Anion–Cation Recognition Pattern, Thermal Stability and DFT-Calculations in the Crystal Structure of H2dap[Cd(HEDTA)(H2O)] Salt (H2dap = H2(N3,N7)-2,6-Diaminopurinium Cation)


 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Crystallography
2.3. Other Physical Measurements
2.4. Synthesis and Relevant IR Spectrum Data
2.5. Theoretical Methods
3. Results and Discussion
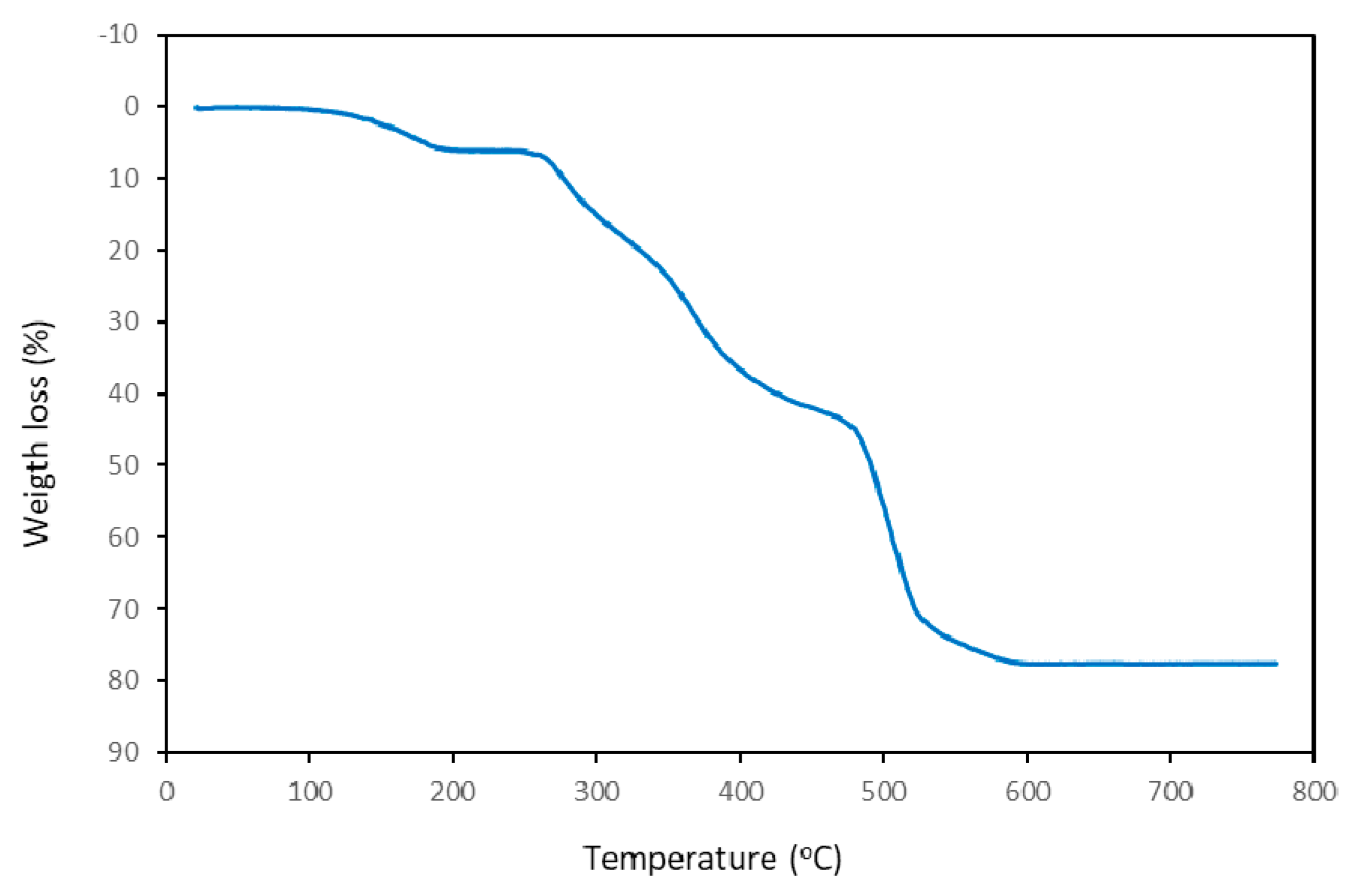
3.1. Thermal Stability
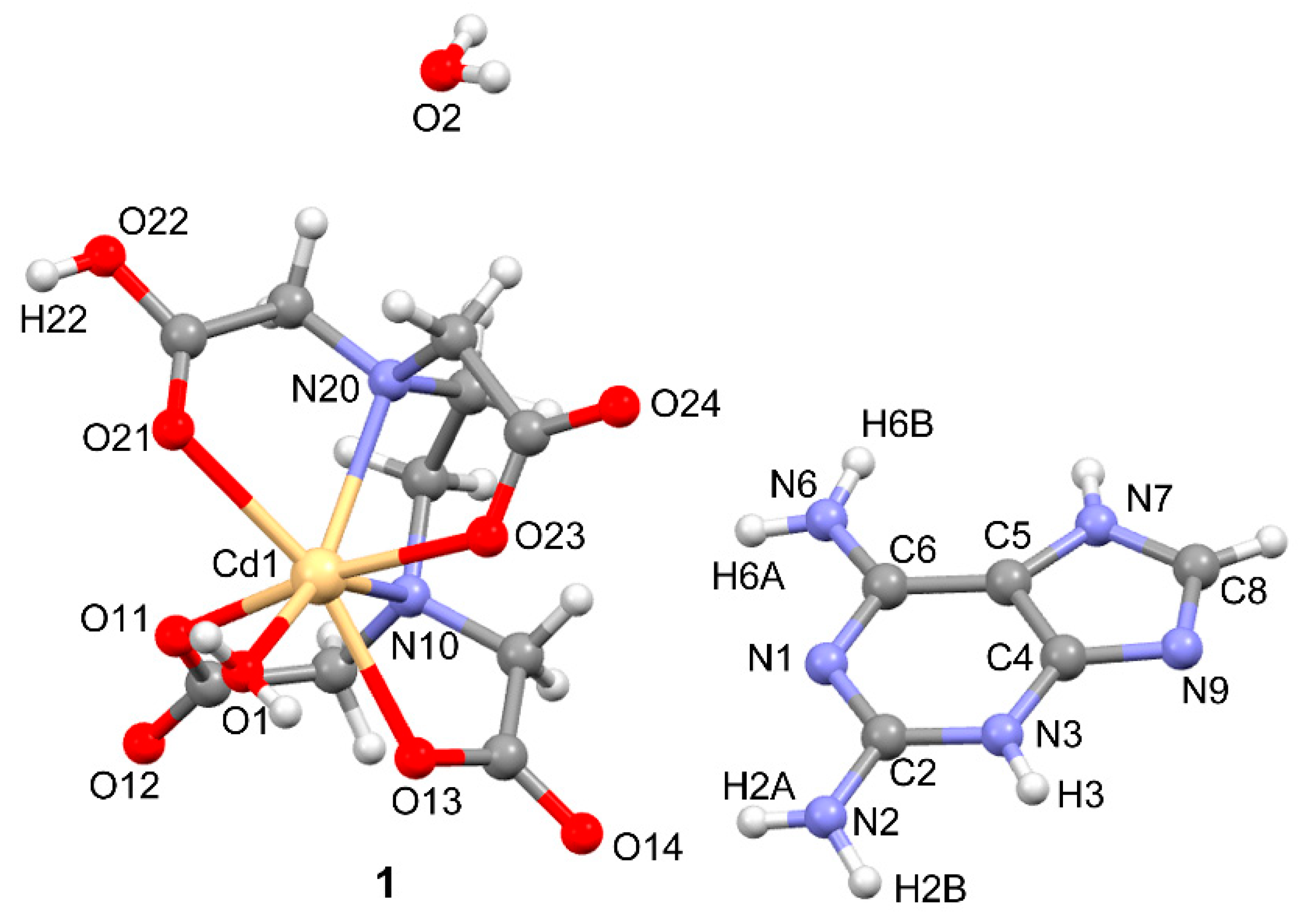
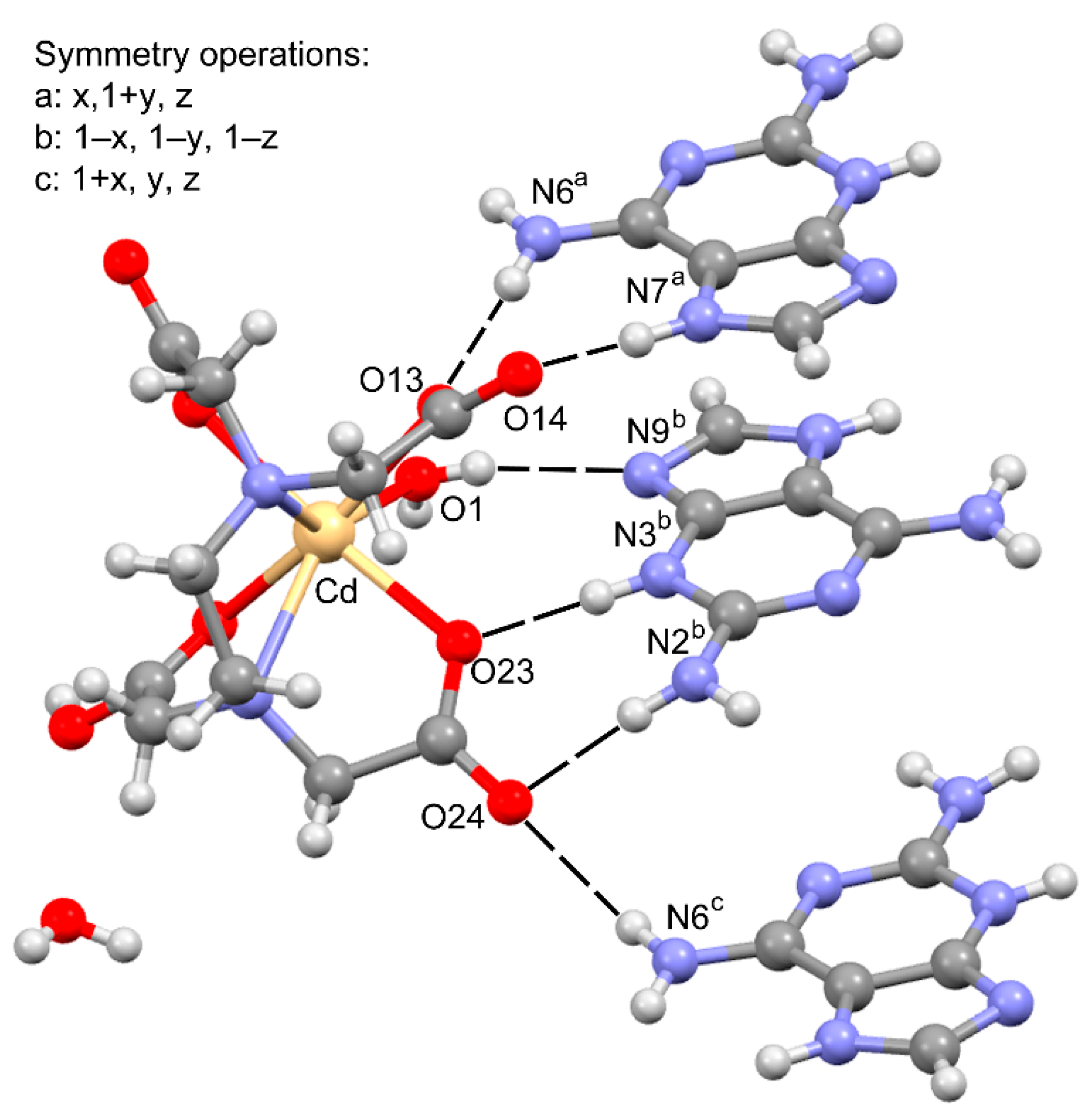
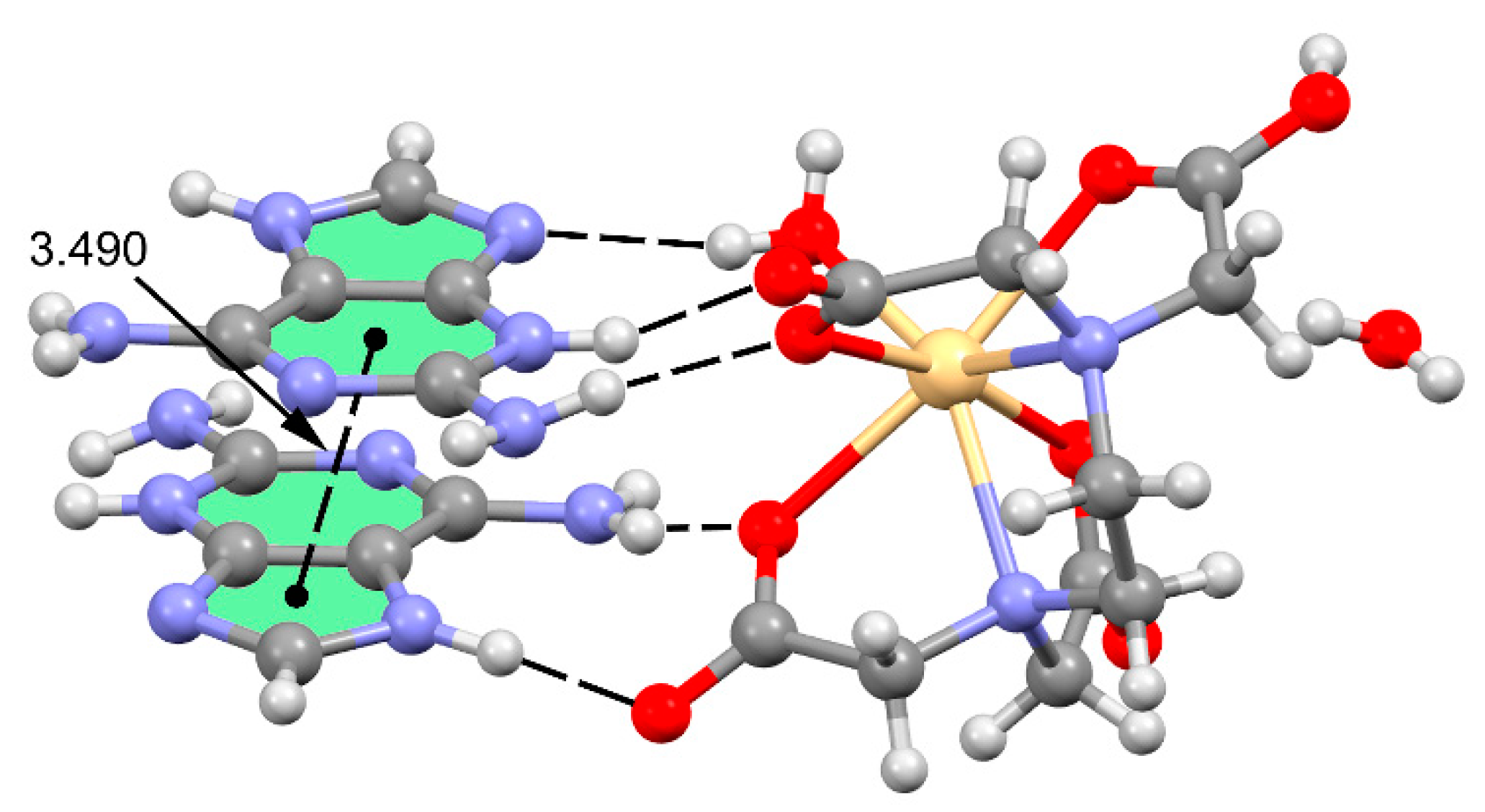
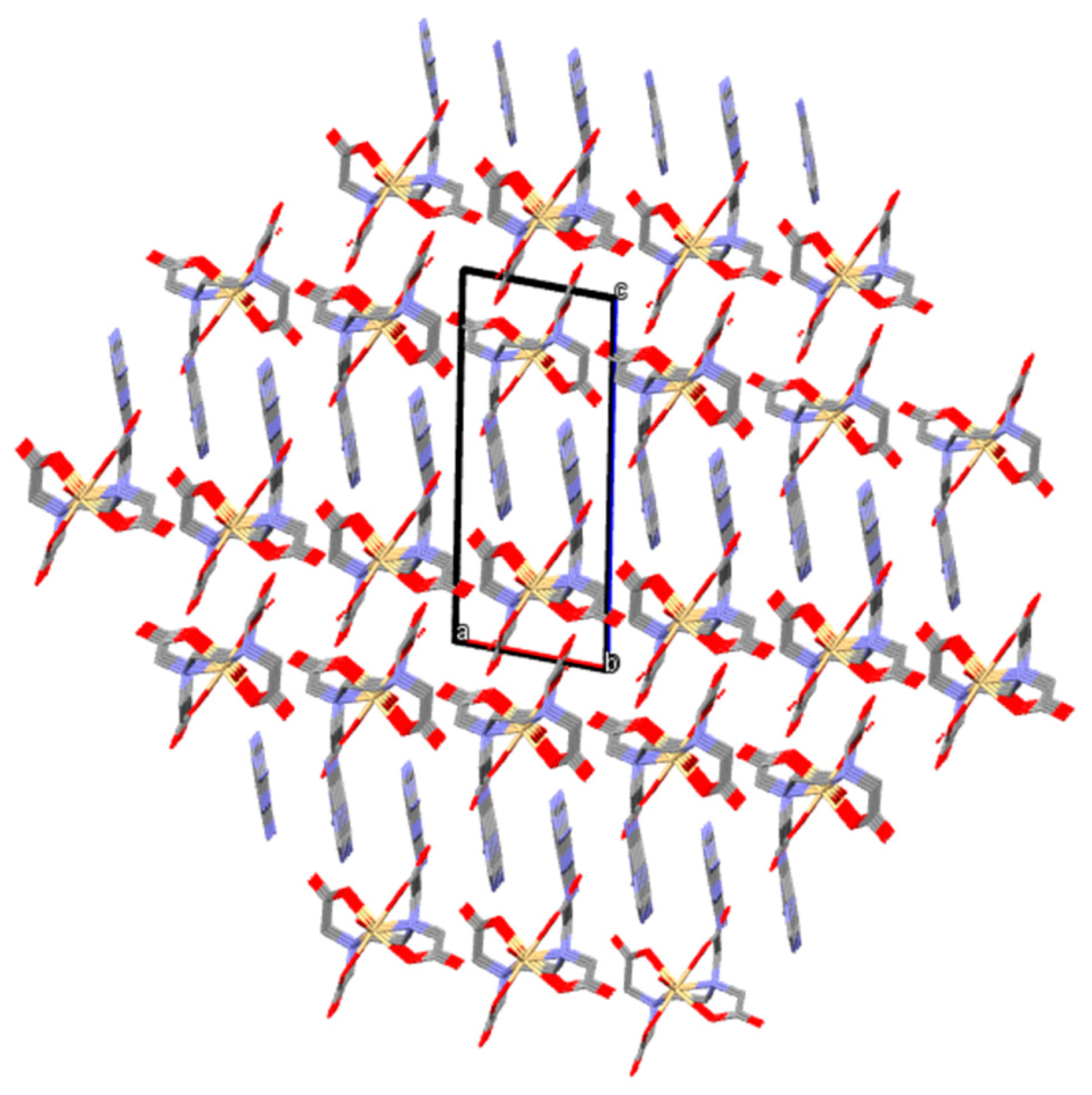
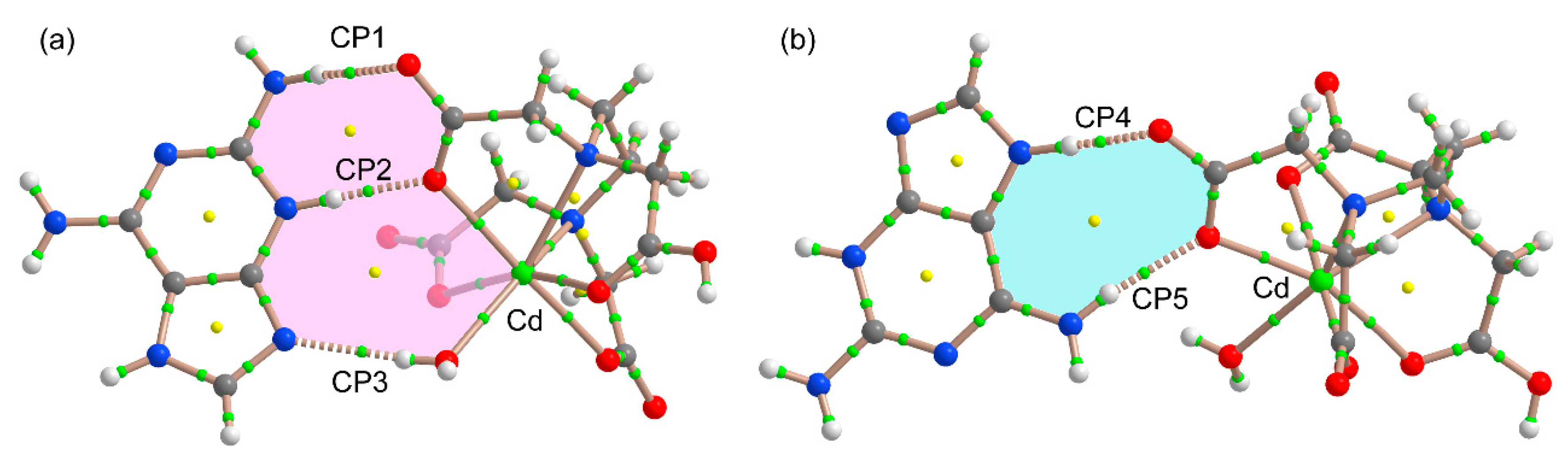
3.2. Crystal Structure and Anion–Cation Recognition Pattern
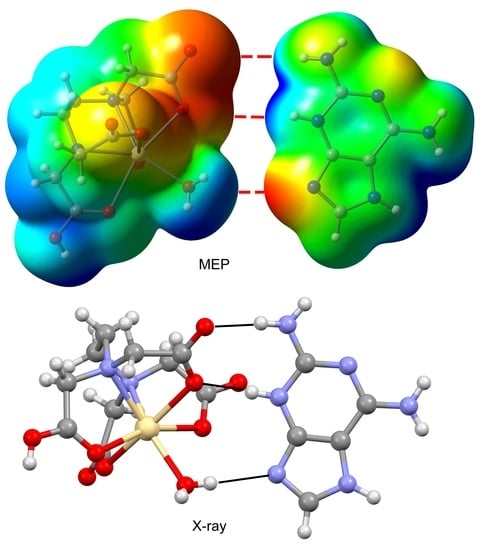
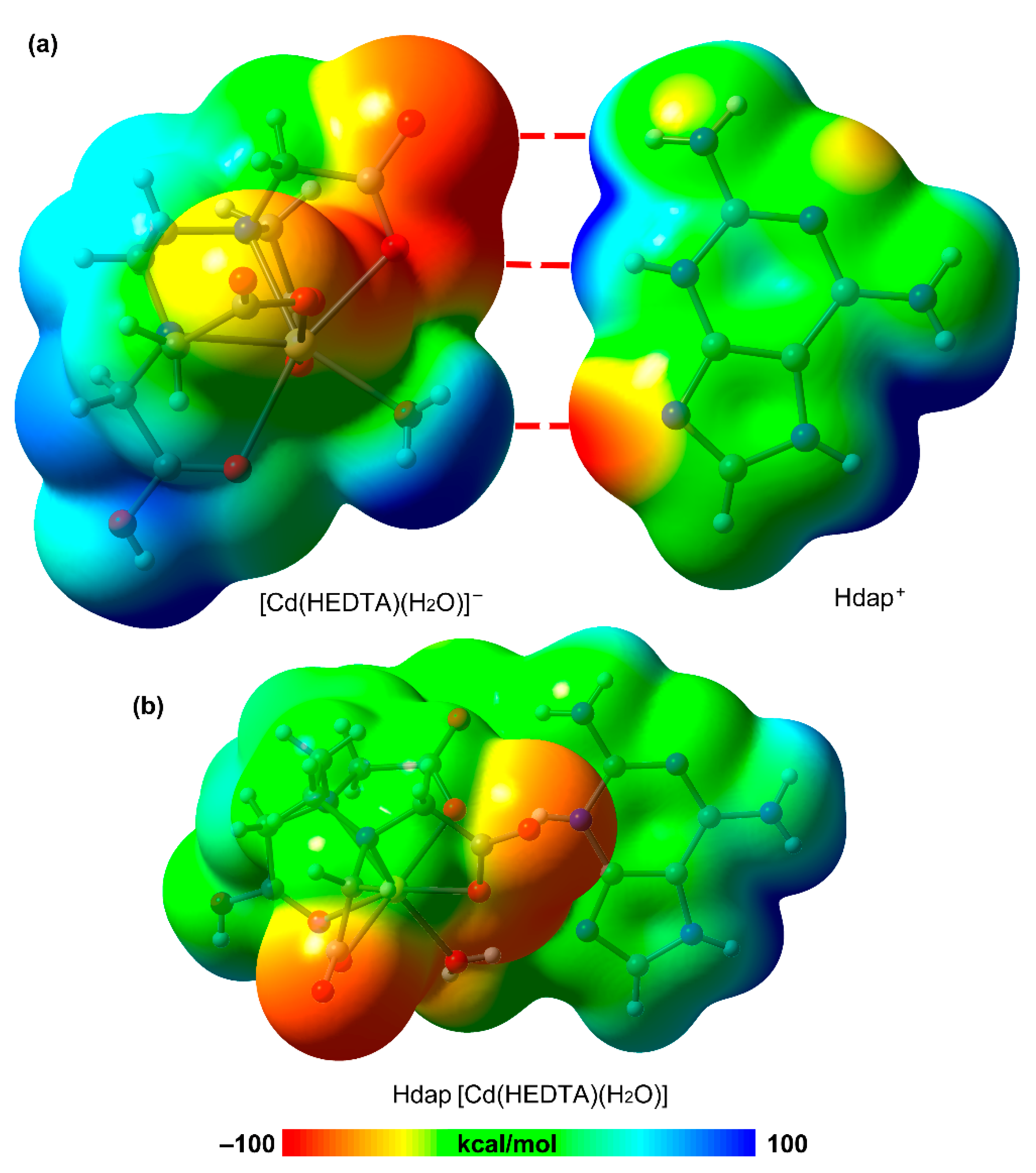
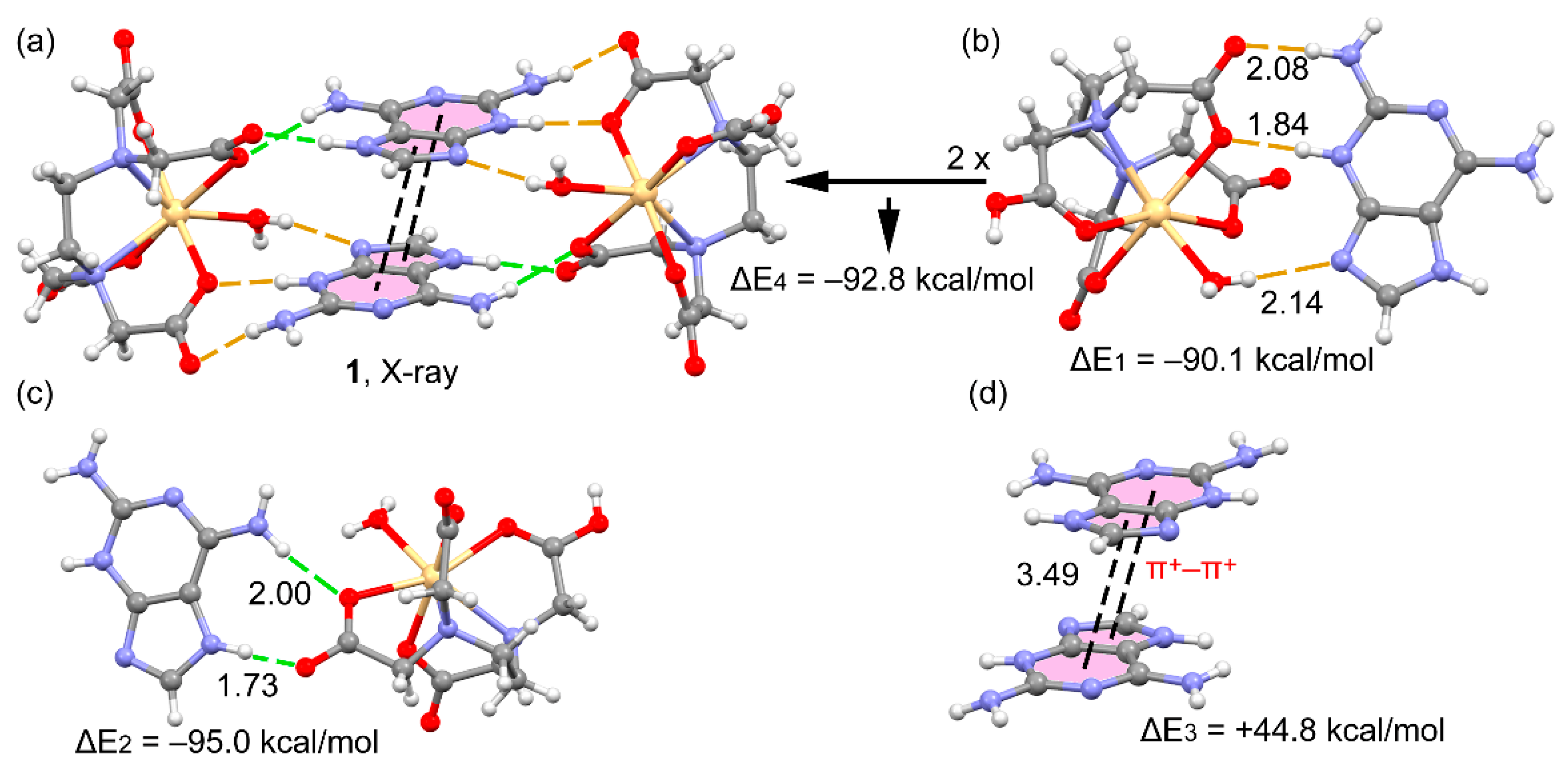
3.3. DFT Calculations
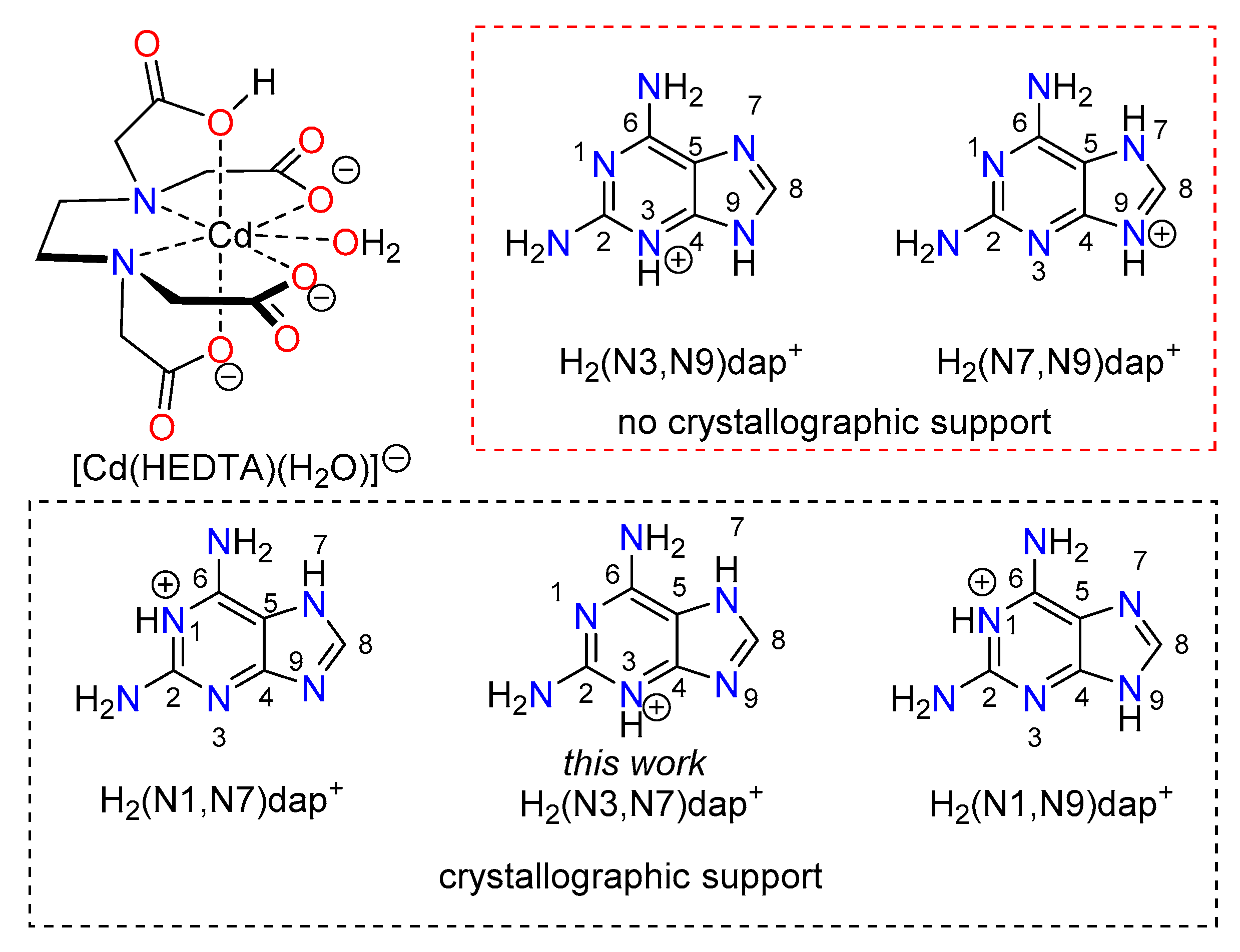
3.4. Structural Insides on N(heterocyclic)-Proton Affinities, H-Tautomerism and Metal Binding Patterns from Hdap and Its Cationic Forms in Salts and Their Metal Complexes.
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Terrón, A.; Fiol, J.J.; García-Raso, A.; Barceló-Oliver, M.; Moreno, V. Biological recognition patterns implicated by the formation and stability of ternary metal ion complexes of low-molecular-weight formed with amino acid/peptides and nucleobases/nucleosides. Coord. Chem. Rev. 2007, 251, 1973–1986. [Google Scholar] [CrossRef]
- Sivakova, S.; Rowan, S.J. Nucleobases as supramolecular motifs. Chem. Soc. Rev. 2005, 34, 9–21. [Google Scholar] [CrossRef]
- Lippert, B. Multiplicity of metal ion binding patterns to nucleobases. Coord. Chem. Rev. 2000, 200–202, 487–516. [Google Scholar] [CrossRef]
- Navarro, J.A.R.; Lippert, B. Simple 1:1 and 1:2 complexes of metal ions with heterocycles as building blocks for discrete molecular as well as polymeric assemblies. Coord. Chem. Rev. 2001, 222, 219–250. [Google Scholar] [CrossRef]
- Choquesillo-Lazarte, D.; Brandi-Blanco, M.P.; García-Santos, I.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Interligand interactions involved in the molecular recognition between copper (II) complexes and adenine or related purines. Coord. Chem. Rev. 2008, 25, 1241–1256. [Google Scholar] [CrossRef]
- Olea, D.; Alexandre, S.S.; Amo-Ochoa, P.; Guijarro, A.; De Jesus, F.; Soler, J.M.; De Pablo, P.J.; Zamora, F.; Gomez-Herrero, J. From Coordination Polymer Macrocrystals to Nanometric Individual Chains. Adv. Mater. 2005, 17, 1761–1765. [Google Scholar] [CrossRef]
