Chiral Conducting Me-EDT-TTF and Et-EDT-TTF-Based Radical Cation Salts with the Perchlorate Anion

 , , and
, , and
Abstract
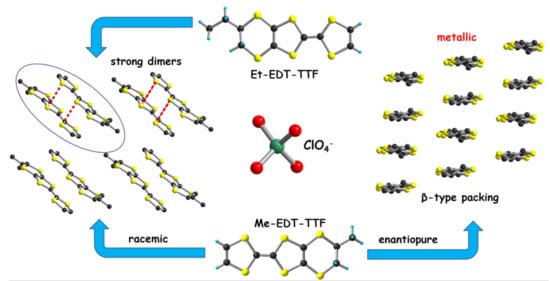
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Solid-State Structures of the Radical Cation Salts
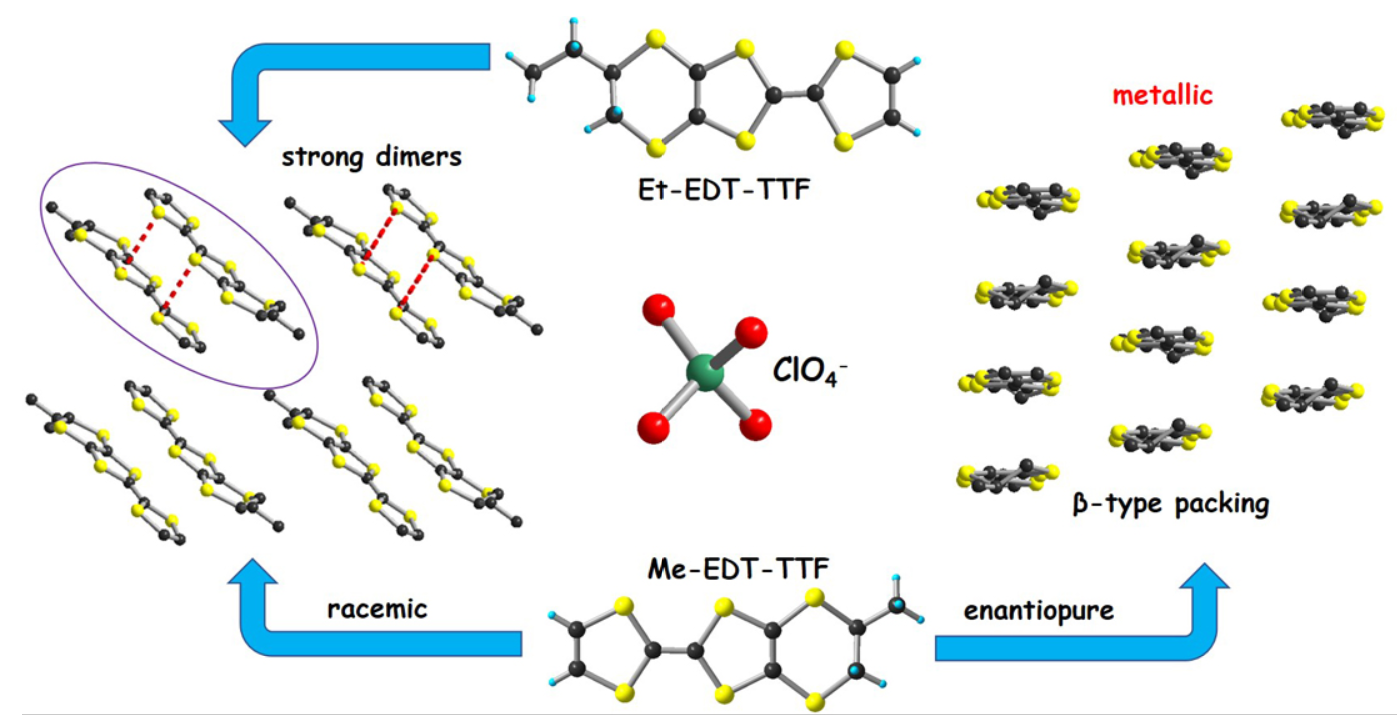
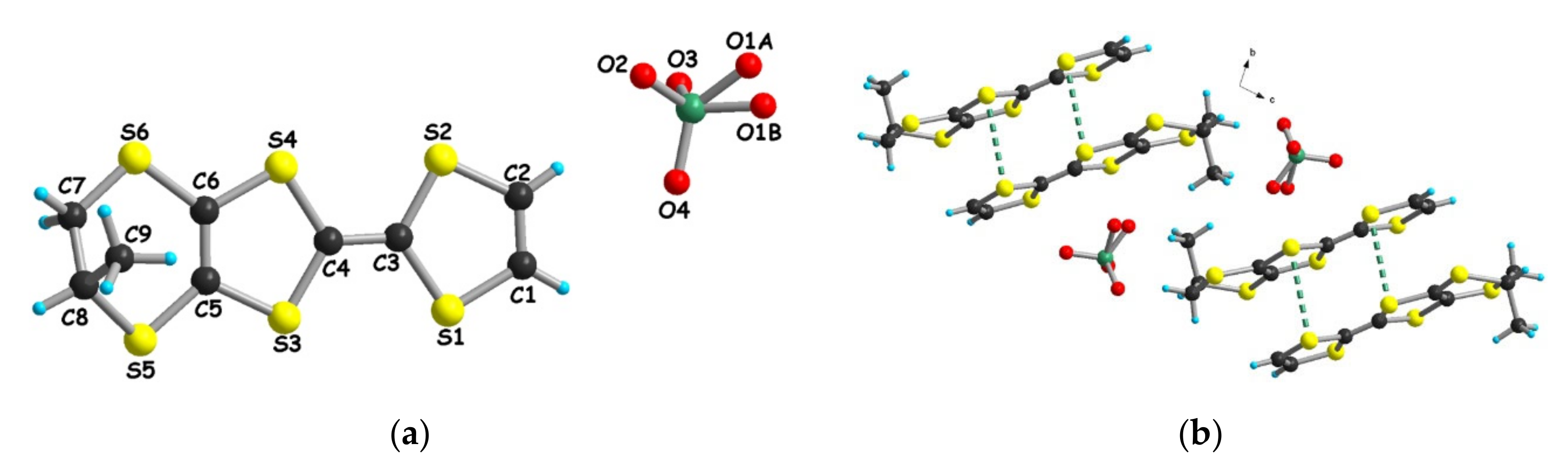
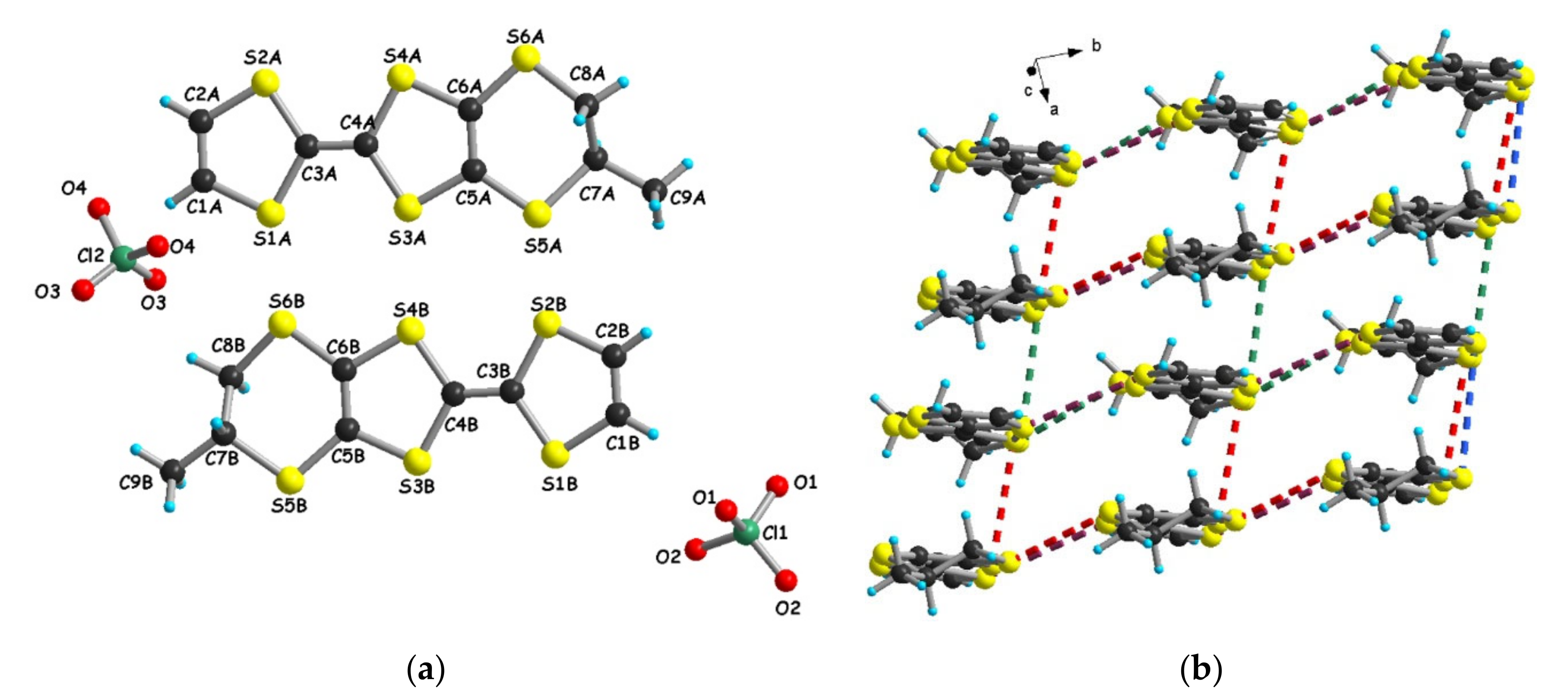
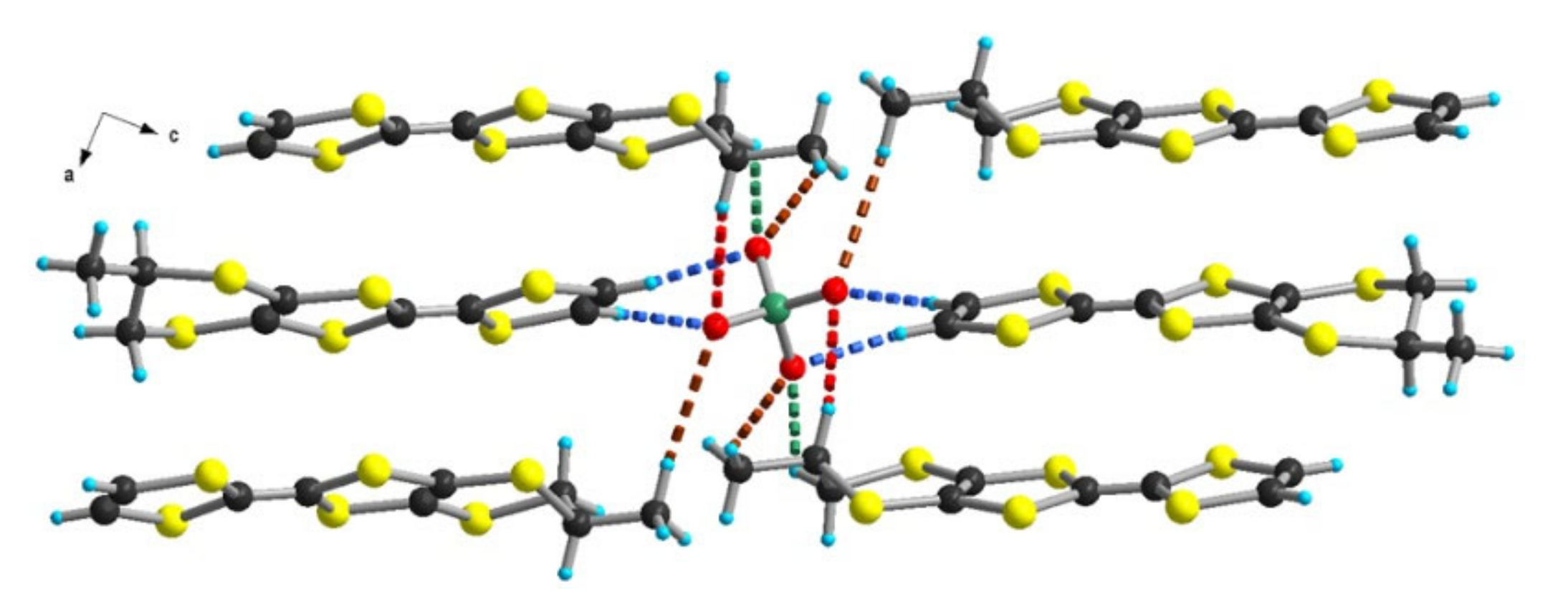
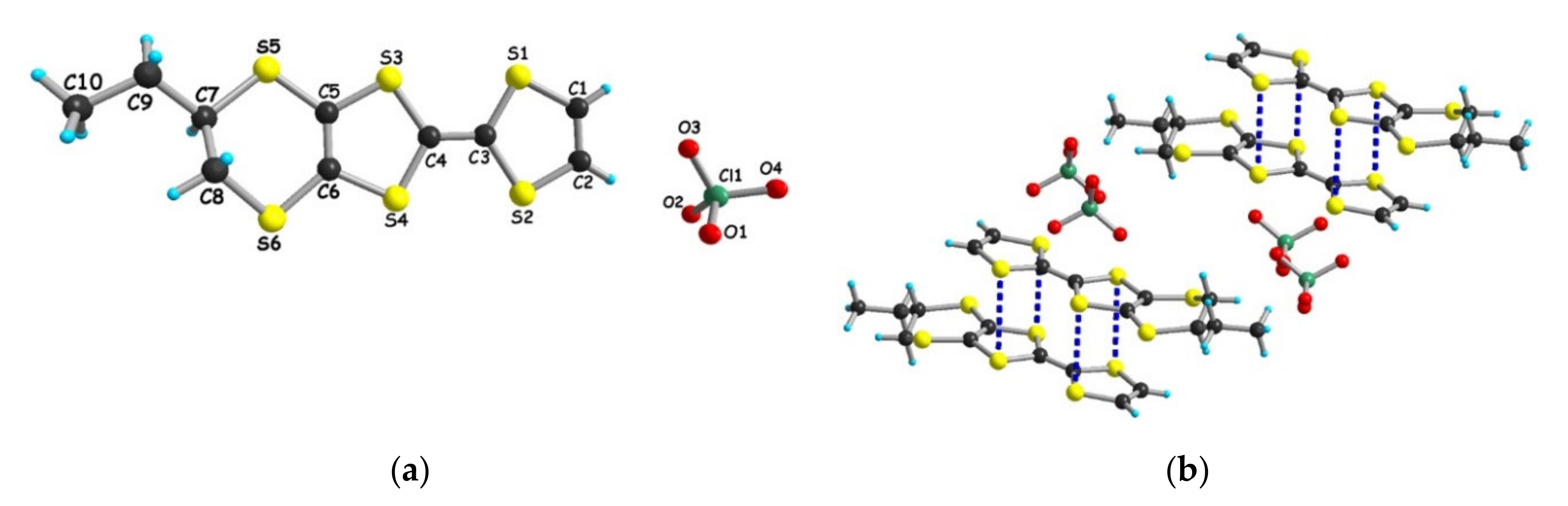
