Author Contributions
Conceptualisation, R.M.A.; Data curation, R.M.A. and E.F.d.S.M.; Formal analysis, R.A.J.; Supervision, M.E.G.V. and R.A.J.; Validation, M.E.G.V. and R.A.J.; Writing—original draft, R.M.A.; Writing—review & editing, M.E.G.V. and R.A.J. All authors have read and agreed to the published version of the manuscript.
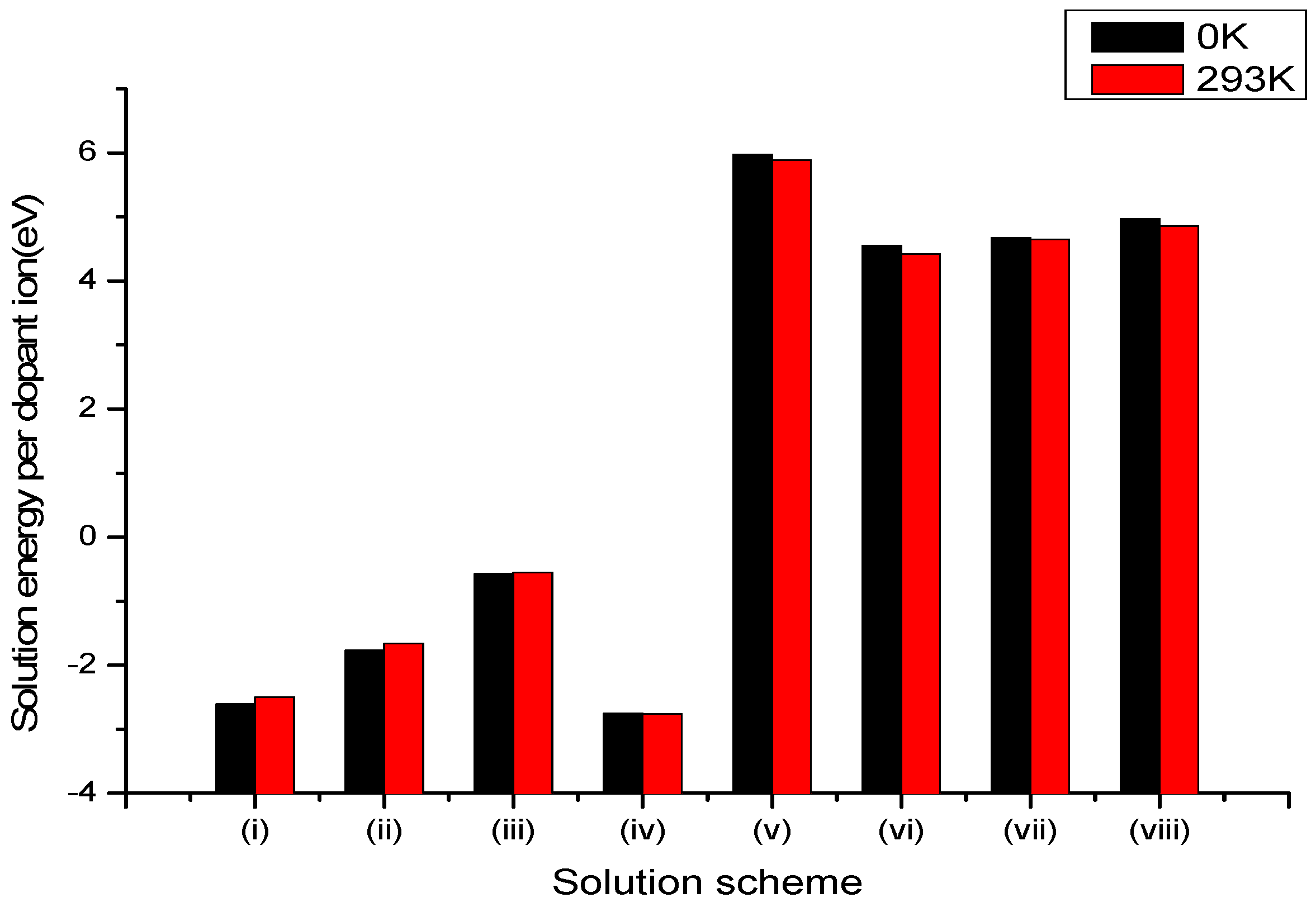
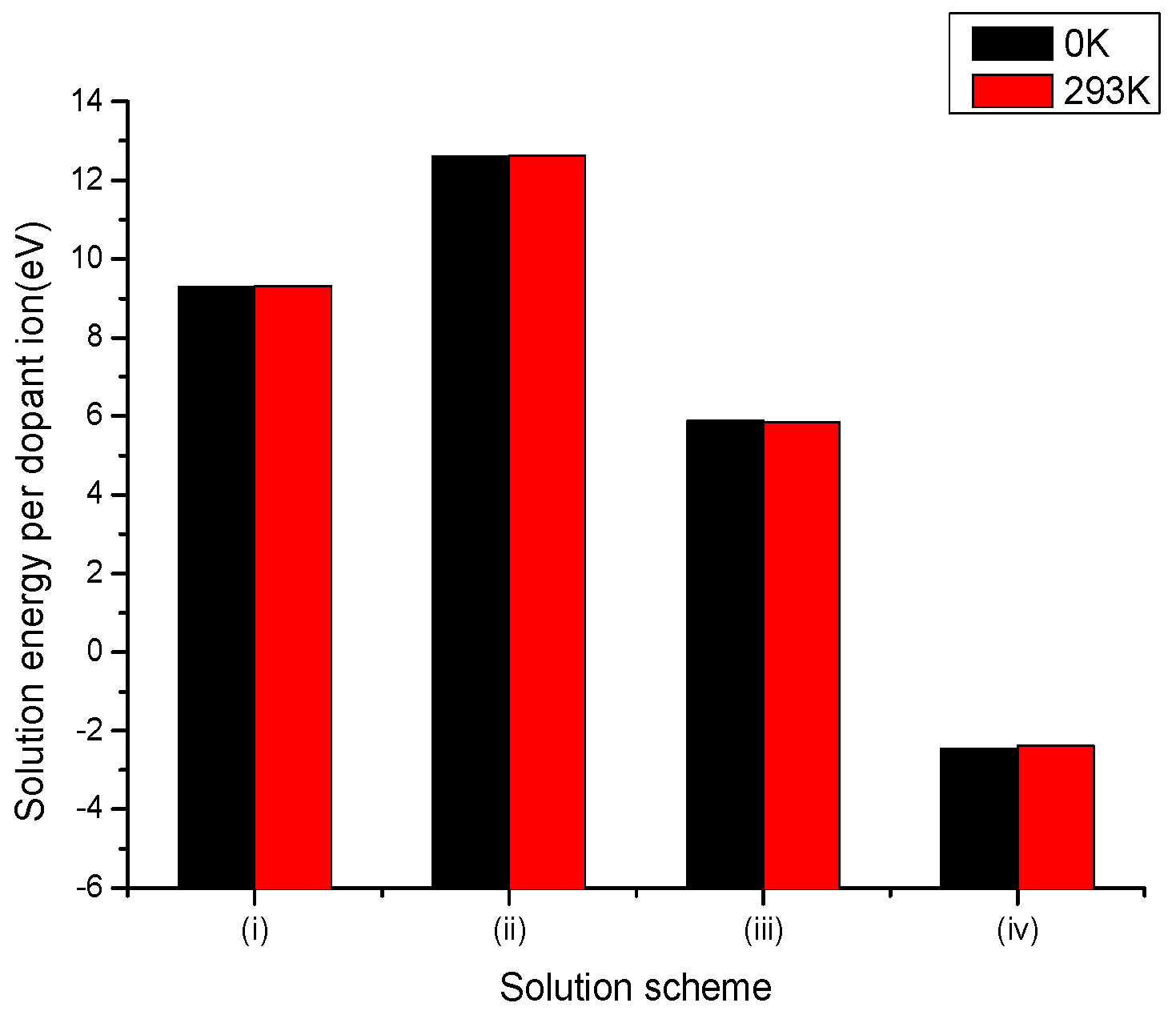
Figure 1.
Bar chart of solution energies vs. solution schemes for divalent dopant (V2+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 1.