- García-Terán, J.P.; Castillo, O.; Luque, A.; Garcıía-Couceiro, U.; Beobide, G.; Román, P. Molecular Recognition of Adeninium Cations on Anionic Metal−Oxalato Frameworks: An Experimental and Theoretical Analysis. Inorg. Chem. 2007, 46, 3593–3602. [Google Scholar] [CrossRef]
- González-Pérez, J.M.; Alarcón-Payer, C.; Castinñiras, A.; Pivetta, T.; Lezama, L.; Choquesillo-Lazarte, D.; Crisponi, G.; Niclós-Gutiérrez, J. A Windmill-Shaped Hexacopper(II) Molecule Built Up by Template Core-Controlled Expansion of Diaquatetrakis(μ2-adeninato-N3,N9)dicopper(II) with Aqua(oxydiacetato)-copper(II). Inorg. Chem. 2006, 45, 877–882. [Google Scholar] [CrossRef]
- Amo-Ochoa, P.; Rodríguez-Tapiador, M.I.; Castillo, O.; Olea, D.; Guijarro, A.; Alexandre, S.S.; Gómez-Herrero, J.; Zamora, F. Assembling of Dimeric Entities of Cd(II) with 6-Mercaptopurine to Afford One-Dimensional Coordination Polymers: Synthesis and Scanning Probe Microscopy Characterization. Inorg. Chem. 2006, 45, 7642–7650. [Google Scholar] [CrossRef]
- García-Terán, J.P.; Castillo, O.; Luque, A.; García-Couceiro, U.; Román, P.; Lloret, F. One-Dimensional Oxalato-Bridged Cu(II), Co(II), and Zn(II) Complexes with Purine and Adenine as Terminal Ligands. Inorg. Chem. 2004, 43, 5761–5770. [Google Scholar] [CrossRef]
- García-Terán, J.P.; Castillo, O.; Luque, A.; García-Couceiro, U.; Román, P.; Lezama, L. An Unusual 3D Coordination Polymer Based on Bridging Interactions of the Nucleobase Adenine. Inorg. Chem. 2004, 43, 4549–4551. [Google Scholar] [CrossRef]
- Rojas-González, P.X.; Catiñeiras, A.; González-Pérez, J.M.; Choquesillo-Lazarte, D.; Niclós-Gutiérrez, J. Interligand Interactions Controlling the μ-N7,N9-Metal Bonding of Adenine (AdeH) to the N-Benzyliminodiacetato(2−) Copper(II) Chelate and Promoting the N9 versus N3 Tautomeric Proton Transfer: Molecular and Crystal Structure of [Cu2(NBzIDA)2(H2O)2(μ-N7,N9-Ade(N3)H)]·3H2O. Inorg. Chem. 2002, 41, 6190–6192. [Google Scholar] [PubMed]
- García-Terán, J.P.; Castillo, O.; Luque, A.; García-Couceiro, U.; Beobide, G.; Román, P. Supramolecular architectures assembled by the interaction of purine nucleobases with metal-oxalato frameworks. Non-covalent stabilization of the 7H-adenine tautomer in the solid-state. Dalton Trans 2006, 902–911. [Google Scholar] [CrossRef]
- Bugella-Altamirano, E.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Sánchez-Moreno, M.J.; Martín-Ramos, R.; Covelo, B.; Carballo, R.; Castiñeiras, A.; Niclós-Gutiérrez, J. Three new modes of adenine-copper(II) coordination: Interligand interactions controlling the selective N3-, N7- and bridging μ-N3,N7–metal-bonding of adenine to different N-substituted iminodiacetato-copper(II) chelates. Inorg. Chim. Acta 2002, 339, 160–170. [Google Scholar] [CrossRef]
