Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor?




1
Department of Chemical System Engineering, School of Engineering, The University of Tokyo 7-3-1, Tokyo 113-8656, Japan
2
The National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba 305-8560, Japan
3
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg 2050, South Africa
*
Author to whom correspondence should be addressed.
Crystals 2020, 10(3), 146; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030146
Submission received: 25 January 2020
/
Revised: 13 February 2020
/
Accepted: 15 February 2020
/
Published: 26 February 2020
(This article belongs to the Special Issue σ- and π-Hole Interactions)
Abstract
:The CH3Cl molecule has been used in several studies as an example purportedly to demonstrate that while Cl is weakly negative, a positive potential can be induced on its axial surface by the electric field of a reasonably strong Lewis base (such as O=CH2). The induced positive potential then has the ability to attract the negative site of the Lewis base, thus explaining the importance of polarization leading to the formation of the H3C–Cl···O=CH2 complex. By examining the nature of the chlorine’s surface in CH3Cl using the molecular electrostatic surface potential (MESP) approach, with MP2/aug-cc-pVTZ, we show that this view is not correct. The results of our calculations demonstrate that the local potential associated with the axial surface of the Cl atom is inherently positive. Therefore, it should be able to inherently act as a halogen bond donor. This is shown to be the case by examining several halogen-bonded complexes of CH3Cl with a series of negative sites. In addition, it is also shown that the lateral portions of Cl in CH3Cl features a belt of negative electrostatic potential that can participate in forming halogen-, chalcogen-, and hydrogen-bonded interactions. The results of the theoretical models used, viz. the quantum theory of atoms in molecules; the reduced density gradient noncovalent index; the natural bond orbital analysis; and the symmetry adapted perturbation theory show that Cl-centered intermolecular bonding interactions revealed in a series of 18 binary complexes do not involve a polarization-induced potential on the Cl atom.
1. Introduction
Clark, Murray, Politzer, and their colleagues have analyzed the surface reactivity of several molecular systems using the molecular electrostatic surface potential (MESP) model [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. They utilized density functional theory (DFT) with a variety of functionals (B3LYP, B3PW91, M06-2X) and a standard double/triple-ζ quality Gaussian basis set to compute the electrostatic potential [1,2,3,4,5,6,7,8,9,10]. They concluded that DFT, together with an 0.001 a.u. isodensity envelope on which to compute the potential, is adequate to reveal the nature of the electrostatic potential on the surface of any atom in a molecule [7,8]. In 1992, some of these authors considered several systems in their study of noncovalent interactions, including molecules such as CH3F and CH3Cl. It was contended in that study that “the potentials of CH3F and CF4 are indicative of fluorine interacting only with electrophiles, as is found experimentally” [1]. In this, and in a later study [2], the authors pondered why a σ-hole is not found when X = F in CF4, as well as in other instances, such as in CH3Cl. (A σ-hole is an electron density deficient region on the outer surface of X along the extension of the R–X bond, where R is remaining part of the molecule [2,7,15,21].) They concluded that the higher electronegativity of fluorine gives it a disproportionately large share of the σ-bonding electrons, which helps to neutralize the σ-hole. This also applies to chlorine in CH3Cl, which does not have a σ-hole and does not halogen bond [2].
A halogen bond is formed when there is a favorable attractive interaction between a positive site (viz. a positive σ-hole) on a halogen in one molecule and a negative site on another molecule [21,22,23,24]. Such a broad view is applicable to other interactions such as the hydrogen bond [25], chalcogen bond [26], pnictogen bond [27], or any other σ-hole interaction [6,15,21] since a positive site on the hydrogen, chalcogen, pnictogen, or halogen atom in the molecule attracts a negative site on the other to form such an interaction.
Contrary to their earlier assertions, Politzer and co-workers have more recently found that the F atom in CH3F molecule does indeed have a σ-hole, but it is negative [7]; similarly, Cl in CH3Cl was also found to have a negative σ-hole [18].
The contention that Cl atom in CH3Cl does not have a σ-hole on its own [2,3], has appeared quite frequently [5,7,10,20,28]. This is sometimes done when proposing that the CH3Cl molecule is a good model system to understand the effect of electrostatic polarization in noncovalent interactions. For example, to explain what causes the formation of a H3C–Cl···O=CH2 complex, it was argued that despite the potential on the outer axial surface of the Cl atom in H3C–Cl being weakly negative in the isolated molecule, this can be transformed and become positive through the electrostatic polarizing effect of the negative site interacting with it [5,7,10,28,29].
Such a provocative view led to the suggestion that the MESP model is superior to other computational methods such as the second-order natural bonding orbital analysis (NBO) [30], the quantum theory of atoms in molecules in molecules (QTAIM) [31,32,33], and the density functional theory symmetry adapted perturbation theory energy decomposition analysis (DFT-SAPT-EDA) [34,35]. While the reliability of these latter methods has been questioned [36,37,38,39], such claims have been rebutted by others [40,41,42,43,44,45,46]. Some of these conflicting views have been briefly highlighted in one of our recent reviews [21].
In contrast with the arguments given by Politzer and co-workers [1,2], some of us have shown that each fluorine in CF4 conceives a positive σ-hole along each of the four C–F bond extensions [47]. CF4 can not only form a 1:1 cluster with Lewis bases such as H2O, NH3, H2C=O, HF, and HCN but also 1:2, 1:3, and 1:4 clusters with the last three (randomly chosen) Lewis bases. There are many known fluorinated compounds in which F conceives a positive or a negative σ-hole that has the ability to engage in a σ-hole centered noncovalent interaction [48,49,50,51,52,53,54,55]. This also applies not only to Cl in H3C–Cl [56,57], but also to O in a variety of molecules as reported recently [58,59], despite claims on several occasions that O does not conceive a σ-hole and does not participate in chalcogen bonding [60,61,62,63,64].
In this study we use the ab initio Møller–Plesset second-order perturbation theory (MP2) method in combination with the Dunning’s correlated consistent aug-cc-pVTZ basis set and the MESP model to investigate the detailed nature of various local potential maxima and minima on the electrostatic surface of a CH3Cl molecule. The critical point (cp) topology of the Laplacian of the charge density is calculated within the QTAIM framework to see whether this model is capable of providing insights into the reactivity of the molecule, and whether these are comparable with the predictions of the MESP model. We consider 10 Lewis bases to examine whether these are capable of sustaining an attractive intermolecular interaction with the axial and/or lateral sites of the Cl atom in CH3Cl. We consider whether the various intermolecular interactions revealed (viz. halogen bonds, chalcogen bonds, hydrogen bonds, and pnictogen bonds) can be unambiguously regarded as σ-hole interactions, as has been claimed [28]. We also explore whether the various arguments advanced [2,3] to support the idea that the positive potential on the Cl atom in CH3Cl can be induced by the electric field of the Lewis base during the course of an intermolecular interaction is tenable.
We utilize the NBO, QTAIM, DFT-SAPT-EDA, and RDG (reduced density gradient) noncovalent index [65] theoretical tools to explore and discuss the reliability of and the agreement between the results of these approaches in elucidating intermolecular interactions in the 18 complexes of H3C-Cl molecules studied. Based on our results, we argue that combining an inappropriate theoretical method with an arbitrarily chosen isodensity envelope can be misleading insofar as the sign of the potential on the axial portion of the Cl atom is concerned, and when such a result is used for the interpretation of the origin of an intermolecular interaction, misleading conclusions can be reached.
2. Computational Details
Using the Gaussian 09 code [66], 10 monomers and 18 binary complexes were fully energy-minimized with MP2 [67] and the aug-cc-pVTZ basis set. A Hessian second derivative calculation was performed for each of them to identify the nature of the structure; positive eigenvalues were found.
To evaluate the effect of the isodensity envelope on the nature of the electrostatic potential, four different isodensity values, viz. 0.0005, 0.0010, 0.0015, and 0.0020 a.u., were chosen on which to compute the electrostatic potential. The local maxima and minima of potential (Vs,max and Vs,min, respectively) on the electrostatic surface of the CH3Cl monomer were identified and characterized. The MP2 energy-minimized geometry of the monomer was used. The positive (Vs,max > 0 or Vs,min > 0) and negative signs (Vs,max < 0 or Vs,min < 0) of the potential on an atom X in a molecule generally represent the electrophilic and nucleophilic regions on any molecule, respectively [47,49,50,51,52,58,59,60]. Regions described by Vs,max > 0 (or Vs,max < 0) on the outer axial portion of the atom X represent a positive (or a negative) σ-hole (as on X in X2 and CX4, where X = F, Cl, Br, I [21,39,68,69] or on F in H–F and H3C–F [51]) and those described by Vs,max = 0 on the outer axial portion of the atom X represent to a neutral σ-hole [2,70].
A selected number of charge density-based descriptors of bonding interaction were evaluated using QTAIM [31,32,33], including the charge density (ρb), the Laplacian of the charge density (∇2ρb), and the total energy density (Hb) at the bond critical points (bcps). The model assumes that an open system is bounded by a surface S(rs) of local zero-flux in the gradient vector field of the charge density ρ(r) (Equation (1), where n(r) is a unit vector normal to the surface at r).