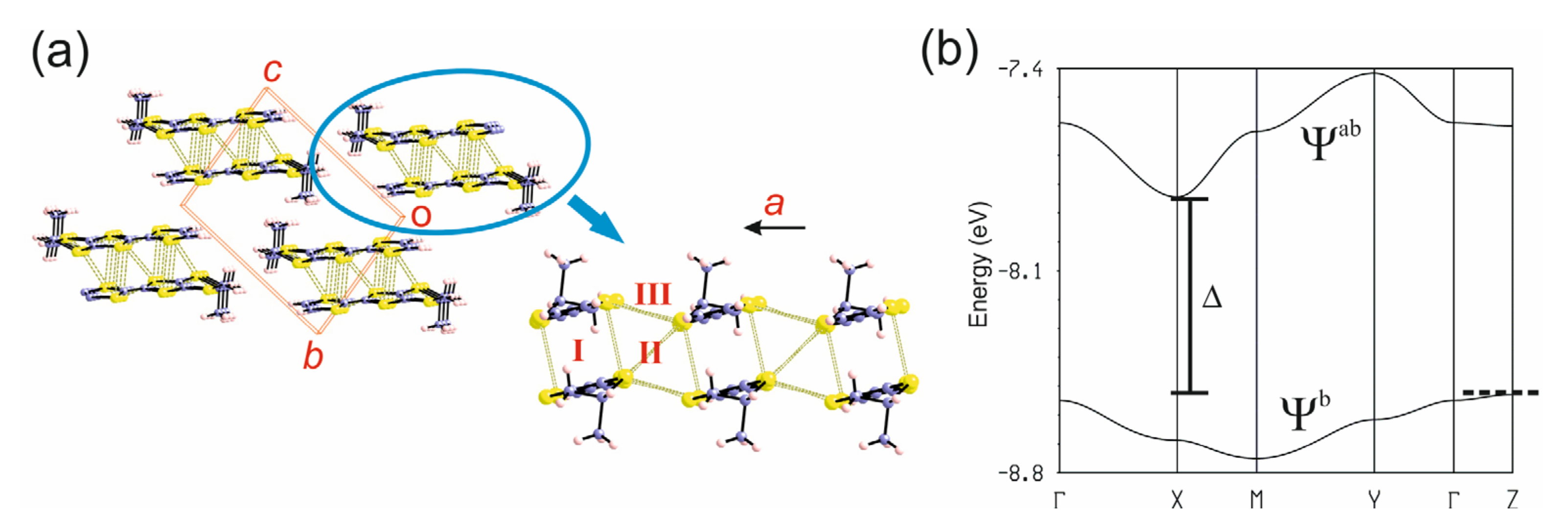
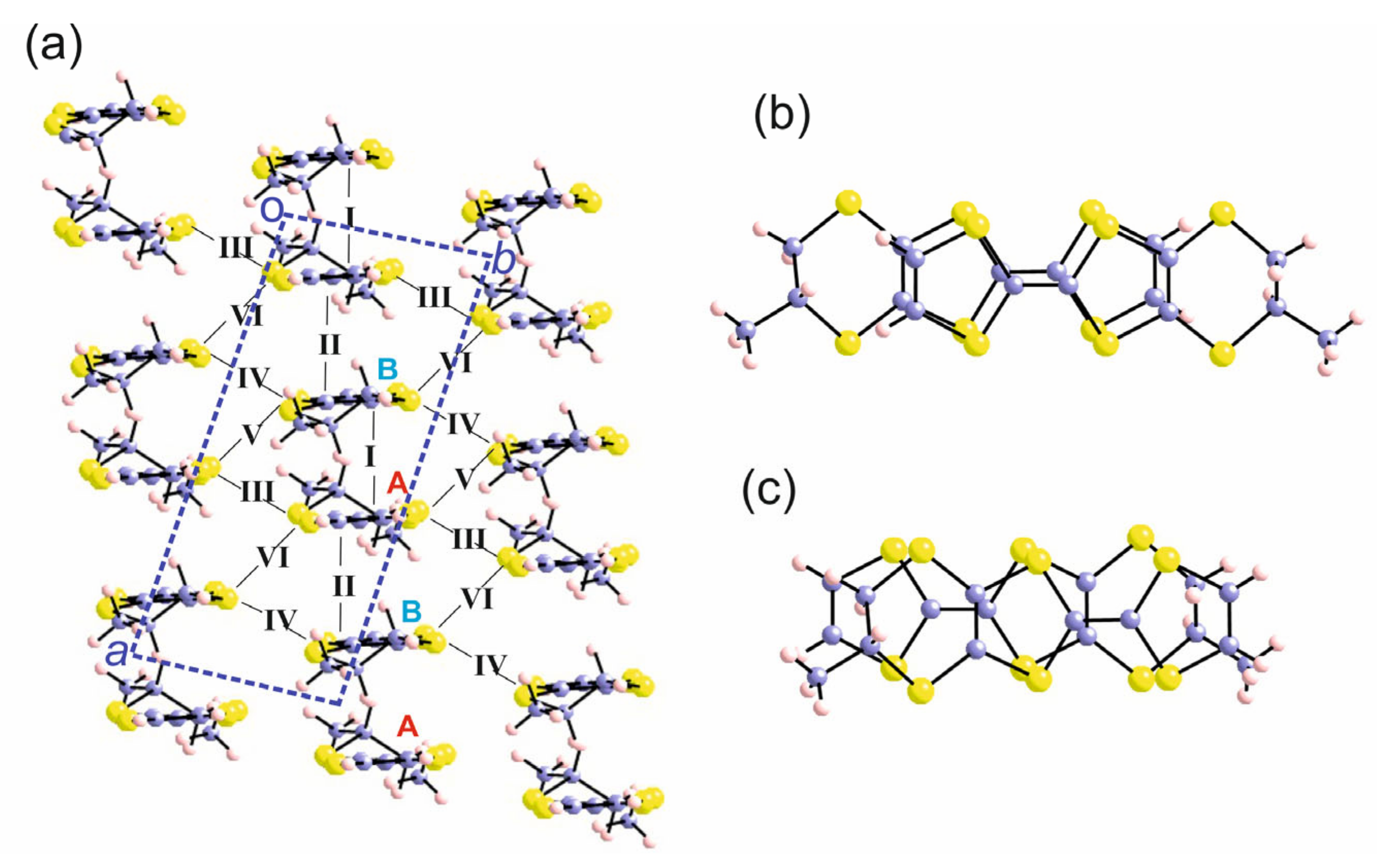
3.1.1. [(rac)-1]ClO4, [(R)-1]2ClO4 and [(S)-1]2ClO4
3.1.2. [(rac)-2]ClO4 and [(R)-2]ClO4
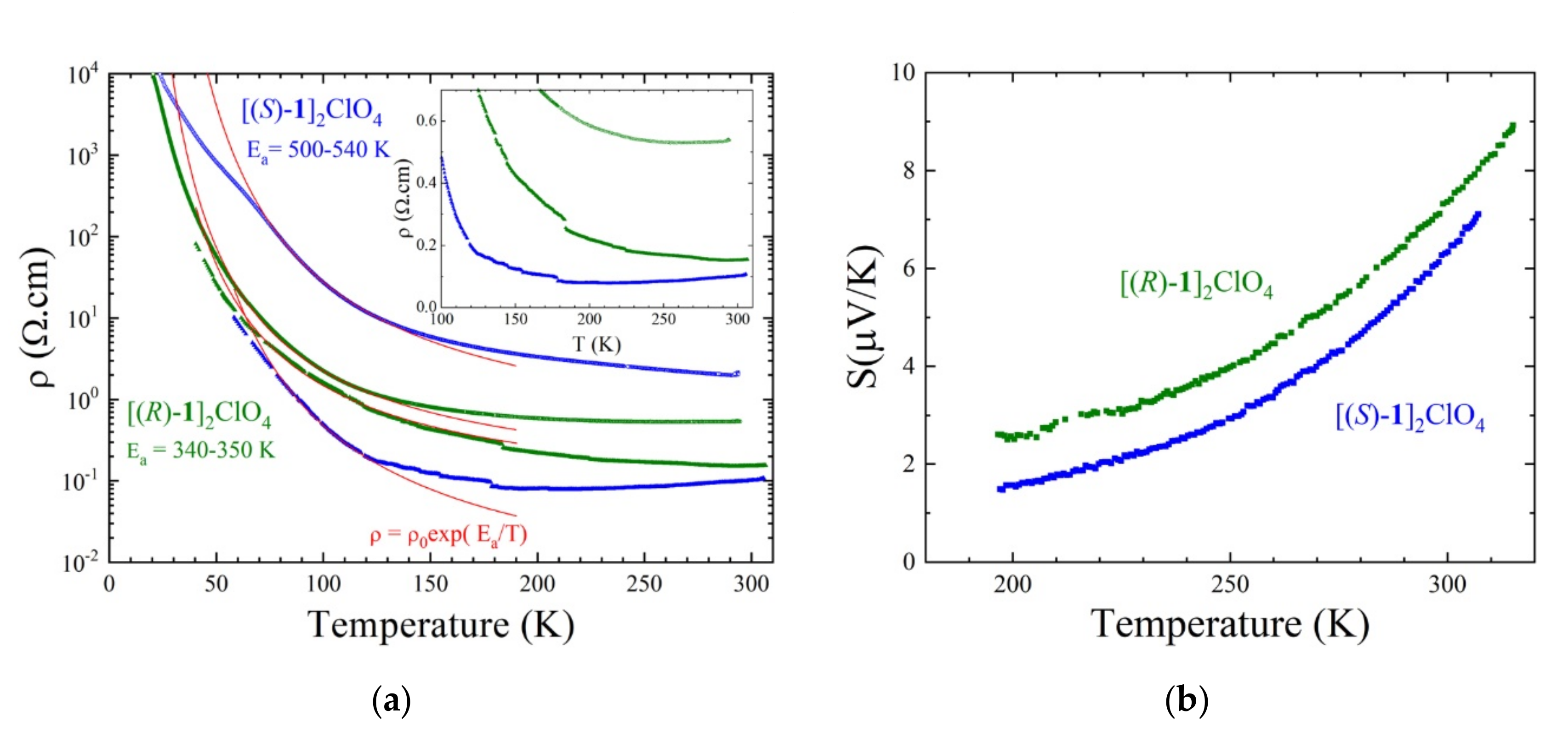
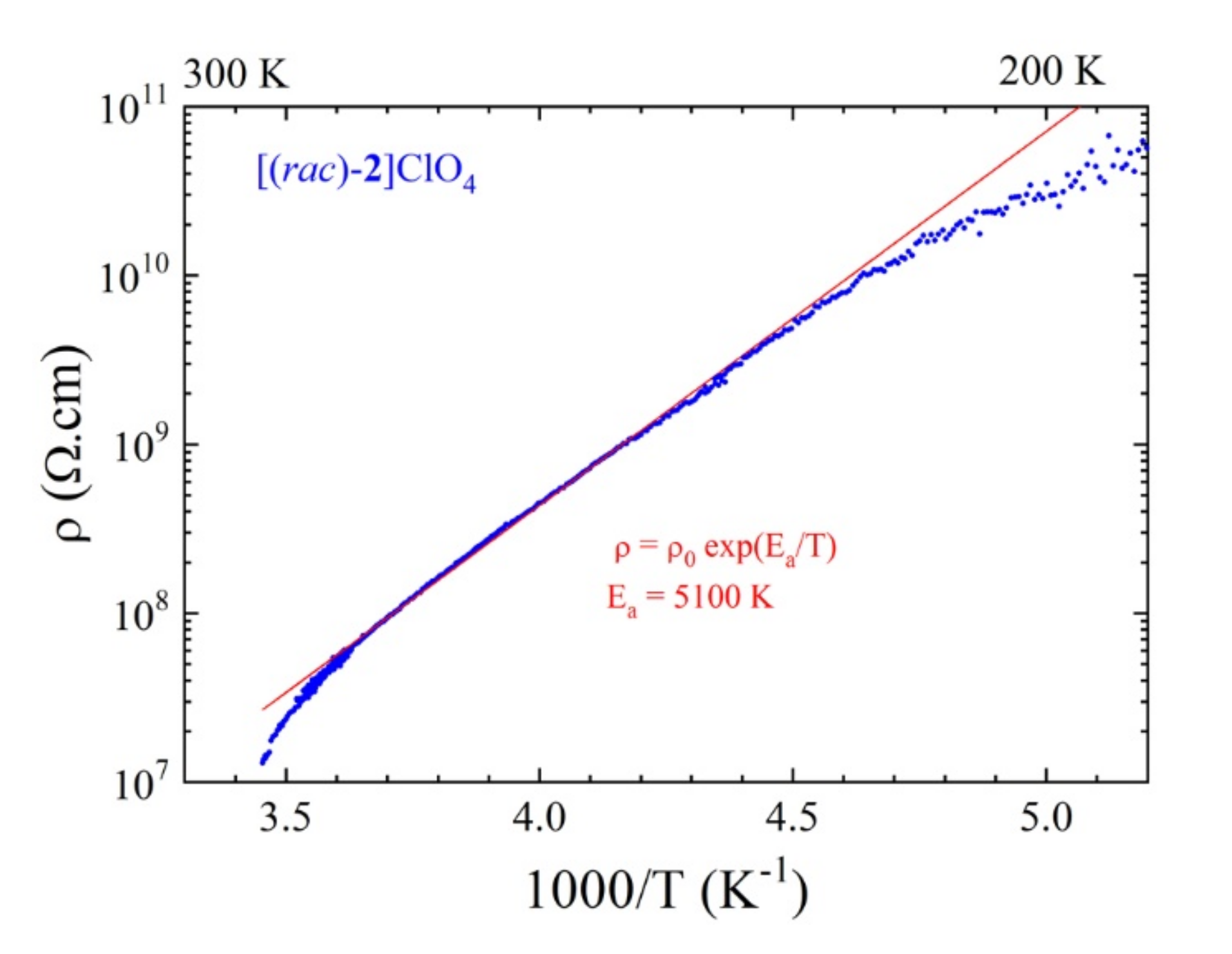
3.2. Single Crystal Conductivity Measurements
3.3. Band Structure Calculations
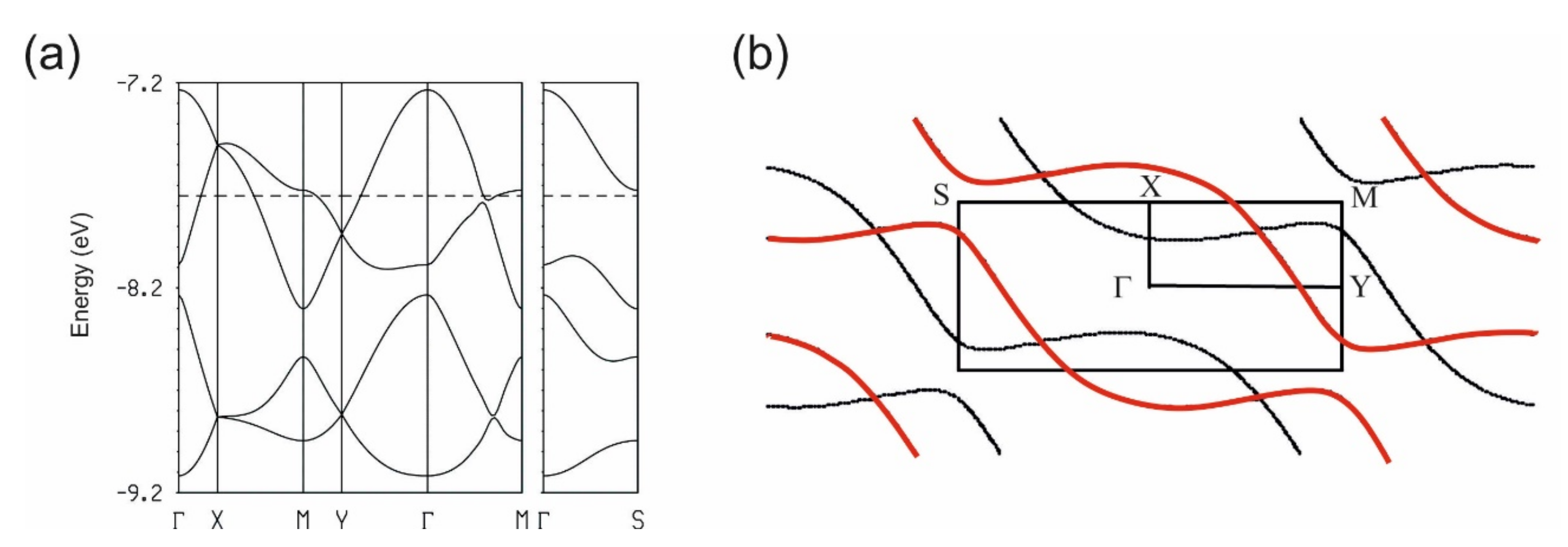
3.3.1. [(rac)-1]ClO4 and [(rac)-2]ClO4