- Suzuki, T.; Hirai, Y.; Monjushiro, H.; Kaizaki, S. Cobalt(III) Complexes of Monodentate N(9)-Bound Adeninate (ade-), [Co(ade-κN9)Cl(en)2]+ (en = 1,2-Diaminoethane): Syntheses, Crystal Structures, and Protonation Behaviors of the Geometrical Isomers. Inorg. Chem. 2004, 43, 6435–6444. [Google Scholar] [CrossRef]
- Yang, E.-C.; Zhao, H.-K.; Feng, Y.; Zhao, X.-J. A Tetranuclear CuII-Based 2D Aggregate with an Unprecedented Tetradentate μ4-N1,N3,N7,N9-Adeninate Nucleobase. Inorg. Chem. 2009, 48, 3511–3513. [Google Scholar] [CrossRef]
- García-Raso, A.; Terrón, A.; Ortega-Castro, J.; Barceló-Oliver, M.; Lorenzo, J.; Rodríguez-Calado, S.; Franconetti, A.; Frontera, A.; Vázquez-López, E.M.; Fiol, J.J. Iridium(III) coordination of N(6) modified adenine derivatives with aminoacid chains. J. Inorg. Biochem. 2020, 205, 111000. [Google Scholar] [CrossRef]
- Ruiz-González, N.; García-Rubiño, M.E.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; Franconetti, A.; Frontera, A.; Castiñeiras, A.; González-Pérez, J.M.; Niclós-Gutiérrez, J. Molecular and supramolecular recognition patterns in ternary copper(II) or zinc(II) complexes with selected rigid-planar chelators and a synthetic adenine-nucleoside. J. Inorg. Biochem. 2020, 203, 110920. [Google Scholar] [CrossRef]
- Martínez, D.; Pérez, A.; Cañellas, S.; Silió, I.; Lancho, A.; García-Raso, A.; Fiol, J.J.; Terrón, A.; Barceló-Oliver, M.; Ortega-Castro, J.; et al. Synthesis, reactivity, X-ray characterization and docking studies of N7/N9-(2-pyrimidyl)-adenine derivatives. J. Inorg. Biochem. 2020, 203, 110879. [Google Scholar] [CrossRef]
- Roitzsch, M.; Lippert, B. Metal Coordination and Imine−Amine Hydrogen Bonding as the Source of Strongly Shifted Adenine pKa Values. J. Am. Chem. Soc. 2004, 126, 2421–2424. [Google Scholar] [CrossRef]
- Añorbe, M.G.; Welzel, T.; Lippert, B. Migration of a cis-(NH3)2PtII Moiety along Two Adenine Nucleobases, from N1 to N6, is Markedly Facilitated by Additional PtII Entities Coordinated to N7. Inorg. Chem. 2007, 46, 8222–8227. [Google Scholar] [CrossRef]
- Purohit, C.S.; Verma, S. A Luminescent Silver−Adenine Metallamacrocyclic Quartet. J. Am. Chem. Soc. 2006, 128, 400–401. [Google Scholar] [CrossRef]
- Purohit, C.S.; Mishra, A.K.; Verma, S. Four-Stranded Coordination Helices Containing Silver−Adenine (Purine) Metallaquartets. Inorg. Chem. 2007, 46, 8493–8495. [Google Scholar] [CrossRef]
- Purohit, C.S.; Verma, S. Patterned Deposition of a Mixed-Coordination Adenine−Silver Helicate, Containing a π-Stacked Metallacycle, on a Graphite Surface. J. Am. Chem. Soc. 2007, 129, 3488–3489. [Google Scholar] [CrossRef]