The analysis of the delocalization indices (DIs) between atom pairs was also performed within the interacting quantum atoms (IQA) model of QTAIM [71,72]. DI is a measure of bond order since it represents the extent of the delocalization of electron pairs between two atomic basins in any closed-shell system [73]. Since noncovalent interactions are a result of very minimal charge density localization between the lump and hole, the DI values are typically small (< 0.05 for weakly bound interactions) [73,74].
The RDG [65] based isosurface plots were evaluated using the MP2 equilibrium geometries of the 18 complexes. This method uses the sign of the second eigenvalue λ2 of the Hessian second derivative charge density matrix to recognize the nature of the chemical interaction. At the same time, it uses the value of charge density ρ to measure the strength of the interaction. As such, the signature sign(λ2) × ρ < 0 represents a closed-shell interaction (attraction). Similarly, sign(λ2) × ρ ≈ 0 represents a van der Waals (attraction) and sign(λ2) × ρ > 0 a steric interaction (repulsion). The AIMAll [75]. Multiwfn [76], and VMD [77] suite of programs, together with some in-house codes, were used for the analysis of the topological properties of the charge density, the RDG isosurfaces, and the electrostatic surface potentials.
The binding energy ΔE for each complex A···B was calculated using the supermolecular procedure proposed by Pople [78], described by Equation (2). The terms ET(A) and ET(B) in Equation (2) are, respectively, the electronic total energies of the two isolated monomers A and B in the complex A···B that has an electronic total energy of ET(A···B). The ΔE was corrected for the basis set superposition error energy, E(BSSE), using the counterpoise procedure proposed by Boys and Bernardi [79]. Equation (3) was used for the calculation of the BSSE corrected energy, ΔE(BSSE).
ΔE(A···B) = ET(A···B) – ET(A) – ET(B)
ΔE(BSSE) = ΔE(A···B) + E(BSSE),
The zeroth-order DFT SAPT-EDA analysis [34,35] was performed using the Psi4 code [80] and the MP2 geometries of the monomers in the complexes. The aug-pVDZ-JKFIT [81] DF basis was used for SCF calculations, whereas the aug-cc-pVDZ-RI DF basis was used for the evaluation of the SAPT0 electrostatics, induction and dispersion components. The frozen core as well as asyncronous I/O was invoked while forming the DF integrals and CPHF coefficients. Equation (4) represents the SAPT0 interaction energy, E(SAPT0), which is the sum of the component energies arising from electrostatics (Eeles), repulsion (Eexch), induction (Eind), and dispersion (Edisp).
E(SAPT0) = Eeles + Eexch + Eind + Edisp,
3. Results and Discussion
3.1. The Reactive Surface Profile of the CH3Cl Monomer
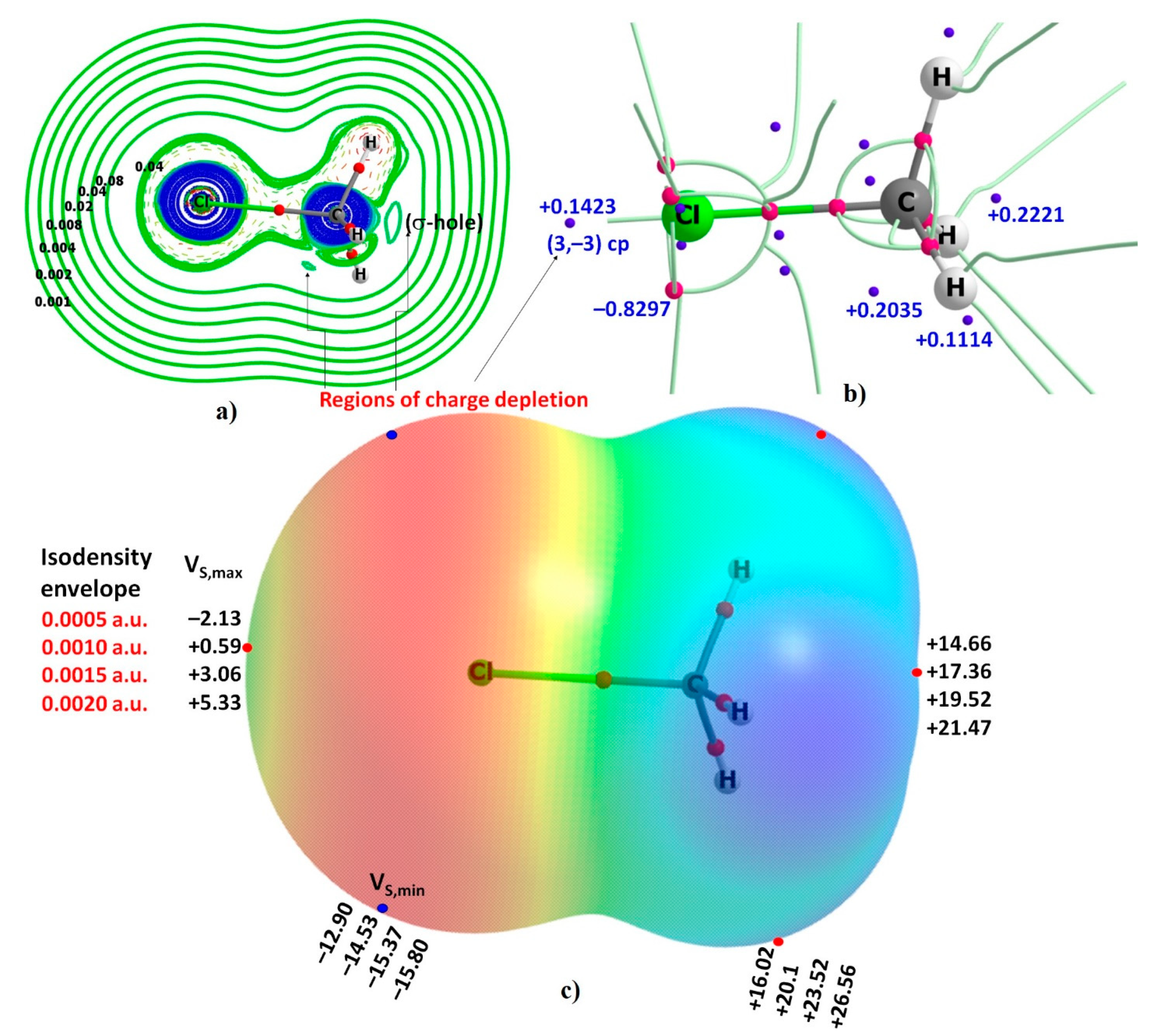
Figure 1a shows the 2D contour plot of the Laplacian of the charge density (∇2ρ) for the CH3Cl molecule, obtained using a Cl-C-H plane. The positive contours (green solid lines) indicate areas of charge depletion, and the negative contours (red dashed lines) indicate areas of charge concentration. As such, the charge depletion is significant near C along the outer extension of the Cl–C covalent bond, thus showing a prominent “hole”. In QTAIM representation, one might call this “hole” a region of valence shell charge depletion (VSCD). The same feature is less noticeable on Cl along the outer extension of the C–Cl bond. One might conclude that there is no “hole” on the Cl atom. We therefore carried out the critical point (cp) analysis of ∇2ρ to provide some insight into the exact nature of charge density concentration and depletion around the Cl atom, since the minimum and maximum of ∇2ρ represent the open- and closed-shell structure, respectively, of any specific region [59,82].
Although many cps of ∇2ρ were identified, only a selected number are illustrated in Figure 1b. The tiny blue spheres represent the (3, –3) cps and are equivalent to the (3, +3) critical points of –∇2ρ. The tiny pink spheres represent the (3, +3) critical point of ∇2ρ and are equivalent to the (3, –3) critical point of –∇2ρ. The (3, –3) critical point of ∇2ρ is a local maximum of ∇2ρ; it is a point of locally maximal “charge depletion” when ∇2ρ > 0, and is a point of locally minimal “charge concentration” when ∇2ρ < 0. Similarly, the (3,+3) cp is a local minimum of ∇2ρ and is a point of locally maximal “charge concentration” when ∇2ρ < 0, and of locally minimal “charge depletion” when ∇2ρ > 0. The ∇2ρ at the (3,–3) cps on the extension of the C–Cl and Cl–C bond are both positive (∇2ρ = +0.1423 a.u. on Cl and +0.2221 a.u. on C); therefore the outer axial regions on the Cl and C atoms in the H3C–Cl molecule are well characterized as regions of VSCD. These are therefore “holes”, which may interact with the lumps localized on Lewis base molecules to form complexes. A similar conclusion might be arrived at when (3, –3) cps of ∇2ρ are analyzed along the C–H bond extensions since (3, –3) critical point of ∇2ρ are all positive (∇2ρ = +0.1114 a.u. on H along the C–H bond extension).