3.3.2. [(R)-1]2ClO4 and [(S)-1]2ClO4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pop, F.; Zigon, N.; Avarvari, N. Main-Group-Based Electro- and Photoactive Chiral Materials. Chem. Rev. 2019, 119, 8435–8478. [Google Scholar] [CrossRef] [PubMed]
- Avarvari, N.; Wallis, J.D. Strategies towards Chiral Molecular Conductors. J. Mater. Chem. 2009, 19, 4061–4076. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Avarvari, N. Chiral Metal-Dithiolene Complexes. Coord. Chem. Rev. 2017, 346, 20–31. [Google Scholar] [CrossRef]
- Mroweh, N.; Auban-Senzier, P.; Vanthuyne, N.; Canadell, E.; Avarvari, N. Chiral EDT-TTF precursors with one stereogenic centre: Substituent size modulation of the conducting properties in the (R-EDT-TTF)2PF6 (R = Me or Et) series. J. Mater. Chem. C 2019, 7, 12664–12673. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Auban-Senzier, P.; Frąckowiak, A.; Ptaszyński, K.; Olejniczak, I.; Wallis, J.D.; Canadell, E.; Avarvari, N. Chirality Driven Metallic versus Semiconducting Behavior in a Complete Series of Radical Cation Salts Based on Dimethyl-Ethylenedithio-Tetrathiafulvalene (DM-EDT-TTF). J. Am. Chem. Soc. 2013, 135, 17176–17186. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Avarvari, N. Anion size control of the packing in the metallic versus semiconducting chiral radical cation salts (DM-EDT-TTF)2XF6 (X = P, As, Sb). Chem. Commun. 2016, 52, 12438–12441. [Google Scholar] [CrossRef] [Green Version]