- Shipman, M.A.; Price, C.; Gibson, A.E.; Elsegood, M.R.J.; Clegg, W.; Houlton, A. Monomer, Dimer, Tetramer, Polymer: Structural Diversity in Zinc and Cadmium Complexes of Chelate-Tethered Nucleobases. Chem. Eur. J. 2000, 6, 4371–4378. [Google Scholar] [CrossRef]
- Kruger, T.; Ruffer, T.; Lang, H.; Wagner, C.; Steinborn, D. Synthesis, Characterization, and Reactivity of [Li(N6,N9-Me2Ade-H)]: A Structurally Characterized Lithiated Adenine. Inorg. Chem. 2008, 47, 1190–1195. [Google Scholar] [CrossRef]
- Amantia, D.; Price, C.; Shipman, M.A.; Elsegood, M.R.J.; Clegg, W.; Houlton, A. Minor Groove Site Coordination of Adenine by Platinum Group Metal Ions: Effects on Basicity, Base Pairing, and Electronic Structure. Inorg. Chem. 2003, 42, 3047–3056. [Google Scholar] [CrossRef]
- Zobi, F.; Spingler, B.; Alberto, R. Structure, Reactivity and Solution Behaviour of [Re(ser)(7-MeG)(CO)3] and [Re(ser)(3-pic)(CO)3]: ‘Nucleoside-mimicking’ Complexes Based on the fac-[Re(CO)3]+ Moiety. Dalton Trans. 2005, 2859–2865. [Google Scholar] [CrossRef]
- Jiang, Q.; Wu, Z.-Y.; Zhang, Y.-M.; Hotze, A.C.G.; Hannon, M.J.; Guo, Z.-J. Effect of Adenine Moiety on DNA Binding Property of Copper(II)–terpyridine Complexes. Dalton Trans 2008, 3054–3060. [Google Scholar] [CrossRef]
- Price, C.; Horrocks, B.R.; Mayeux, A.; Elsegood, M.R.J.; Clegg, W.; Houlton, A. Self-Complementary Metal Complexes Containing a DNA Base Pair. Angew. Chem. Int. Ed. 2002, 41, 1047–1049. [Google Scholar] [CrossRef]
- Dobrzynska, D.; Jerzykiewicz, L.B. Adenine Ribbon with Watson− Crick and Hoogsteen Motifs as the ‘Double-Sided Adhesive Tape’ in the Supramolecular Structure of Adenine and Metal Carboxylate. J. Am. Chem. Soc. 2004, 126, 11118–11119. [Google Scholar] [CrossRef]
- Serrano Padial, E.; Choquesillo-Lazarte, D.; Bugella Altamirano, E.; Castiñeiras, A.; Carballo, R.; Niclós Gutiérrez, J. New Copper(II) Compound having Protonated forms of Ethylenediamine-tetraacetate(4-) ion (EDTA) and Adenine (AdeH): Synthesis, Crystal Structure, Molecular Recognition and Physical Properties of (AdeH2)[Cu(HEDTA)(H2O)]·2H2O. Polyhedron 2002, 21, 1451–1457. [Google Scholar] [CrossRef]
- Bruker. APEX3 Software; v2018.7-2; Bruker AXS Inc.: Madison, WI, USA, 2018. [Google Scholar]
- Sheldrick, G.M. SADABS. Program for Empirical Absorption Correction of Area Detector Data; University of Goettingen: Goettingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.C. International Tables of Crystallography; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; Volume C. [Google Scholar]