Bar chart of solution energies vs. solution schemes for divalent dopant (V2+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
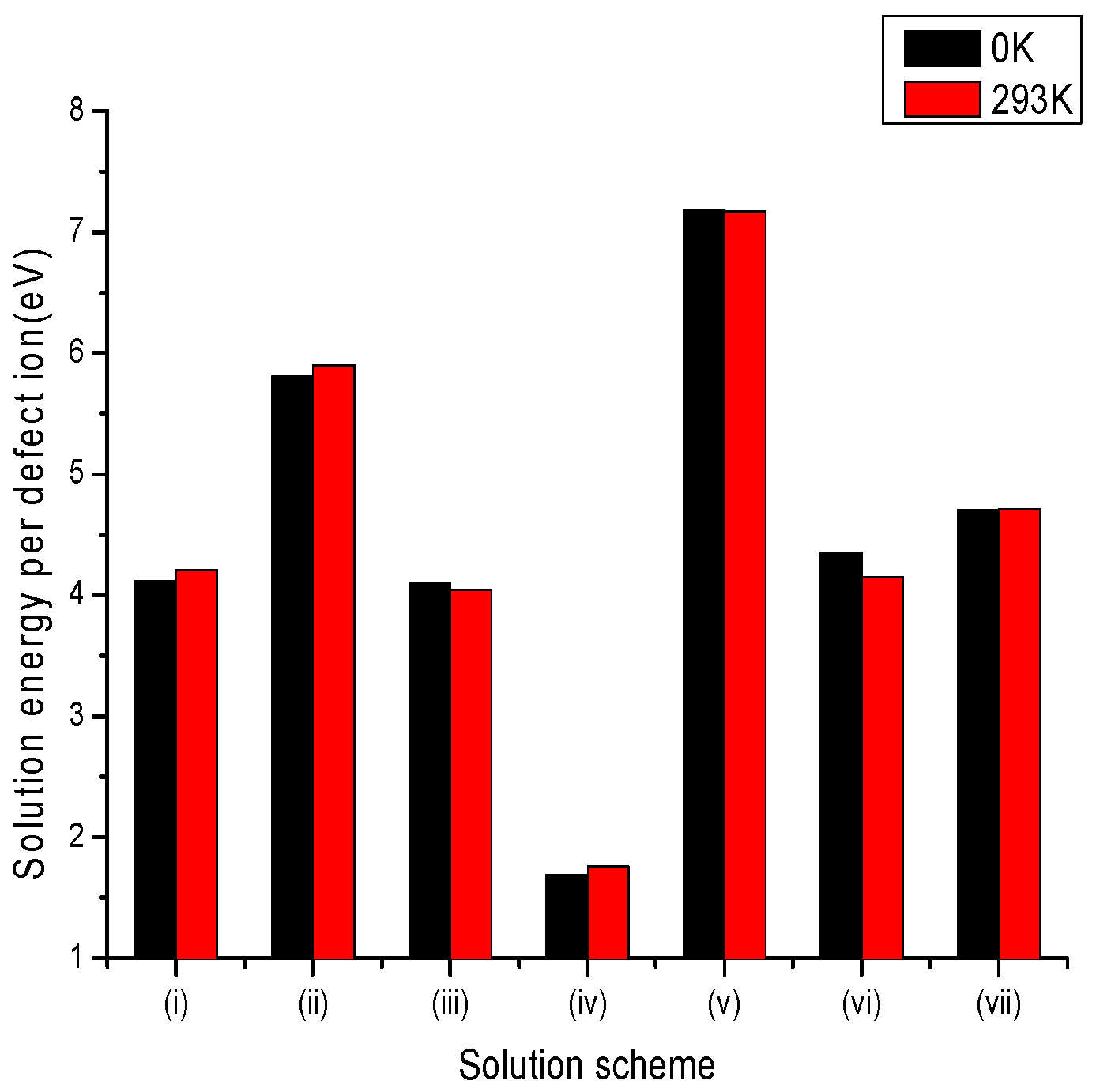
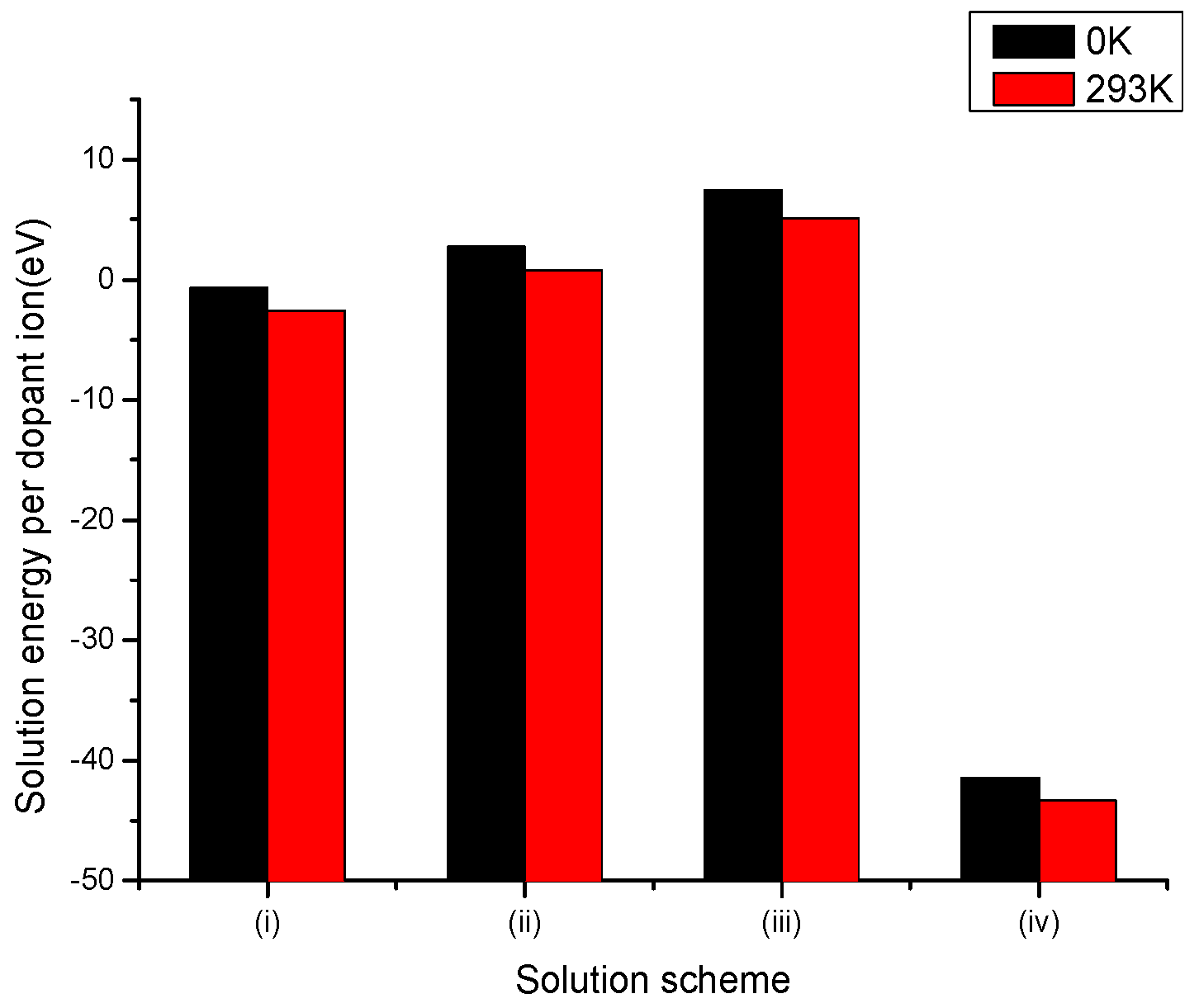