By contrast, the lateral portions of the Cl atom in the H3C–Cl molecule are characterized by three (3, +3) cps of ∇2ρ. These are all negative (∇2ρ = –0.8297 a.u. each). They are associated with the lone-pairs on Cl; these “lumps” may have the ability to attract “holes” on an interacting molecule.
The insight gained from an evaluation of the cps of ∇2ρ is virtually no different from what might be inferred from the results of the MESP model. Figure 1c depicts the 0.001 a.u. (electrons bohr−3) isodensity envelope mapped potential on the electrostatic surface of the H3C–Cl molecule. It shows the axial outer portion of the Cl atom has a positive potential VS,max of +0.59 kcal mol−1. This potential is associated with what might be called a positive, albeit weak, “σ-hole”. The σ-hole region is surrounded by a belt of negative potential. The local minima associated with the lateral portions of the atom are characterized by a VS,min of –14.53 kcal mol−1.
Passing from the 0.0010 a.u. through the 0.0015 a.u. to the 0.0020 a.u. isodensity envelope did not change the nature (sign) of the potential on the outer surface of the Cl atom in H3C–Cl noted above, although the negative sites on Cl became more negative and the positive site becomes more positive. This is expected given that on moving closer to the nucleus of the atom one generally comes up with a relatively tiny electron density deficient surface.
The 0.0005 a.u. isodensity envelope mapped potentials are also included in Figure 1c. Passing from the 0.0005 a.u to the 0.0010 a.u. isodentiy surface has indeed had a notable effect on both the sign and magnitude of potential on Cl along the C–Cl bond extension. For instance, the VS,max was computed to be –2.13 kcal mol−1 on the 0.0005 a.u. isodensity envelope, which is completely different from that of +0.59 kcal mol−1 computed on the 0.001 a.u. isodensity envelope. This result unequivocally shows that the choice of the isodensity surface is arbitrary, which can lead to change in the sign of the potential. There is a somewhat less negative potential (VS,min = –12.90 kcal mol−1) on the lateral portions of the same atom.
The negative potential on the axial portion of the Cl atom may be misleading given the 0.0005 a.u. isodensity envelope does not totally encompass the van der Waals surface of the molecule. This is consistent with the views of Bader et al. [83] and others [44,84], who have advocated the use of two contour values (0.0010 and 0.0020 a.u.) that should be large enough to encompass > 96% of a molecule’s electronic charge density.
Based on the concern of a reviewer, and to confirm the reliability of [MP2/aug-cc-pVTZ] results above, we examined the nature of the local most potentials on the Cl atom using the aug-cc-pV(T + d)Z basis set. We used 10 different computational models, including the CCSD and nine popular density functionals. The results summarized in Table 1 demonstrate that the axial and lateral portions of the Cl atom on the C–Cl bond extensions are always positive and negative, respectively. Except for the PBE1 (PBE1PBE) functional, all other DFT and DFT-D3 functionals slightly underestimated the magnitude of VS,max on Cl compared to that obtained with CCSD. In addition, both the H and C atoms along the C–H and Cl–C bond extensions are positive, indicating that these can be sites for hydrogen bond and chalcogen bond formation when placed in close proximity to negative sites on another molecule.
3.2. Geometries of Intermolecular Complexes of H3C–Cl with 10 Lewis Bases
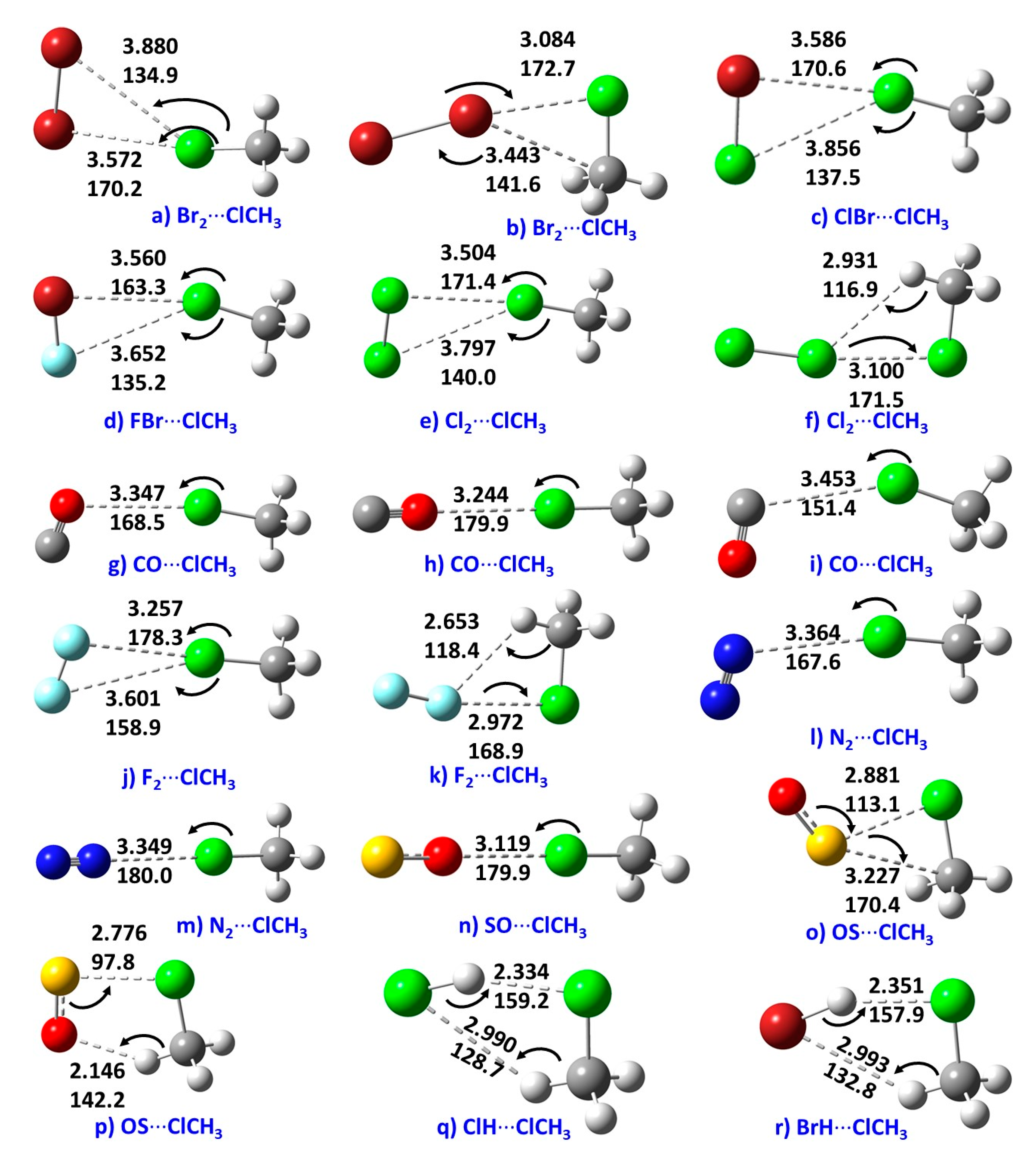
Figure 2 shows the optimized geometries of 18 binary complexes formed between H3C–Cl and nine Lewis bases. In many of these complexes, both the axial and lateral portions of the Cl atom in H3C–Cl are involved in the attractive engagement with negative and positive sites, respectively, on the bases. The behavior of the Cl atom in H3C–Cl towards the acids and bases in the interacting monomers is clearly similar in all cases. This is consistent with the reactivity profile predicted by cps of ∇2ρ (viz. a “lump” attracts a “hole” and vice-versa), and that predicted using the MESP model. Since the potential on the Cl atom is essentially positive, there is certainly no transformation (induction) from a negative potential to a positive potential when the axial portion of the Cl atom is in close proximity to the negative site of the Lewis base. This result clearly negates the suggestion that a positive potential is induced on the Cl atom by the electric field of the interacting partner to promote a mutual Coulomb-type attractive engagement between them [2,3]. It should be pointed out that the previous studies used a lower-level of theory and a double-ζ Gaussian basis set to compute the electrostatic potential on the surface of the H3C–Cl molecule [2,3]. This combination predicted an incorrect (negative) potential associated with the σ-hole of the Cl atom in H3C–Cl and led the authors to offer a different interpretation of the nature of the surface reactivity of the molecule, thus exaggerating the importance of the idea of “electrostatic polarization” in complex formation.
From the intermolecular geometries shown in Figure 2 between the monomers of the complexes, it is apparent that the Cl atom in CH3Cl forms directional interactions with Br2 (a); ClBr (c); FBr (d); Cl2 (e); CO (g−i); F2 (j);N2 (l−m); and SO (n). The directionality of each contact is realized based on the angles of approach of the electrophile on the Cl atom, viz., 170.2, 170.6, 163.3, 171.4, 168.5, 179.9, 151.4, 178.3, 167.6, 180.0, and 179.9° for these complexes, respectively. These angles vary between 150 and 180°, and are typical of Type II contacts [21].