- Spek, A.L. PLATON. A multipurpose Crystallographic tool. Acta Crystallogr. 2009, 65, 148–155. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hazari, A.; Das, L.K.; Kadam, R.M.; Bauza, A.; Frontera, A.; Ghosh, A. Unprecedented structural variations in trinuclear mixed valence Co (II/III) complexes: Theoretical studies, pnicogen bonding interactions and catecholase-like activities. Dalton Trans. 2015, 44, 3862–3876. [Google Scholar] [CrossRef]
- Mitra, M.; Manna, P.; Bauzá, A.; Ballester, P.; Seth, S.K.; Ray Choudhury, S.; Frontera, A.; Mukhopadhyay, S. 3-Picoline Mediated Self-Assembly of M (II)–Malonate Complexes (M= Ni/Co/Mn/Mg/Zn/Cu) Assisted by Various Weak Forces Involving Lone Pair− π, π–π, and Anion··· π–Hole Interactions. J. Phys. Chem. B 2014, 118, 14713–14726. [Google Scholar] [CrossRef]
- Kolaria, K.; Sahamies, J.; Kalenius, E.; Novikov, A.S.; Kukushkin, V.Y.; Haukka, M. Metallophilic interactions in polymeric group 11 thiols. Solid State Sci. 2016, 60, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.S.; Ivanov, D.M.; Bikbaeva, Z.M.; Bokach, N.A.; Kukushkin, V.Y. Noncovalent Interactions Involving Iodofluorobenzenes: The Interplay of Halogen Bonding and Weak lp(O)···π-Holearene Interactions. Cryst. Growth Des. 2018, 18, 7641–7654. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Novikov, A.S.; Chernyshev, A.N.; Suslonov, V.V. Intermolecular hydrogen bonding H···Cl− in the solid palladium(II)-diaminocarbene complexes. Z. Kristallogr. Cryst. Mater. 2017, 232, 299–305. [Google Scholar] [CrossRef]
- Baykov, S.V.; Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Boyarskiy, V.P. Pt/Pd and I/Br Isostructural Exchange Provides Formation of C–I···Pd, C–Br···Pt, and C–Br···Pd Metal-Involving Halogen Bonding. Cryst. Growth Des. 2018, 18, 5973–5980. [Google Scholar] [CrossRef]
- Usoltsev, A.N.; Adonin, S.A.; Novikov, A.S.; Samsonenko, D.G.; Sokolov, M.N.; Fedina, V.P. One-dimensional polymeric polybromotellurates(iv): Structural and theoretical insights into halogen···halogen contacts. CrystEngComm 2017, 19, 5934–5939. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 13.05.06); TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations with Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef]
- Domínguez-Martín, A.; Brandi-Blanco, M.P.; Matilla-Hernández, A.; El Bakkali, H.; Nurchi, V.M.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Unraveling the Versatile Metal Binding Modes of Adenine: Looking at Molecular Recognition Patterns of Deaza- and Aza-adenines in Mixed Ligand Metal Complexes. Coord. Chem. Rev. 2013, 257, 2814–2838. [Google Scholar]