Figure 2.
Bar chart of solution energies vs. solution schemes for trivalent dopant (V3+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 2.
Bar chart of solution energies vs. solution schemes for trivalent dopant (V3+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
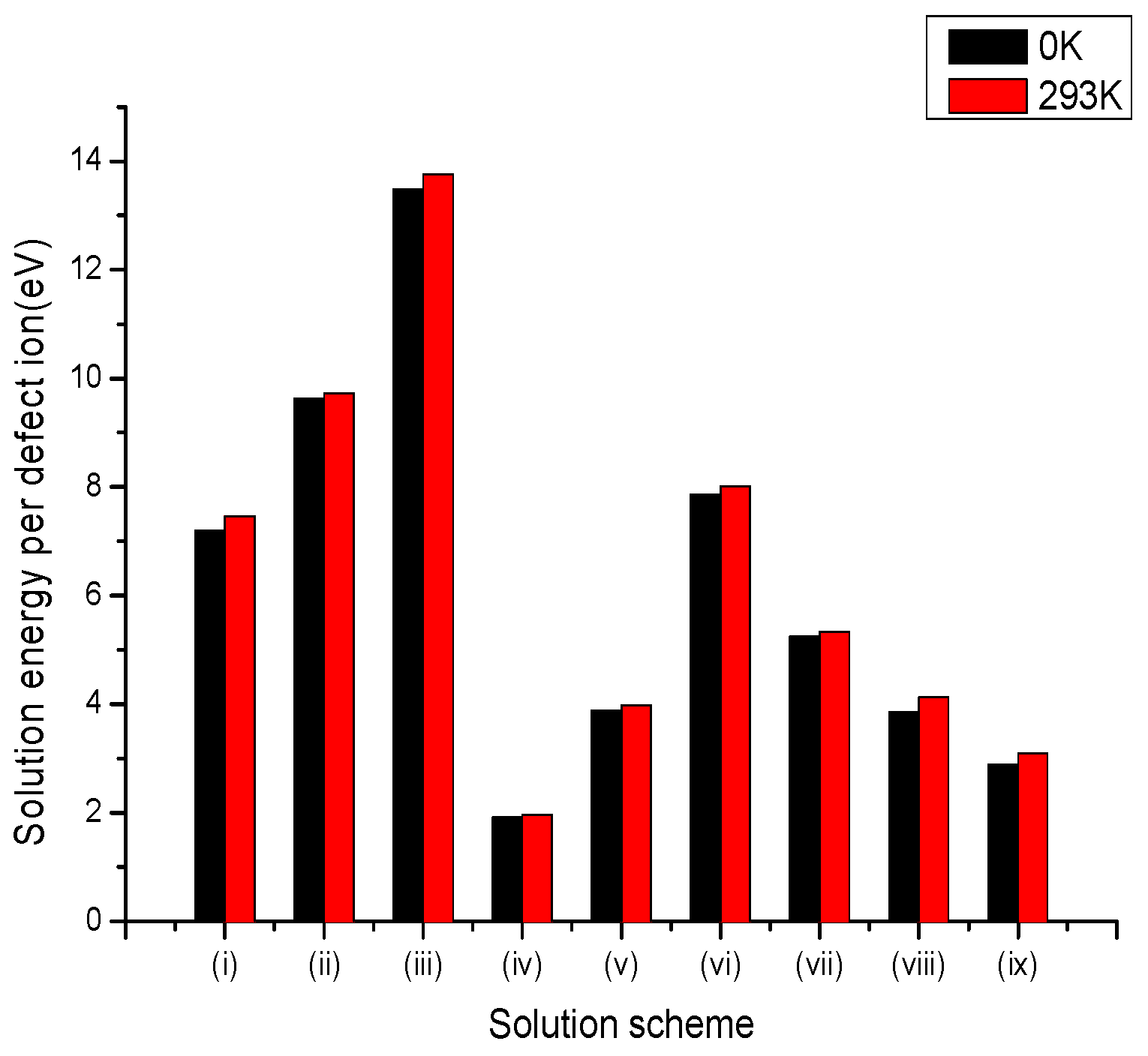
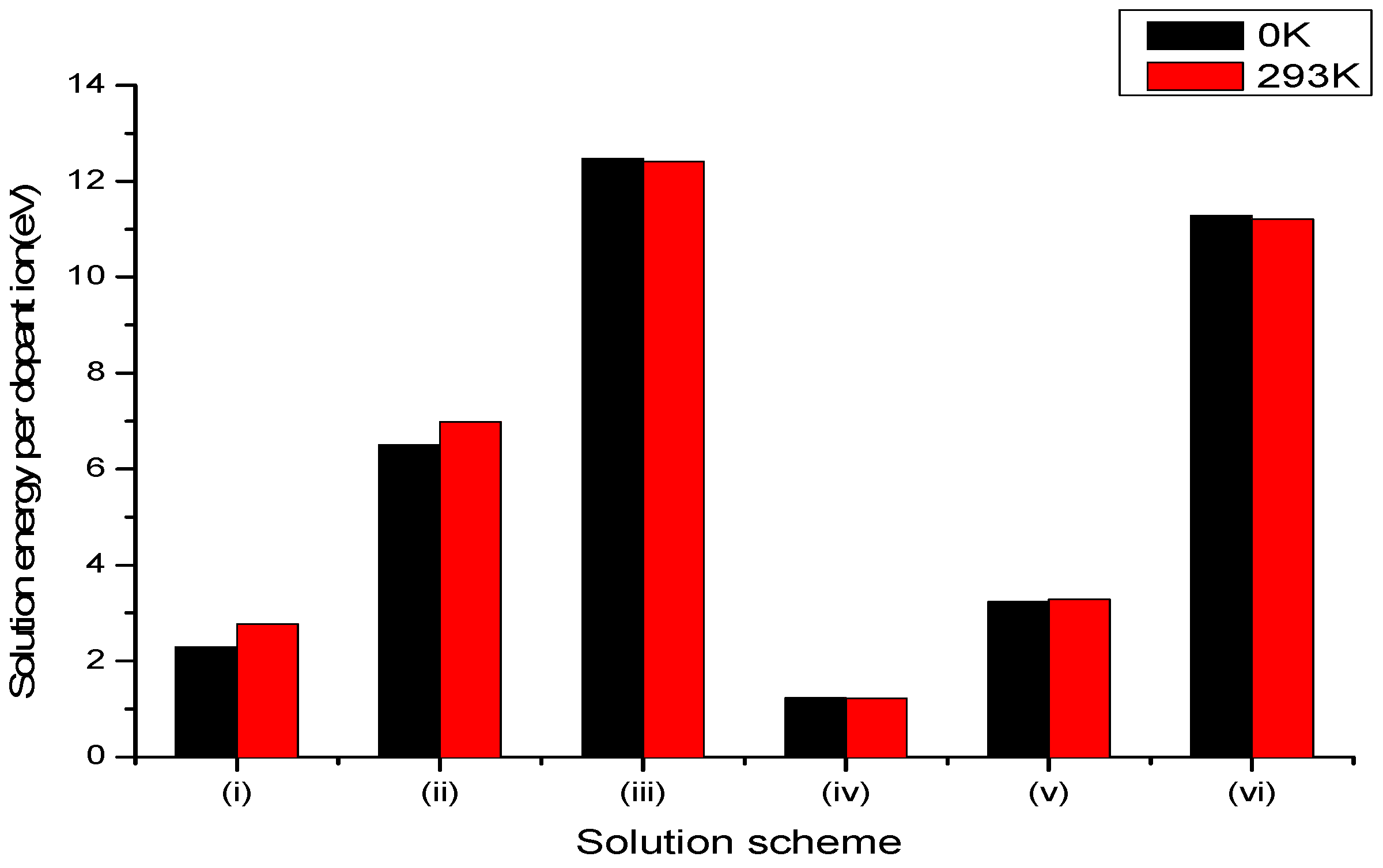
Figure 3.