The intermolecular contact distances in all the complexes of Figure 2 are less than the sum of the van der Waals radii of the respective bonded atomic basins, (rvdW (H) = 1.20 Å; rvdW (F) = 1.46 Å; rvdW (N) = 1.66 Å; rvdW (O) = 1.50 Å; rvdW (S) = 1.89 Å; rvdW (Cl) = 1.82 Å; and rvdW (Br) = 1.86 Å) [85]. This is consistent with the geometry-based criterion recommended for hydrogen bonding [25], halogen bonding [24], and chalcogen bonding [26]. For instance, the IUPAC recommendation advises that in “a chalcogen-bonded complex R–Ch···A, the interatomic distance between the chalcogen donor atom Ch and the nucleophilic site in the acceptor A tends to be less than the sum of the van der Waals radii and more than the sum of covalent radii” [26].
For the complexes H3C–Cl···Br2, H3C–Cl···Cl–Cl and H3C–Cl···F–F shown in Figure 2b,f,k, respectively, the lateral portion of the Cl atom in H3C–Cl acts as a lump for making an attractive engagement with the “hole” on the partner molecules. The attraction is arguably due to the outer axial surfaces of the halogen atoms in the Br–Br, Cl–Cl, and F–F molecules, characterized by positive electrostatic potentials [86], interacting with the lateral negative site on the Cl atom in H3C–Cl in the aforementioned complexes, resulting in the formation of the Cl···Br–Br, Cl···Cl–Cl, and Cl···F–F halogen bond interactions, respectively. The intermolecular distances associated with these interactions are 3.084, 3.100, and 2.972 Å, respectively, while the intermolecular angles are 172.7, 17.5, and 168.9°, respectively. The angular feature indicates not only the presence of Type II contacts, but also clarifies why the intermolecular distances are smaller than the sum of the van der Waals radii of the interacting atomic basins. For example, the intermolecular distances 3.084 (Cl···Br), 3.100 (Cl···Cl) and 2.972 Å (F···Cl) in Figure 2b,e,k are less than the sum of the van der Waals radii of 3.68, 3.64, and 3.28 Å, respectively. Clearly, the feasibility of positive potentials on the Br, Cl, and F atoms in Br2, Cl2, and F2 causing the formation of these three complexes is certainly not developed by induction caused by the electric field of the lumps of the Cl atom in H3C–Cl. The potentials on the bimolecular halogen atoms are inherently positive (as observed on Cl in H3C–Cl), thus helping with the development of the intermolecular interaction with the lumps of the Cl atom in H3C–Cl.
The intermolecular bonding features shown in the complexes (a), (c–e), (g–j), and (l−n) can also be regarded as halogen bonding. However, the only difference between these and the above set of three complexes (b, e, k) is that the Cl atom in H3C–Cl acts as an electrophile in the former complexes but as a nucleophile in the latter. The results provide evidence of the amphoteric nature of the charge density profile on the surface of the Cl atom in H3C–Cl, in excellent agreement with the nature of the surface reactivity predicted by the cp topology of ∇2ρ.
The complexes H3CCl···SO and H3CCl···SO shown in (o) and (p) are not the consequence of halogen bonding. They both feature a Cl···S intermolecular contact. For this, the lump on the lateral portion of the Cl atom in H3C–Cl interacts with the S atom in SO. The intermolecular distances associated with the Cl···S contacts are very short, with r(Cl···S) of 2.881 Å and 2.776 Å for complexes in (o) and (p), respectively. These are significantly smaller than the sum of the van der Waals radii of the Cl and S atoms, 3.71 Å (rvdW (S) = 1.89 Å; rvdW (Cl) = 1.82 Å). Moreover, an examination of the intermolecular angular geometry suggests that Type I bonding topologies promote the formation of these contacts. Type I contacts are generally characterized by a contact angle that varies between 90° and 150°, and the participating atoms that form the contact are generally either both positive or both negative [21]. Previous studies have demonstrated that Type I contacts are dispersion driven [21,23,87]. This view has been advanced because the σ-hole model fails to provide true insight into the origin of this interaction; in this case, the Coulombic model description of noncovalent interactions [10,17,18,28] does not work very well.
The chalcogen bonded contacts identified in the H3CCl···SO complexes provide unequivocal evidence that the newly identified Type I contact can be formed not only between sites of opposite polarity, but also feature the fact that the Coulomb description (viz. positive site attracts a negative one!) can be utilized for its effective realization.
For the H3CCl···HCl and H3CCl···HBr complexes shown in (q) and (r), respectively, the “hole” on the hydrogen atom in HX (X = Cl, Br), which is described by the (3, –3) cp of ∇2ρ (Figure 1b), interacts with the lump of the Cl in H3CCl. The intermolecular distance associated with the resulting X···H (X = Cl, Br) contact (Cl···H = 2.334 Å and Br···H = 2.351 Å) is shorter than the sum of van der Waals radii of the X and H atoms (rvdW (Cl + H) = 3.02 Å and and rvdW (Br + H) = 3.06 Å). The approach angle of the electrophile identifies the interaction to be of Type II (∠Cl···H–Cl = 159.2o and ∠Cl···H–Br = 157.9o in the respective complexes). These signify the presence of hydrogen bonding in the complexes of H3CCl···HCl and H3CCl···HBr.
3.3. QTAIM Description of Intermolecular Bonding Interactions in the Complexes of H3C–Cl
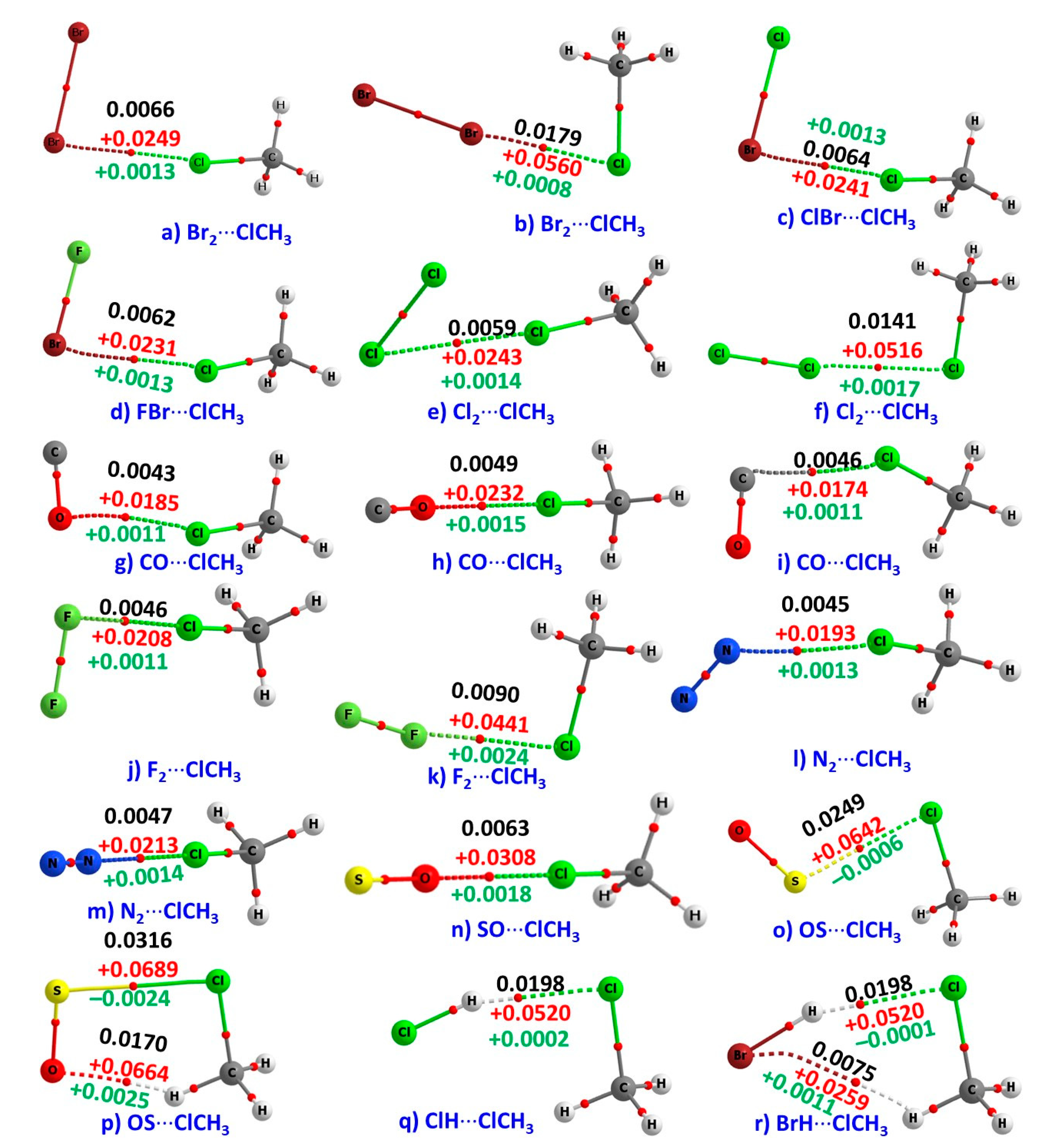
The QTAIM molecular graphs of the 18 binary complexes of CH3Cl studied are shown in Figure 3. They confirm the presence of primary interactions between the monomers in the complexes, as discussed above; there are well-defined bond paths and (3, –1) bond critical points between the bonded atomic basins in each complex. This is in good agreement with the recommendation of IUPAC [24,25,26]. The molecular graphs also indicate the possibility of secondary interactions in two cases: SO···H in (p) and Br···H in (r).