- Vílchez-Rodríguez, E.; Pérez-Toro, I.; Bauzá, A.; Matilla-Hernández, A. Structural and Theoretical Evidence of the Depleted Proton Affinity of the N3-Atom in Acyclovir. Crystals 2016, 6, 193. [Google Scholar]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; Vílchez-Rodríguez, E.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Lights and Shadows in the Challenge of Binding acyclovir, a synthetic Purine-like Nucleoside with Antiviral Activity, at an Apical-Distal Coordination Site in Copper-Polyamine Chelates. J. Inorg. Biochem. 2015, 148, 84–92. [Google Scholar] [CrossRef]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Highest Reported Denticity of a Synthetic Nucleoside in the Unprecedented Tetradentate Mode of Acyclovir. Cryst. Growth Des. 2018, 18, 4282–4286. [Google Scholar] [CrossRef]
- Bats, J.W.; Nasiri, H.R. CSD Database, reference code NULCO. Private Communication, 2015. [Google Scholar]
- Atria, A.M.; Corsini, G.; Herrera, N.; Garland, M.T.; Baggio, R. Two Isomorphous Transition Metal Complexes Containing a Protonated Diaminopurine Ligand: Diaquabis(2,6-diamino-7H-purin-1-ium-κN9) bis(homophthalato-κO)nickel(II) Tetrahydrate and the Cobalt(II) Analogue. Acta Crystallogr. 2011, 67, m169–m172. [Google Scholar] [CrossRef]
- Atria, A.M.; Garland, M.T.; Baggio, R. 2,6-Diamino-9H-purine Monohydrate and Bis(2,6-diamino-9H- purin-1-ium) 2-(2-Carboxylatophenyl)acetate Heptahydrate: Two Simple Structures with Very Complex Hydrogen-bonding Schemes. Acta Crystallogr. 2010, 66, o547–o552. [Google Scholar] [CrossRef]
- Belletire, J.L.; Schneider, S.; Shackelford, S.A.; Peryshkov, D.V.; Strauss, S.H. Pairing Hetherocycic Cations with closo-Dodecafluorododecaborate(-). Synthesis of Binary Heterocyclium(1+) Salts and Ag4(hetherocycle)84+ Salt of B11F122−. J. Fluor. Chem. 2011, 132, 925–936. [Google Scholar] [CrossRef]
- Yang, E.-C.; Chan, Y.-N.; Liu, H.; Wang, Z.-C.; Zhao, X.J. Unusual Polymeric ZnII/CdII Complexes with 2,6-Diaminopurine by Synergistic Coordination of Nucleobases and Polycarboxylate Anions: Binding Behavior, Self-Assembled Pattern of the Nucleobase, and Luminescent Properties. Cryst. Growth Des. 2009, 9, 4933–4944. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Dong, H.-M.; Wang, X.G.; Zhao, X.-J.; Yang, E.-C. Three Purine Containing Metal Complexes with Discrete Binuclear and Polymeric Chain Motifs: Synthesis, Crystal Structure and Luminescence. Inorg. Chim. Acta 2014, 416, 135–141. [Google Scholar] [CrossRef]
- Atria, A.M.; Parada, J.; Moreno, Y.; Suárez, S.; Baggio, R.; Peña, O. Synthesis, Crystal Structure and Magnetic Properties of Diaquabis(2,6-diamino-7H-purin-1-ium-κN9)bis(4,4’-oxydibenzoato-κO)cobalt(II) Dihydrate. Acta Crystallogr. 2018, 74, 37–44. [Google Scholar]