Bar chart of solution energies vs. solution schemes for trivalent dopant (Mo3+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 3.
Bar chart of solution energies vs. solution schemes for trivalent dopant (Mo3+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
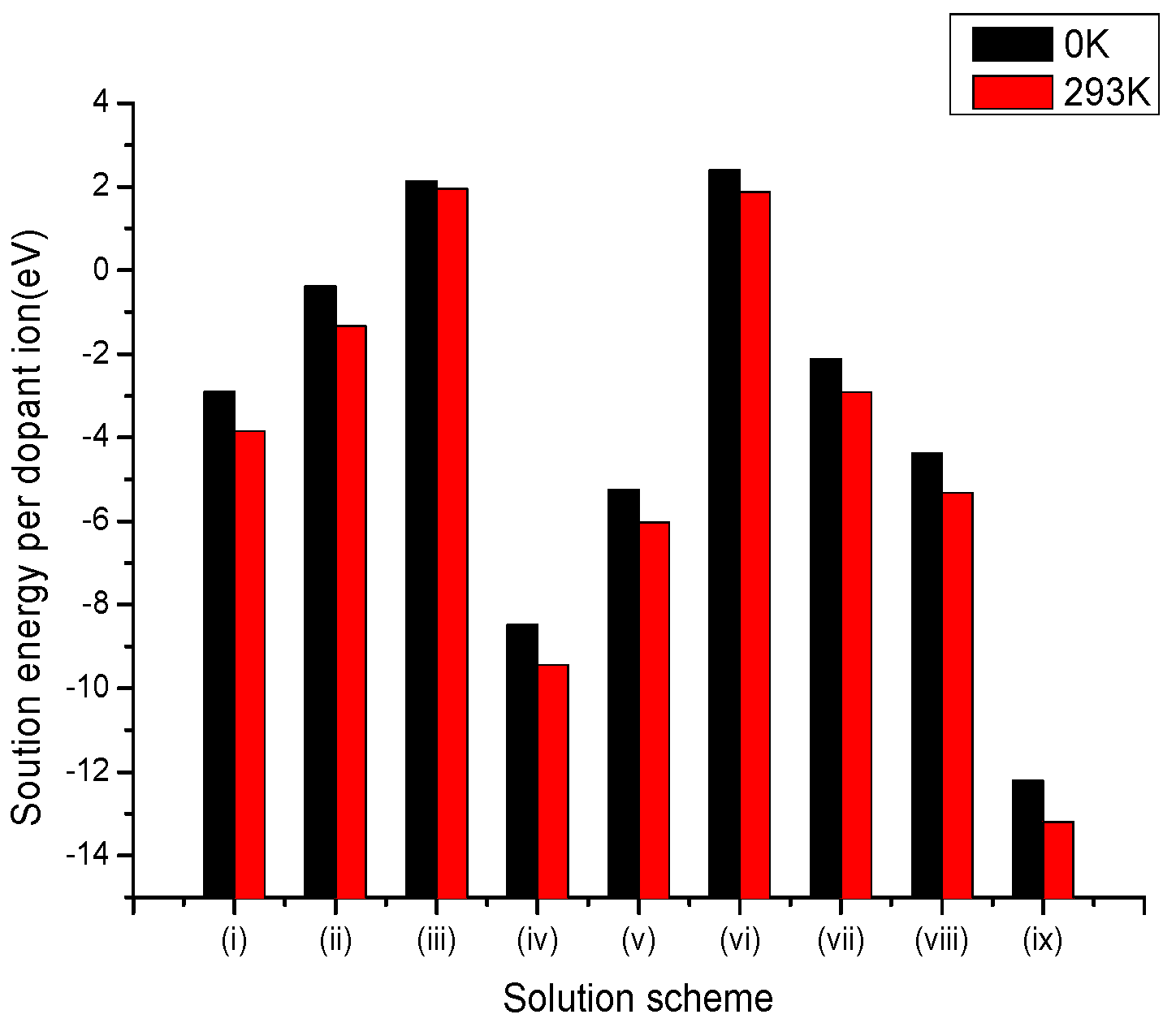
Figure 4.
