, again by numerically differentiating analytical gradients. CASSCF calculations can be an important source of insight in the electronic structure of a molecule; however, the results depend strongly on the choice of the active space, which therefore introduces some elements of arbitrariness. The choice of the active orbitals can be motivated by chemical considerations, as done by Kalita et al. [1], who include in the active space all the orbitals involved in Fe-C bonds, resulting in a CASSCF calculation with 12 correlated electrons in 12 orbitals. Here, we adopt a different strategy, based on an automatic, a priori strategy to select the active orbitals. In particular, we select the active space according to the Unrestricted-Natural-Orbitals (UNO) criterion [20,21]. In this procedure, symmetry-broken unrestricted HF solutions are first found. An average density matrix is computed using all the independent UHF solution and then diagonalized to obtain the UNO. The active space is built by including all the UNO with occupation between 0.01 and 1.99. The Restricted HF wavefunction of FeC(CO) presents six triplet instabilities; the resulting active space consists in 10 electrons in 10 orbitals, whose representation is given in Figure 1. The active space mostly involves the d orbitals of the metal and the p orbitals of the investigated C atom.
Looking at the orbitals and at their occupation numbers, obtained by diagonalizing the final CASSCF one-body density matrix at the CASSCF equilibrium geometry, we note that the system definitely exhibits a relevant static correlation. Looking closer at the CASSCF wavefunction, we find that the contribution of the HF determinant has a weight of just 0.64, confirming the multireference nature of the molecule. This is in contrast to what was found by Kalita et al., who found a CASSCF wavefunction dominated by the HF reference (they report a coefficient of 0.952). We believe that this contradiction could be due to the fact that their calculation was performed on the DFT-optimized geometry, while we relaxed the geometry. Furthermore, our choice of the active space, resulting from a well-established automated procedure, is different, and in particular slightly more compact. While this may accentuate the multireference character of our CASSCF description, we believe that our choice of active space is robust, which is confirmed empirically by the smooth convergence of the CASSCF wavefunction optimization, which is performed using a second-order method. The CASSCF calculations predict a Fe-C distance of 1.573 Å and an associated harmonic stretching frequency of 1016 cm, which is in qualitative agreement with the CC results.
We believe that our results show that it is too soon to conclude that there exists a quadruple Fe-C bond in FeC(CO). The evanescent nature of the concept of bond order itself has stimulated many discussions in the literature [24,25], which makes the topic particularly interesting. However, assessing something as unprecedented as the existence of a quadruple Fe-C bond requires a very careful validation of the results. Therefore, we conclude that the possibility of such a quadruple bond can, at the moment, be neither confirmed nor denied, and further, more accurate calculations are required.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/computation9090095/s1.
Author Contributions
Conceptualization, F.L.; methodology, F.L.; software, T.N. and F.L.; validation, T.N.; formal analysis, T.N. and F.L.; investigation, T.N. and F.L.; resources, F.L.; data curation, T.N.; writing—original draft preparation, F.L.; writing—review and editing, T.N.; supervision, F.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in the Supporting Information.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kalita, A.J.; Rohman, S.S.; Kashyap, C.; Ullah, S.S.; Guha, A.K. Transition metal carbon quadruple bond: Viability through single electron transmutation. Phys. Chem. Chem. Phys. 2020, 22, 24178–24180. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 1007, 4572–4585. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc–Zn. J. Chem. Phys. 2005, 123, 064107. [Google Scholar] [CrossRef] [Green Version]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Pople, J.A.; Krishnan, R.; Schlegel, H.B.; Binkley, J.S. Derivative studies in hartree-fock and møller-plesset theories. Int. J. Quantum Chem. 1979, 16, 225–241. [Google Scholar] [CrossRef]
- Scheiner, A.C.; Scuseria, G.E.; Rice, J.E.; Lee, T.J.; Schaefer, H.F. Analytic evaluation of energy gradients for the single and double excitation coupled cluster (CCSD) wave function: Theory and application. J. Chem. Phys. 1987, 87, 5361–5373. [Google Scholar] [CrossRef]
- Scuseria, G.E. Analytic evaluation of energy gradients for the singles and doubles coupled cluster method including perturbative triple excitations: Theory and applications to FOOF and Cr2. J. Chem. Phys. 1991, 94, 442–447. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G. CFOUR, Coupled-Cluster techniques for Computational Chemistry, a quantum-chemical program package. With contributions from A.A. Auer, R.J. Bartlett, U. Benedikt, C. Berger, D.E. Bernholdt, S. Blaschke, Y. J. Bomble, S. Burger, O. Christiansen, D. Datta, F. Engel, R. Faber, J. Greiner, M. Heckert, O. Heun, M. Hilgenberg, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, T. Kirsch, K. Klein, G.M. KopperW.J. Lauderdale, F. Lipparini, T. Metzroth, L.A. Mück, D.P. O’Neill, T. Nottoli, D.R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J.D. Watts and the integral packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. Available online: http://www.cfour.de (accessed on 29 August 2021).