- Atria, A.M.; Morel, M.; Garland, M.T.; Baggio, R. Bis(2,6-diamino-1H-purin-3-ium) Di-µ-croconato- κ3O,O’:O”;κ3O:O’,O”-bis[tetraaqua(croconato-κ2O,O’)-neodymium(III)]. Acta Crystallogr. 2011, 67, m17–m21. [Google Scholar] [CrossRef]
- Atria, A.M.; Garland, M.T.; Baggio, R. Tetraaquabis(2,6-diamine-7H-κN9)cobalt(II) benzene-1,2,4,5- tetracarboxylate tetrahydrate. Acta Crystallogr. 2011, 67, m275–m278. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C15H24CdN8O10 |
|---|---|
| Formula weight | 588.82 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Triclinic, P-1 |
| Unit cell dimensions | a = 7.4924(3) Å, α = 81.9310(10)° |
| b = 9.0078(4) Å, β = 78.0170(10)° | |
| c = 17.2884(6) Å, γ = 70.545(2)° | |
| Volume | 1072.99(8) Å3 |
| Z, Calculated density | 2, 1.822 Mg/m3 |
| Absorption coefficient | 1.090 mm−1 |
| F(000) | 596 |
| Crystal size | 0.160 × 0.030 × 0.020 mm |
| Theta range for data collection | 2.405 to 30.507° |
| Limiting indices | −10 ≤ h ≤ 10, −12 ≤ k ≤ 12, −24 ≤ l ≤ 24 |
| Reflections collected / unique | 88812 / 6551 [R(int) = 0.0556] |
| Completeness to θ = 25.242 | 99.9% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 1.000 and 0.962 |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 6551 / 0 / 307 |
| Goodness-of-fit on F2 | 1.073 |
| Final R indices [I > 2σ(I)] | R1 = 0.0222, wR2 = 0.0454 |
| R indices (all data) | R1 = 0.0273, wR2 = 0.0477 |
| Largest diff. peak and hole | 0.588 and −0.469 e.Å−3 |
| CCSD refcode | 1992206 |
| Step or R | Temperature (°C) | Time (min) | Weight (%) | Evolved Gases or Residue (R) | |
|---|---|---|---|---|---|
| Experimental | Calulated | ||||
| 1 | 55–220 | 2.5–21 | 6.056 | 6.159 * | 2 H2O, CO2 (t) |
| 2 | 220–315 | 21–31 | 12.071 | - | CO2, H2O, CO, |
| 3 | 315–450 | 31–43 | 23.569 | - | CO2, H2O, CO, NH3, N2O, NO, NO2, CH4 |
| 4 | 450–560 | 43–53 | 33.071 | - | CO2, H2O, CO, NH3, N2O, NO, NO2, CH4 |
| 5 | 560–600 | 53–70 | 2.676 | - | CO2, H2O, NH3, N2O, NO, NO2 |
| R | 600 | - | 22.557 | 21.808 | CdO |
| R | 675 | - | 22.462 | 21.808 | CdO |
| Atoms | Distance or Angle | Atoms | Distance or Angle |
|---|---|---|---|
| Cd(1)-O(1) | 2.2672(11) | Cd(1)-N(10) | 2.4111(13) |
| Cd(1)-O(11) | 2.2984(11) | Cd(1)-O(21) | 2.4400(11) |
| Cd(1)-O(23) | 2.3010(11) | Cd(1)-N(20) | 2.4585(13) |
| Cd(1)-O(13) | 2.3748(11) | O(1)-Cd(1)-O(11) | 94.13(4) |
| O(1)-Cd(1)-O(23) | 91.28(4) | O(11)-Cd(1)-O(21) | 81.61(4) |
| O(11)-Cd(1)-O(23) | 168.52(4) | O(23)-Cd(1)-O(21) | 109.06(4) |