The ρb values (ρb < 0.0198 a.u.) at the X···Cl (X = Cl, Br, F), Cl···X (X = Cl, Br, F), O···S, O···H, and Br···H bcps, are small for all the complexes (see Figure 3 for exact values). At all bcps ∇2ρb > 0. These signatures indicate the closed-shell nature of the intermolecular interactions [42,43,47,48,49,50,51,52,57,58,59,88,89,90,91].
The total energy density, Hb, is another topological descriptor of bonding interactions; it is the sum of the “gradient” kinetic energy density and potential energy density (i.e., Hb = Gb + Vb) [88,89,90,91]. Hb > 0 indicates that Gb > Vb while Hb < 0 implies Vb > Gb. These are considered to be signatures of stabilizing and destabilizing interactions, respectively [88,89,90,91]. The Hb values are were found to be negative at the S···Cl and H···Br bcps of the complexes shown in (o), (p), and (r), respectively. This means that these interactions include partial shared (covalent) character. On the other hand, the Hb values were positive at the X···Cl (X = Cl, Br, F), Cl···X (X = Cl, Br, F), O···H, and H···Cl bcps of the remaining complexes of Figure 2, which is indicative of closed-shell ionic interactions.
It was recently argued [28] that many classical and non-classical interactions, variously referred to as proper and improper, blue-shifted and red-shifted, dihydrogen and anti-hydrogen, resonance-assisted and polarization-assisted, and so on, are straightforward σ-hole interactions. What then can be said about the Cl···S interactions identified in the H3CCl···SO complexes (Figure 3o,p)? It would be misleading to refer to them as σ-hole interactions. The results of the MESP model suggests that the lateral portion of the S atom in SO is described by four extrema of potential. Two of them are positive, each with the VS,max of +34.2 kcal mol−1. The other two are negative, each with a VS,min of –8.9 kcal mol−1. There is no extremum of positive potential identified on the S atom along the outer extension of the O–S bond. The site on S that is interacting with the negative lateral site on Cl in H3CCl is positive, thus forming the OS···ClCH3 complexes. Since VS,max on S is a result of the depopulation of a π-type orbital, its engagement with the Cl atom in H3CCl does not lead to the formation of a σ-hole interaction. As indicated above, the interaction cannot be regarded as a Type II interaction (∠Cl···S–O is 113.1° in (o) and in 97.8° in (p)).
To provide further insight into the orbital origin of the Cl···S interaction, we carried out an analysis of the second-order perturbative estimates of “donor-acceptor” (bond-antibond) interaction energies using the NBO approach [30]. Our results suggest that the Cl···S interaction in (o) is described by the combined effects of n(3)Cl → σ*(S–O) and n(3)Cl → π*(S–O) charge transfer delocalizations, where n refers to the lone-pair bonding orbital, and σ* and π* are the anti-bonding σ- and π-type orbitals, respectively. These charge transfer delocalizations are accompanied by second order perturbative lowering energy E(2) of 2.4 and 8.7 kcal mol−1, respectively. Similarly, the E(2) for the charge transfer delocalizations responsible for the formation of the Cl···S interaction in (p) were found to be 0.4 and 23.9 kcal mol−1, respectively. These results demonstrate that the origin of the Cl···S interactions in (o) and (p) cannot be understood by the oversimplified Coulombic arguments of the MESP model.
3.4. RDG Isosurface Topologies of the Complexes of H3C–Cl
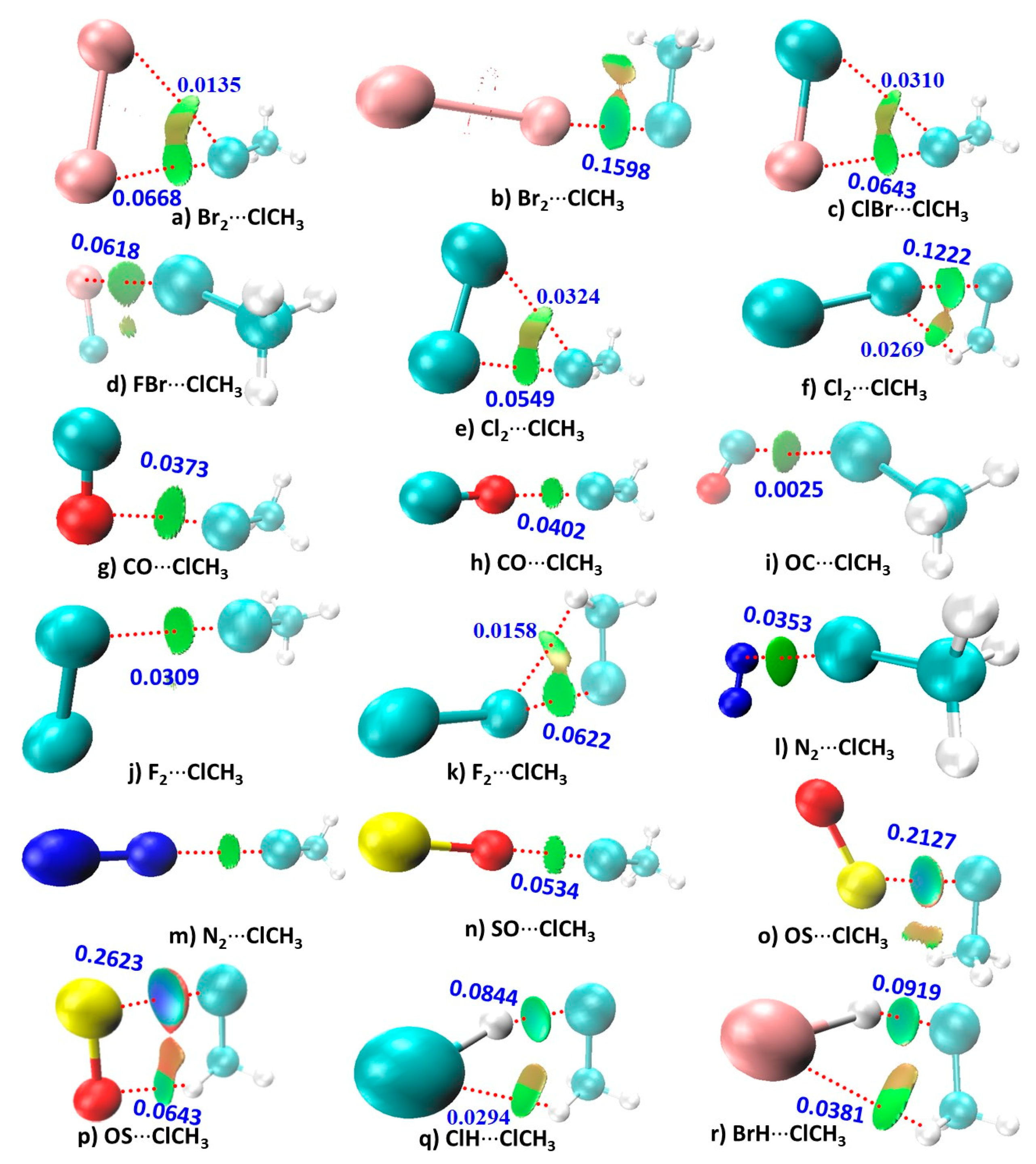
The results of the RDG isosurface analysis, summarized in Figure 4, show that the intermolecular bonding region in each complex is characterized by one (or two) RDG isosurface domain(s). These domains are colored either in bluish-green, green, light brown, or dark red. The coloring scheme is based on the combined effect of the extent of the electron density delocalization between the atomic basins and the sign of the second eigenvalue λ2 of the Hessian second derivative charge density matrix. The signature sign (λ2) × ρ < 0 represents an attractive interaction; sign (λ2) × ρ ≈ 0 represents a van der Waals interaction; and sign (λ2) × ρ > 0 represents a repulsive interactions. The spread of the isosurface (volume) is tuned by the extent of the charge density delocalization around the critical bonding region.
RDG predicts the presence of both primary and secondary contacts between the monomers in 14 complexes, except for (g)–(j) and (l)–(n). From the values of the angles of interaction shown in Figure 2, it is clear that the secondary interactions identified in most of the complexes follow the Type I topology of bonding and hence are dispersion driven.