- Matthews, D.A.; Cheng, L.; Harding, M.E.; Lipparini, F.; Stopkowicz, S.; Jagau, T.C.; Szalay, P.G.; Gauss, J.; Stanton, J.F. Coupled-cluster techniques for computational chemistry: The CFOUR program package. J. Chem. Phys. 2020, 152, 214108. [Google Scholar] [CrossRef]
- Jensen, H.J.A.; Jorgensen, P. A direct approach to second-order MCSCF calculations using a norm extended optimization scheme. J. Chem. Phys. 1984, 80, 1204–1214. [Google Scholar] [CrossRef]
- Lipparini, F.; Gauss, J. Cost-Effective Treatment of Scalar Relativistic Effects for Multireference Systems: A CASSCF Implementation Based on the Spin-free Dirac–Coulomb Hamiltonian. J. Chem. Theory Comput. 2016, 12, 4284–4295. [Google Scholar] [CrossRef] [PubMed]
- Pulay, P.; Hamilton, T.P. UHF natural orbitals for defining and starting MC-SCF calculations. J. Chem. Phys. 1988, 88, 4926–4933. [Google Scholar] [CrossRef]
- Tóth, Z.; Pulay, P. Comparison of Methods for Active Orbital Selection in Multiconfigurational Calculations. J. Chem. Theory Comput. 2020, 16, 7328–7341. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G. Intrinsic atomic orbitals: An unbiased bridge between quantum theory and chemical concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knizia, G.; Klein, J.E. Electron flow in reaction mechanisms—Revealed from first principles. Angew. Chem. Int. Ed. 2015, 54, 5518–5522. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Frenking, G. The Chemical Bond in C2. Chem. Eur. J. 2016, 22, 4100–4108. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Rzepa, H.S.; Hoffmann, R. One Molecule, Two Atoms, Three Views, Four Bonds? Angew. Chem. Int. Ed. 2013, 52, 3020–3033. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The (10,10) initial active space as obtained from the UNO procedure. Below the labels, the fractional occupations of the converged CASSCF natural orbitals. Carbon atoms are shown in grey, oxygens in red, and the iron atom in orange. Orbitals are visualized with the IboView software [22,23].
Figure 1.
The (10,10) initial active space as obtained from the UNO procedure. Below the labels, the fractional occupations of the converged CASSCF natural orbitals. Carbon atoms are shown in grey, oxygens in red, and the iron atom in orange. Orbitals are visualized with the IboView software [22,23].


{kind=link}
Table 1.
Equilibrium Fe-C distance (, in Å) and harmonic vibrational Fe-C stretching frequencies (in cm) computed with various DFT exchange-correlation functionals. The values reported with a “*” are taken from Reference [1] and reported here for comparison.
Table 1.
Equilibrium Fe-C distance (, in Å) and harmonic vibrational Fe-C stretching frequencies (in cm) computed with various DFT exchange-correlation functionals. The values reported with a “*” are taken from Reference [1] and reported here for comparison.
| Method | ||
|---|---|---|
| M062X | 1.482 | 1208 |
| M062X * | 1.481 | 1211 |
| B3LYP | 1.526 | 1113 |
| MN15 | 1.501 | 1165 |
| PBE0 | 1.514 | 1146 |
| B97X-D | 1.510 | 1154 |
Table 2.
Equilibrium Fe-C distance (, in Å) and harmonic vibrational Fe-C stretching frequencies (in cm) computed at systematically more accurate levels of theory.
Table 2.
Equilibrium Fe-C distance (, in Å) and harmonic vibrational Fe-C stretching frequencies (in cm) computed at systematically more accurate levels of theory.
| Method | ||
|---|---|---|
| HF | 1.489 | 1041 |
| MP2 | 1.654 | 591 |
| CCSD | 1.539 | 1039 |
| CCSD(T) | 1.559 | 926 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nottoli, T.; Lipparini, F. Is There a Quadruple Fe-C Bond in FeC(CO)3? Computation 2021, 9, 95. https://0-doi-org.brum.beds.ac.uk/10.3390/computation9090095
AMA Style
Nottoli T, Lipparini F. Is There a Quadruple Fe-C Bond in FeC(CO)3? Computation. 2021; 9(9):95. https://0-doi-org.brum.beds.ac.uk/10.3390/computation9090095
Chicago/Turabian StyleNottoli, Tommaso, and Filippo Lipparini. 2021. "Is There a Quadruple Fe-C Bond in FeC(CO)3?" Computation 9, no. 9: 95. https://0-doi-org.brum.beds.ac.uk/10.3390/computation9090095
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.