Bar chart of solution energies vs. solution schemes for tetravalent dopant (V4+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 4.
Bar chart of solution energies vs. solution schemes for tetravalent dopant (V4+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 5.
Bar chart of solution energies vs. solution schemes for tetravalent dopant (Mo4+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 5.
Bar chart of solution energies vs. solution schemes for tetravalent dopant (Mo4+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 6.
Bar chart of solution energies vs. solution schemes for pentavalent dopant (V5+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 6.
Bar chart of solution energies vs. solution schemes for pentavalent dopant (V5+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 7.
Bar chart of solution energies vs. solution schemes for pentavalent dopant (Mo5+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 7.
Bar chart of solution energies vs. solution schemes for pentavalent dopant (Mo5+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 8.
Bar chart of solution energies vs. solution schemes for hexavalent dopant (Mo6+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Figure 8.
Bar chart of solution energies vs. solution schemes for hexavalent dopant (Mo6+) at the Li and Nb sites, considering the first neighbours in relation to the c axis.
Table 1.
Interionic potentials obtained from a fit to the VO, V2O3, VO2, V2O5, LiMoO2, Li2MoO3, Li3MoO4 and Li2MoO4 structures.
Table 1.
Interionic potentials obtained from a fit to the VO, V2O3, VO2, V2O5, LiMoO2, Li2MoO3, Li3MoO4 and Li2MoO4 structures.
| Interaction | Aij(eV) | ρij(Å) | Cij(Å6 eV) |
|---|
| Licore-Oshell | 950.0 | 0.2610 | 0.0 |
| Vcore-Oshell | 293.240087 | 0.475181 | 0.0 |
| Mocore-Licore | 573.532325 | 0.369602 | 0.0 |
| Mocore-O2−shell | 3003.79 | 0.3474 | 0.0 |
| Mocore-Ocore | 600.263736 | 0.328558 | 0.0 |
| O2−shell-O2−shell | 22764.0 | 0.1490 | 27.88 |
| Harmonic | k(eV Å2) | ro(Å) | |
| Vcore-Ocore | 46.997833 | 1.942956 | |
| Mocore-Ocore | 385.638986 | 2.073074 | |
| Species | | | Y(e) |
| Mocore | | | 3.0 4.0 5.0 6.0 |
| Vcore | | | 2.0 3.0 4.0 5.0 |
| Ocore | | | 0.9 |
| Oshell | | | −2.9 |
| Spring | | | k(Å−2 eV) |
| Ocore-Ooore | | | 70.0 |
Table 2.
Comparison of calculated (calc.) and experimental (expt.) lattice parameters.
Table 2.
Comparison of calculated (calc.) and experimental (expt.) lattice parameters.
| Oxide | Lattice Parameter | Exp. | Calc. (0 K) | Δ% | Calc. (293 K) | Δ% |
| VO | a(Å) = b(Å) = c(Å) | 4.067800 | 4.108237 | 0.99 | 4.10683 | 0.98 |
| V2O3 | a(Å) = b(Å) = c(Å) | 9.393000 | 9.304757 | 0.90 | 9.346331 | 0.94 |
| VO2 | a (Å) = b(Å) | 4.556100 | 4.569483 | 0.20 | 4.566212 | 0.22 |
| c(Å) | 2.859800 | 2.866421 | 0.23 | 2.857861 | 0.07 |
| V2O5 | a(Å) | 11.971900 | 11.99652 | 0.20 | 12.01247 | 0.33 |
| b(Å) | 4.701700 | 4.722561 | 0.44 | 4.660343 | 0.88 |
| c(Å) | 5.325300 | 5.355671 | 0.57 | 5.371149 | 0.86 |
| Lithium Molybdates | Lattice Parameter | Exp. | Calc. (0 K) | Δ% | Calc. (293 K) | Δ% |
| LiMoO2 | a(Å) = b(Å) | 2.866300 | 2.880528 | 0.50 | 2.887246 | 0.73 |
| c(Å) | 15.474300 | 15.409390 | 0.42 | 15.595024 | 0.78 |
| Li2MoO3 | a(Å) = b(Å) | 2.878000 | 2.854443 | 0.82 | 2.859809 | 0.63 |
| c(Å) | 14.91190 | 15.002886 | 0.61 | 15.04632 | 0.90 |
| Li3MoO4 | a(Å) = b(Å) = c(Å) | 4.1389 | 4.107762 | 0.75 | 4.106941 | 0.77 |
| Li2MoO4 | a(Å) = b(Å) | 14.330000 | 14.301305 | 0.20 | 14.384501 | 0.38 |
| c(Å) | 9.584 | 9.492067 | 0.96 | 9.632413 | 0.96 |
Table 3.