The primary interactions in 14 of the 18 complexes are characterized by green isosurfaces. The interactions in the other four complexes are characterized by bluish RDG domains, including (o)–(r). The isosurface representing these interactions between the S and Cl atoms in OS···ClCH3 (in (o) and (p)), and that between the H and Cl atoms in ClH···ClCH3, (q), as well as that between the H and Br atoms in BrH···ClCH3, (r), is bluish-green. It indicates that the strength of the intermolecular interaction in these four complexes is stronger than those in the remaining 14 complexes. This is consistent with the ρb, ∇ρb, Hb, and DI values predicted for these interactions (see Figure 3 and Figure 4 for values), suggesting that the stability of the intermolecular interaction in these four complexes is in the order S···Cl (p) >> S···Cl (o) > H···Cl (r) > H···Cl (q). Similarly, the preferential stability of the hydrogen bonds in the complexes (p), (q), and (r) is in the order H···O (p) > H···Br (r) > H···Cl (q). The positive VS,max on the donor atoms of the monomers responsible for these interactions predicted by the MESP model fail to provide such an insight, suggesting that the extrema of potential may not be reliable as a measure of bond stability.
QTAIM based bond path features shown in Figure 3 are in reasonable agreement with the RDG isosurface topologies for most of the complexes. The only discrepancy between them is in the complexes of ClCH3 with Br2 (k), ClBr (c), Cl2 ((e) and (f)), F2 (k), and ClH (q). This is apparently because the RDG method predicts the possibility of secondary interactions between interacting monomers in these complexes, but QTAIM does not recognize these as interactions since the bond path topologies between the bonded atomic basins are missing. The mismatch is not very surprising given that QTAIM sometimes underestimates weakly bound interactions in molecular complexes [58,59]. Even so, the QTAIM based delocalization results summarized in Figure 4 are in good agreement with the RDG’s isosurface topologies of secondary interactions since the former recognizes all the interactions inferred by the latter.
3.5. Energy Stability
Table 2 summarizes the MP2 calculated binding energies for the 18 complexes of ClCH3 examined in this study. As indicated above, the Cl-bonded complexes of ClCH3 with X2 (X = F, Cl, Br) are weaker than the X bonded complexes of ClCH3. For example, the ΔE of the complex in (b) is –3.07 kcal mol−1 larger than that of complex (a) and of the complex (f) is –1.96 kcal mol−1 larger than that of (e). Similarly, the ΔE of complex (k) is –0.97 kcal mol−1 larger than that of (j). These results suggest that the weaker σ-hole on the Cl atom in ClCH3 forms weaker complexes compared to those formed by the relatively stronger σ-holes on X in X2. While this conclusion is rather qualitative, it has to be appreciated that the energy due to the secondary interactions does play a role to determine the overall strength of each of these complexes.
The OS···ClCH3 complex (p), on the other hand, is found to be most stable in the series, with the ΔE of –6.79 kcal mol−1. The complexes H3CCl···Br2 (b), H3CCl···Cl2 (f), SO···ClCH3 (n), H3CCl···HCl (q), and H3CCl···HBr (r) are of intermediate strength, with the ΔE of –3.23, –4.52, –4.30, and –4.59 kcal mol−1, respectively.
The BSSE has a significant effect on the binding energies of all the complexes. It is as large as 1.47, 1.08, and 1.25 kcal mol−1 for complexes (b), (p), and (r), respectively. Nevertheless, the BSSE corrected MP2 binding energies, ΔE(BSSE), are found to be comparable with the corresponding DFT-SAPT interaction energies (E(SAPT0)) for the 18 complexes. The marginal discrepancy between them can be attributed to the level of correlation effect accounted for by the DFT-SAPT formalism, together with the basis set utilized. The largest difference of 0.3 kcal mol−1 between E(SAPT0) and ΔE(BSSE) is found for the complexes of Br2 with ClCH3 ((a) and (b)). There is no obvious relationship between E(SAPT0) (or ΔE(BSSE)) and the extrema of the electrostatic potential responsible for the formation of the 18 complexes examined.
The interaction energies for nine of the 18 complexes were found to be smaller than –1.0 kcal mol−1. Does this mean the complexes are unbound? Should one actually consider the link between the monomers in these complexes as an attraction? Since the interaction energy is negative, the answer to the first question is certainly “no”, since a negative interaction energy provides a clear and unequivocal signature for any bound state. The answer to the second question is “yes”. The obvious reason for this is that van der Waals complexes usually have a weak binding energy of less than –1.0 kcal mol−1 [92,93,94,95,96,97,98]. The importance of such weakly bound interactions have been much appreciated in many fields including polymer science, biology, and crystal engineering [92,93,94,95,96,97,98]. For instance, van der Waals interactions are always weaker than any other chemical interaction and are the determinant of structure of proteins or even the overall shape of polymer structures [92,95,96,98] and the significance of such weakly bound interactions cannot be overlooked thus assuming that only strong interactions are significant for materials design and weak interactions do not play an important role in the field of noncovalent interactions.
The results of the DFT-SAPT based decomposed energy components summarized in Table 2 suggest that dispersive attraction (Edisp) does not tend to exceed the electrostatic and polarization components (Eeles and Eind, respectively) for 12 of the 18 complexes. These include complexes (a)–(f), (i), (k), (q), and (r). The formation of these complexes is not strictly electrostatically driven, but the contributions due to dispersion and repulsion also play a significant role in determining their overall interaction energies and hence promoting their overall stability.
By contrast, the dispersive attraction tends to exceed the electrostatic and polarization components in the other six complexes, viz. (a), (c), (d), (e), (g), (h), (j), (l), (m), and (o). This might prompt the suggestion that the weak attraction that does exist in these complexes is less the result of a specific interatomic interaction, and more a general, non-specific, fairly isotropic, attraction that would occur between any pair of molecules. However, one should not forget that the overall interaction energy in these six complexes is the sum of four specific interaction types, and that these interactions collectively work to determine and explain the directionality of the intermolecular interactions identified, as has been pointed out before [99]. There should be no ambiguity in the origin of the attractive forces that lead to the formation of the 18 complexes examined in this study.
4. Conclusions
This study has shown that the analysis of the critical points of the Laplacian of the charge density could be informative in revealing the actual nature of the surface reactivity of the chlorine atom in CH3Cl. This is in line with the nature of the local extrema of electrostatic potential identified on the surface of the Cl atom in CH3Cl using the MESP model. In particular, it is shown that the combination of a suitable isodensity envelope with an appropriate theoretical method is important to correctly identify the electrophilic nature the Cl atom in CH3Cl.
The electronic charge density distributions around the lateral and axial sites of Cl in CH3Cl is not isotropic, indicating the amphiphilic nature of the Cl atom. The negative lateral sites on the Cl are shown to display sufficient ability to attract positive sites on the interacting atoms to form halogen bonds, or chalcogen bonds, or hydrogen bonds.
The attractive interaction of the positive “hole” on the Cl atom in CH3Cl with various “lumps” in the interacting bases has led to the conclusion that the positive electrostatic potential on the Cl is certainly not induced by the electric field of the interacting species as others have suggested [2,3,10,28]. Rather, it is an inherent property of this atom in the molecule.
The bond path and critical point topologies of QTAIM associated with the primary bonding interactions in the 18 complexes are shown to be consistent with an RDG isosurface analysis. Although these topologies did not appear between the weakly bound atoms in some complexes, the results of QTAIM’s delocalization analysis were shown to be concordant with those of RDG.
The supermolecular and SAPT interaction energies were shown to be in agreement. The dispersion interaction was also shown to be one the most important driving forces responsible for the formation of the 18 complexes investigated.
As shown for the complexes between CH3Cl and SO, all types of intermolecular contacts cannot be regarded as σ-hole interactions.