| O(1)-Cd(1)-O(13) | 79.59(4) | O(13)-Cd(1)-O(21) | 161.48(4) |
| O(11)-Cd(1)-O(13) | 91.09(4) | N(10)-Cd(1)-O(21) | 123.95(4) |
| O(23)-Cd(1)-O(13) | 79.93(4) | O(1)-Cd(1)-N(20) | 138.89(4) |
| O(1)-Cd(1)-N(10) | 145.66(4) | O(11)-Cd(1)-N(20) | 111.24(4) |
| O(11)-Cd(1)-N(10) | 73.31(4) | O(23)-Cd(1)-N(20) | 70.22(4) |
| O(23)-Cd(1)-N(10) | 96.63(4) | O(13)-Cd(1)-N(20) | 129.28(4) |
| O(13)-Cd(1)-N(10) | 69.10(4) | N(10)-Cd(1)-N(20) | 74.65(4) |
| O(1)-Cd(1)-O(21) | 83.96(4) | O(21)-Cd(1)-N(20) | 69.17(4) |
| H-bond | D···A (Å) | Angle (°) |
|---|---|---|
| O(1)-H(1WA)···N(9)#1 | 2.9017(17) | 166.6 |
| O(1)-H(1WB)···O(12)#2 | 2.7398(16) | 169.0 |
| O(22)-H(22)···O(11)#3 | 2.5552(16) | 175.5 |
| N(2)-H(2A)···O(14) | 2.8158(18) | 165.9 |
| N(2)-H(2B)···O(24)#1 | 2.9784(18) | 169.5 |
| N(3)-H(3)···O(23)#1 | 2.7123(17) | 179.5 |
| N(6)-H(6A)···O(24)#4 | 2.9898(18) | 144.6 |
| N(6)-H(6B)···O(13)#5 | 2.8307(17) | 167.7 |
| N(7)-H(7)···O(14)#5 | 2.6746(18) | 177.2 |
| O(2)-H(2WA)···O(12)#6 | 2.7517(17) | 163.4 |
| O(2)-H(2WB)···O(11)#7 | 2.9970(18) | 131.5 |
| CP# | ρr | Vr | Gr | Edis (−0.5 × Vr) | Edis (0.429 × Gr) |
|---|---|---|---|---|---|
| 1 | 0.0186 | −0.0122 | 0.0155 | 3.83 | 4.17 |
| 2 | 0.0328 | −0.0297 | 0.0308 | 9.32 | 8.29 |
| 3 | 0.0171 | −0.0110 | 0.0148 | 3.45 | 3.98 |
| 4 | 0.0429 | −0.0421 | 0.0380 | 12.2 | 10.2 |
| 5 | 0.0224 | −0.0172 | 0.0207 | 6.49 | 5.57 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belmont-Sánchez, J.C.; Ruiz-González, N.; Frontera, A.; Matilla-Hernández, A.; Castiñeiras, A.; Niclós-Gutiérrez, J. Anion–Cation Recognition Pattern, Thermal Stability and DFT-Calculations in the Crystal Structure of H2dap[Cd(HEDTA)(H2O)] Salt (H2dap = H2(N3,N7)-2,6-Diaminopurinium Cation). Crystals 2020, 10, 304. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10040304
Belmont-Sánchez JC, Ruiz-González N, Frontera A, Matilla-Hernández A, Castiñeiras A, Niclós-Gutiérrez J. Anion–Cation Recognition Pattern, Thermal Stability and DFT-Calculations in the Crystal Structure of H2dap[Cd(HEDTA)(H2O)] Salt (H2dap = H2(N3,N7)-2,6-Diaminopurinium Cation). Crystals. 2020; 10(4):304. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10040304
Chicago/Turabian StyleBelmont-Sánchez, Jeannette Carolina, Noelia Ruiz-González, Antonio Frontera, Antonio Matilla-Hernández, Alfonso Castiñeiras, and Juan Niclós-Gutiérrez. 2020. "Anion–Cation Recognition Pattern, Thermal Stability and DFT-Calculations in the Crystal Structure of H2dap[Cd(HEDTA)(H2O)] Salt (H2dap = H2(N3,N7)-2,6-Diaminopurinium Cation)" Crystals 10, no. 4: 304. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10040304