Types of defects considered due to M = V2+ incorporation in LiNbO3.
Table 3.
Types of defects considered due to M = V2+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Li+ and Nb5+ | Self-Compensation | (iv) |
| Nb5+ | Lithium Vacancies
and Anti-site () | (v) |
| | Anti-site () | (vi) |
| | | (vii) |
| | Oxygen Vacancies | (viii) |
Table 4.
Types of defects considered due to V3+ incorporation in LiNbO3.
Table 4.
Types of defects considered due to V3+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Li+ and Nb5+ | Self-Compensation | (iv) |
| Nb5+ | Oxygen Vacancies | (v) |
| | Anti-site () | (vi) |
| | Lithium Vacancies and Anti-site () | (vii) |
Table 5.
Types of defects considered due to Mo3+ incorporation in LiNbO3.
Table 5.
Types of defects considered due to Mo3+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Li+ and Nb5+ | Self-Compensation | (iv) |
| Nb5+ | Oxygen Vacancies | (v) |
| Nb5+ | Anti-site () | (vi) |
| Nb5+ | Lithium Vacancies and Anti-site () | (vii) |
Table 6.
Types of defects considered due to M = V4+ incorporation in LiNbO3.
Table 6.
Types of defects considered due to M = V4+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Li+ and Nb5+ | Self-Compensation | (iv) |
| Nb5+ | Anti-site () | (v) |
| | Lithium Vacancies and Anti-site () | (vi) |
| | | (vii) |
| | | (viii) |
| | Oxygen Vacancies | (ix) |
Table 7.
Types of defects considered due to M=Mo4+ incorporation in LiNbO3.
Table 7.
Types of defects considered due to M=Mo4+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Li+ and Nb5+ | Self-Compensation | (iv) |
| Nb5+ | Anti-site () | (v) |
| | Lithium Vacancies
and Anti-site () | (vi) |
| | | (vii) |
| | | (viii) |
| | Oxygen Vacancies | (ix) |
Table 8.
Types of defects considered due to M = V5+ incorporation in LiNbO3.
Table 8.
Types of defects considered due to M = V5+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Nb5+ | No Charge
Compensation | (iv) |
Table 9.
Types of defects considered due to Mo5+ incorporation in LiNbO3.
Table 9.
Types of defects considered due to Mo5+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Nb5+ | No Charge
Compensation | (iv) |
Table 10.
Types of defects considered due to Mo6+ incorporation in LiNbO3.
Table 10.
Types of defects considered due to Mo6+ incorporation in LiNbO3.
| Site | Charge Compensation | Reaction |
|---|
| Li+ | Lithium Vacancies | (i) |
| | Niobium Vacancies | (ii) |
| | Oxygen Interstitial | (iii) |
| Nb5+ | Lithium Vacancies | (iv) |
| | Niobium Vacancies | (v) |
| | Oxygen Interstitial | (vi) |
Table 11.
Lattice energies used in the solution energy calculations (eV).
Table 11.
Lattice energies used in the solution energy calculations (eV).
| Compound | Lattice Energy | Lattice Energy |
|---|
| 0 K | 293 K |
|---|
| LiNbO3 | −174.45 | −174.66 |
| Li2O | −33.16 | −32.92 |
| Nb2O5 | −314.37 | −313.39 |
| VO | −22.06 | −22.07 |
| V2O3 | −124.37 | −124.39 |
| VO2 | −111.54 | −111.57 |
| V2O5 | −315.65 | −274.18 |
| LiMoO2 | −98.07 | −97.09 |
| Li2MoO3 | −150.38 | −149.10 |
| Li3MoO4 | −181.28 | −178.88 |
| Li2MoO4 | −234.06 | −234.12 |

 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}