Author Contributions
Conceptualization and Project Design, P.R.V. and A.V.; Investigation, P.R.V. and A.V.; Supervision, P.R.V.; Writing—Original Draft, P.R.V. and A.V.; Writing—Review and Editing, P.R.V., A.V., and H.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
This work was entirely conducted using the various facilities provided by the University of Tokyo. P.R.V. is currently affiliated with AIST and thanks Koichi Yamashita for support. H.M.M. thanks the National Research Foundation, Pretoria, South Africa, and the University of the Witwatersrand for funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Conch, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Concha, M.C. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The s-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Murray, J.S.; Resnati, G.; Politzer, P. Close contacts and noncovalent interactions in crystals. Faraday Discuss. 2017, 203, 113–130. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Mathematical modeling and physical reality in noncovalent interactions. J. Mol. Model. 2015, 21, 52. [Google Scholar] [CrossRef]
- Clark, T.; Politzer, P.; Murray, J.S. Correct electrostatic treatment of noncovalent interactions: The importance of polarization. WIREs Comput. Mol. Sci. 2015, 5, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Huheey, J.E.; Murray, J.S.; Grodzicki, M. Electronegativity and the concept of charge capacity. J. Mol. Str. THEOCHEM 1992, 259, 99–120. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Politzer, P. The σ-Hole Coulombic Interpretation of Trihalide Anion Formation. ChemPhysChem 2018, 19, 3044–3049. [Google Scholar] [CrossRef]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on Halogen Bonding and Other Sigma-Hole Interactions: Lex Parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2−8. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. σ-Hole Bonding: A Physical Interpretation. In Halogen Bonding I: Impact on Materials Chemistry and Life Sciences; Metrangolo, P., Resnati, G., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 19–42. [Google Scholar]
- Clark, T. σ-Holes. WIREs Comput. Mol. Sci. 2013, 3, 13–20. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The Hellmann-Feynman theorem: A perspective. J. Mol. Model. 2018, 24, 266. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Lane, P.; Concha, M.C. Electrostatically driven complexes of SiF4 with amines. Int. J. Quant. Chem. 2009, 109, 3773–3780. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Murray, J.S.; Macaveiu, L.; Politzer, P. Factors affecting the strengths of σ-hole electrostatic potentials. J. Comput. Sci. 2014, 5, 590–596. [Google Scholar] [CrossRef]
- Murray, J.S.; Riley, R.E.; Politzer, P.; Clark, T. Directional weak intermolecular interactions: Sigma-hole bonding. Aust. J. Chem. 2010, 63, 1598–1607. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Metrangolo, P.; Resnati, G. Halogen Versus Hydrogen. Science 2008, 321, 918–919. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Shing Ho, P.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, R.G.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An Overview of Strengths and Directionalities of Noncovalent Interactions: σ-Holes and π-Holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Murray, J.S.; Politzer, P. Role of Polarization in Halogen Bonds. Austr. J. Chem. 2014, 67, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Bader, R.F.W.; Nguyen-Dang, T.T.; Tai, Y. A topological theory of molecular structure. Rep. Prog. Phys. 1981, 44, 893–947. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. In Encyclopedia of Computational Chemistry; Schleyer, P.V., Ed.; John Wiley and Sons: Chichester, UK, 1998; Volume 1, pp. 64–86. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Politzer, P. A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys. Chem. Chem. Phys. 2018, 20, 30076–30082. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. A look at bonds and bonding. Struct. Chem. 2019, 30, 1153–1157. [Google Scholar] [CrossRef]
- Clark, T.; Heßelmann, A. The coulombic σ-hole model describes bonding in CX3I⋯Y− complexes completely. Phys. Chem. Chem. Phys. 2018, 20, 22849–22855. [Google Scholar] [CrossRef]
- Andrés, J.; Ayers, P.W.; Boto, R.A.; Carbó-Dorca, R.; Chermette, H.; Cioslowski, J.; Contreras-García, J.; Cooper, D.L.; Frenking, G.; Gatti, C.; et al. Nine questions on energy decomposition analysis. J. Comp. Chem. 2019, 40, 2248–2283. [Google Scholar] [CrossRef]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Nature of halogen-centered intermolecular interactions in crystal growth and design: Fluorine-centered interactions in dimers in crystalline hexafluoropropylene as a prototype. J. Comp. Chem. 2019, 40, 1836–1860. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Can Combined Electrostatic and Polarization Effects Alone Explain the F···F Negative-Negative Bonding in Simple Fluoro-Substituted Benzene Derivatives? A First-Principles Perspective. Computation 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Sánchez–Sanz, G.; Alkorta, I.; Elguero, J. Theoretical Study of Intramolecular Interactions in Peri-Substituted Naphthalenes: Chalcogen and Hydrogen Bonds. Molecules 2017, 22, 227. [Google Scholar] [CrossRef] [Green Version]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The many faces of halogen bonding: A review of theoretical models and methods. WIREs: Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Fluorines in tetrafluoromethane as halogen bond donors: Revisiting address the nature of the fluorine’s σ-hole. Int. J. Quantum Chem. 2015, 115, 453–470. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Unusual bonding modes of perfluorobenzene in its polymeric (dimeric, trimeric and tetrameric) forms: Entirely negative fluorine interacting cooperatively with entirely negative fluorine. Phys. Chem. Chem. Phys. 2015, 17, 31624–31645. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Can an entirely negative fluorine in a molecule, viz. perfluorobenzene, interact attractively with the entirely negative site (s) on another molecule (s)? Like liking like! RSC Adv. 2016, 6, 19098–19110. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Comment on “Extended Halogen Bonding between Fully Fluorinated Aromatic Molecules: Kawai et al., ACS Nano, 2015, 9, 2574–2583”. arXiv 2018, arXiv:1802.09995. [Google Scholar]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Revealing Factors Influencing the Fluorine-Centered Non-Covalent Interactions in Some Fluorine-substituted Molecular Complexes: Insights from First-Principles Studies. ChemPhysChem 2018, 19, 1486–1499. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Theoretical study on σ- and π-hole carbon⋯carbon bonding interactions: Implications in CFC chemistry. Phys. Chem. Chem. Phys. 2016, 18, 32155–32159. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Electrostatically enhanced F···F interactions through hydrogen bonding, halogen bonding and metal coordination: An ab initio study. Phys. Chem. Chem. Phys. 2016, 18, 20381–20388. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, K.; Lesani, M. Does fluorine participate in halogen bonding? Chem. Eur. J. 2015, 21, 4739–4746. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Halogen bonding interaction of chloromethane withseveral nitrogen donating molecules: Addressing thenature of the chlorine surface σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 19573–19589. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R. Does Oxygen Feature Chalcogen Bonding? Molecules 2019, 24, 3166. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; MacDougall, P.J. The chalcogen bond: Can it be formed by oxygen? Phys. Chem. Chem. Phys. 2019, 21, 19969–19986. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Mukherjee, A.J.; Zade, S.S.; Singh, H.B.; Sunoj, R.B. Organoselenium Chemistry: Role of Intramolecular Interactions. Chem. Rev. 2010, 110, 4357–4416. [Google Scholar] [CrossRef]
- Huang, H.; Yang, L.; Facchetti, A.; Marks, T.J. Organic and Polymeric Semiconductors Enhanced by Noncovalent Conformational Locks. Chem. Rev. 2017, 117, 10291–10318. [Google Scholar] [CrossRef]
- Benz, S.; Mareda, J.; Besnard, C.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds: Neutral benzodiselenazole scaffolds with high-precision selenium donors of variable strength. Chem. Sci. 2017, 8, 8164–8169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallinford, UK, 2009. [Google Scholar]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2017, 56, 488–502. [Google Scholar] [CrossRef]
- Brinck, T.; Borrfors, A.N. Electrostatics and polarization determine the strength of the halogen bond: A red card for charge transfer. J. Mol. Model. 2019, 25, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Outeiral, C.; Vincent, M.A.; Martín Pendás, Á.; Popelier, P.L.A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. [Google Scholar] [CrossRef] [Green Version]
- Hugas, D.; Guillaumes, L.; Duran, M.; Simon, S. Delocalization indices for non-covalent interaction: Hydrogen and diHydrogen bond. Comput. Theor. Chem. 2012, 998, 113–119. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (version 17.01.25); TK Gristmill Software: Overland Park, KS, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pople, J.A. The Lennard-Jones lecture. Intermolecular binding. Faraday Discuss. 1982, 73, 7–17. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An open-source ab initio electronic structure program. WIREs Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Auxiliary Basis Sets. Available online: http://www.psicode.org/psi4manual/master/basissets_byfamily.html (accessed on 11 February 2020).
- Popelier, P.L.A. On the full topology of the Laplacian of the electron density. Coord. Chem. Rev. 2000, 197, 169–189. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. On the Importance of Halogen–Halogen Interactions in the Solid State of Fullerene Halides: A Combined Theoretical and Crystallographic Study. Crystals 2017, 7, 191. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. A DFT assessment of some physical properties of iodine-centered halogen bonding and other non-covalent interactions in some experimentally reported crystal geometries. Phys. Chem. Chem. Phys. 2018, 20, 15316–15329. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density—Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Chemical Implications of Local Features of the Electron Density Distribution; Springer: Heidelburg, Germary, 1990; Volume 2, pp. 457–542. [Google Scholar]
- Zou, W.; Zhang, X.; Dai, H.; Yan, H.; Cremer, D.; Kraka, E. Description of an unusual hydrogen bond between carborane and a phenyl group. J. Organomet. Chem. 2018, 865, 114–127. [Google Scholar] [CrossRef]
- Sarkhel, S.; Desiraju, G.R. N–H…O, O–H…O, and C–H…O hydrogen bonds in protein–ligand complexes: Strong and weak interactions in molecular recognition. Proteins Struct. Funct. Bioinform. 2004, 54, 247–259. [Google Scholar] [CrossRef]
- Schneider, H.-J. Crystallographic searches for weak interactions–the limitations of data mining. Acta Cryst. 2018, B74, 322–324. [Google Scholar] [CrossRef]
- Steiner, T.; R. Desiraju, G. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 891–892. [Google Scholar] [CrossRef]
- Ege, S. Organic Chemistry: Structure and Reactivity, 4th ed.; Houghton Mifflin College: Boston, MA, USA, 2003. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Emsley, J. Very strong hydrogen bonding. Chem. Soc. Rev. 1980, 9, 91–124. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology (International Union of Crystallography Monographs on Crystallography, 9); Oxford University Press: Oxford, UK; New York, NY, USA, 1999. [Google Scholar]
- Stone, A.J. Are Halogen Bonded Structures Electrostatically Driven? J. Am. Chem. Soc. 2013, 135, 7005–7009. [Google Scholar] [CrossRef]
Figure 1.
(a) The Laplacian of the charge density plot of CH3Cl. (b) Selected critical points of the Laplacian of the charge density (values in a.u.). (c) The 0.001 a.u. isodensity envelope mapped potential on the surface of the CH3Cl molecule. Values of potential extrema (VS,min and VS,min in kcal mol−1) obtained via mapping with various isodensity envelopes are also shown.
Figure 1.
(a) The Laplacian of the charge density plot of CH3Cl. (b) Selected critical points of the Laplacian of the charge density (values in a.u.). (c) The 0.001 a.u. isodensity envelope mapped potential on the surface of the CH3Cl molecule. Values of potential extrema (VS,min and VS,min in kcal mol−1) obtained via mapping with various isodensity envelopes are also shown.

Figure 2.
MP2/aug-cc-pVTZ energy-minimized geometries of the 18 binary complexes of CH3Cl examined in this study. The intermolecular distance in Å (upper entry) and the angle of approach in degree (lower entry) between the monomers in each complex are given.
Figure 2.
MP2/aug-cc-pVTZ energy-minimized geometries of the 18 binary complexes of CH3Cl examined in this study. The intermolecular distance in Å (upper entry) and the angle of approach in degree (lower entry) between the monomers in each complex are given.

Figure 3.
MP2/aug-cc-pVTZ calculated quantum theory of atoms in molecules in molecules (QTAIM) molecular graphs of the 18 binary complexes of CH3Cl studied. The bond paths (solid and dotted lines) and the bond critical points (bcps) (tiny red spheres) are shown between the atomic basins. The charge density (ρb/a.u,), the Laplacian of the charge density (∇2ρb/a.u.), and the total energy density (Hb/a.u.) at the bcps of the intermolecular interactions are shown in black, red, and green fonts, respectively.
Figure 3.
MP2/aug-cc-pVTZ calculated quantum theory of atoms in molecules in molecules (QTAIM) molecular graphs of the 18 binary complexes of CH3Cl studied. The bond paths (solid and dotted lines) and the bond critical points (bcps) (tiny red spheres) are shown between the atomic basins. The charge density (ρb/a.u,), the Laplacian of the charge density (∇2ρb/a.u.), and the total energy density (Hb/a.u.) at the bcps of the intermolecular interactions are shown in black, red, and green fonts, respectively.

Figure 4.
The MP2/aug-cc-pVTZ computed reduced density gradient (RDG) isosurface topologies for the 18 binary complexes of CH3Cl studied. The delocalization indices (DIs) corresponding to selected atom–atom pairs are shown for each complex. The blue, green, and brownish isosurfaces represent strong, medium-strength, and weakly bound attractive interactions, respectively, whereas that in red represents repulsive interactions.
Figure 4.
The MP2/aug-cc-pVTZ computed reduced density gradient (RDG) isosurface topologies for the 18 binary complexes of CH3Cl studied. The delocalization indices (DIs) corresponding to selected atom–atom pairs are shown for each complex. The blue, green, and brownish isosurfaces represent strong, medium-strength, and weakly bound attractive interactions, respectively, whereas that in red represents repulsive interactions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The 0.001 a.u. isodensity envelope mapped electrostatic potential on the outer surface of various bonded atoms in CH3Cl, computed using various computational approaches in conjunction with the aug-cc-pV(T + d)Z basis set.
Table 1.
The 0.001 a.u. isodensity envelope mapped electrostatic potential on the outer surface of various bonded atoms in CH3Cl, computed using various computational approaches in conjunction with the aug-cc-pV(T + d)Z basis set.
| Method/Basis Set | VS,max | VS,min | VS,max | VS,max |
|---|---|---|---|---|
| C−Cl | C−Cl | Cl−C | C−H | |
| [CCSD/aug-cc-pV(T + d)Z] | 0.86 | −14.78 | 17.52 | 20.17 |
| [MP2/aug-cc-pV(T + d)Z] | 0.71 | −14.58 | 17.44 | 20.19 |
| [PBE0/aug-cc-pV(T + d)Z] | 0.72 | −14.74 | 17.03 | 20.37 |
| [PBE1/aug-cc-pV(T + d)Z] | 1.00 | −14.16 | 16.74 | 19.70 |
| [M062X/aug-cc-pV(T + d)Z] | 0.50 | −14.83 | 17.25 | 20.57 |
| [wB97XD/aug-cc-pV(T + d)Z] | 0.52 | −15.12 | 17.62 | 20.88 |
| [B97D3/aug-cc-pV(T + d)Z] | 0.61 | −14.56 | 17.68 | 19.74 |
| [B3PW91/aug-cc-pV(T + d)Z] | 0.69 | −14.83 | 17.18 | 20.26 |
| [B3LYP/aug-cc-pV(T + d)Z] | 0.47 | −14.71 | 17.83 | 19.94 |
| [B3LYP-D3/aug-cc-pV(T + d)Z] | 0.49 | −14.71 | 17.91 | 19.96 |
Table 2.
Comparison of the MP2/aug-cc-pVTZ computed binding energies with the density functional theory symmetry adapted perturbation theory (DFT-SAPT) interaction energies for the 18 binary complexes of CH3Cl a.
Table 2.
Comparison of the MP2/aug-cc-pVTZ computed binding energies with the density functional theory symmetry adapted perturbation theory (DFT-SAPT) interaction energies for the 18 binary complexes of CH3Cl a.
| Figure 2 | Complex | ΔE | ΔE(BSSE) | Eeles | Eexch | Eind | Edisp | E(SAPT0) |
|---|---|---|---|---|---|---|---|---|
| (a) | H3CCl···Br2 | −1.83 | −1.07 | −0.87 | 2.77 | −0.30 | −2.37 | −0.78 |
| (b) | H3CCl···Br2 | −4.90 | −3.43 | −6.49 | 10.88 | −2.86 | −4.66 | −3.13 |
| (c) | H3CCl···BrCl | −1.58 | −1.02 | −0.73 | 2.33 | −1.10 | −2.10 | −0.76 |
| (d) | H3CCl···BrF | −1.21 | −0.71 | −0.70 | 1.96 | −0.24 | −1.56 | −0.53 |
| (e) | Cl2···ClCH3 | −1.27 | −0.96 | −0.59 | 1.97 | −0.20 | −1.94 | −0.76 |
| (f) | H3CCl···Cl2 | −3.23 | −2.68 | −4.13 | 6.91 | −1.74 | −3.50 | −2.46 |
| (g) | OC···ClCH3 | −0.78 | −0.61 | −0.47 | 0.97 | −0.08 | −0.99 | −0.57 |
| (h) | OC···ClCH3 | −0.73 | −0.5 | −0.10 | 0.82 | −0.09 | −0.94 | −0.30 |
| (i) | OC···ClCH3 | −0.72 | −0.56 | −0.62 | 1.08 | −0.15 | −1.00 | −0.70 |
| (j) | F2···ClCH3 | −0.55 | −0.37 | −0.21 | 0.68 | −0.04 | −0.77 | −0.34 |
| (k) | H3CCl···F2 | −1.52 | −1.15 | −1.47 | 2.82 | −0.58 | −1.66 | −0.89 |
| (l) | N2···ClCH3 | −0.68 | −0.45 | −0.29 | 0.90 | −0.09 | −0.97 | −0.45 |
| (m) | N2···ClCH3 | −0.80 | −0.6 | −0.37 | 1.07 | −0.09 | −1.09 | −0.49 |
| (n)b | SO···ClCH3 | −4.52 | −3.74 | --- | --- | --- | --- | --- |
| (o) | OS···ClCH3 | −0.74 | −0.48 | 0.08 | 1.36 | −0.24 | −1.45 | −0.25 |
| (p)b | OS···ClCH3 | −6.79 | −5.71 | --- | --- | --- | --- | --- |
| (q) | H3CCl···HCl | −4.30 | −3.65 | −5.29 | 7.72 | −2.70 | −3.27 | −3.55 |
| (r) | H3CBr···HBr | −4.59 | −3.34 | −5.37 | 8.86 | −2.83 | −3.80 | −3.13 |
a Values in kcal mol−1. b DFT-SAPT calculations could not be performed for these two complexes because of the convergence issues associated with the Psi4 code.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor? Crystals 2020, 10, 146. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030146
AMA Style
Varadwaj PR, Varadwaj A, Marques HM. Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor? Crystals. 2020; 10(3):146. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030146
Chicago/Turabian StyleVaradwaj, Pradeep R., Arpita Varadwaj, and Helder M. Marques. 2020. "Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor?" Crystals 10, no. 3: 146. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030146
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.