- Mroweh, N.; Pop, F.; Mézière, C.; Allain, M.; Auban-Senzier, P.; Vanthuyne, N.; Alemany, P.; Canadell, E.; Avarvari, N. Combining chirality and hydrogen bonding in methylated ethylenedithio-tetrathiafulvalene primary diamide precursors and radical cation salts. Cryst. Growth Des. 2020, 20, 2516–2526. [Google Scholar] [CrossRef]
- Wallis, J.D.; Karrer, A.; Dunitz, J.D. Chiral metals? A chiral substrate for organic conductors and superconductors. Helv. Chim. Acta 1986, 69, 69–70. [Google Scholar] [CrossRef]
- Mroweh, N.; Mézière, C.; Allain, M.; Auban-Senzier, P.; Canadell, E.; Avarvari, N. Conservation of structural arrangements and 3:1 stoichiometry in a series of crystalline conductors of TMTTF, TMTSF, BEDT-TTF, and chiral DM-EDT-TTF with the oxo-bis[pentafluorotantalate(V)] dianion. Chem. Sci. 2020, 11, 10078–10091. [Google Scholar] [CrossRef]
- Matsumiya, S.; Izuoka, A.; Sugawara, T.; Taruishi, T.; Kawada, Y. Effect of Methyl Substitution on Conformation and Molecular Arrangement of BEDT-TTF Derivatives in the Crystalline Environment. Bull. Chem. Soc. Jpn. 1993, 66, 513–522. [Google Scholar] [CrossRef]
- Matsumiya, S.; Izuoka, A.; Sugawara, T.; Taruishi, T.; Kawada, Y.; Tokumoto, M. Crystal Structure and Conductivity of Chiral Radical Ion Salts (Me2ET)2X. Bull. Chem. Soc. Jpn. 1993, 66, 1949–1954. [Google Scholar] [CrossRef]
- Pop, F.; Allain, M.; Auban-Senzier, P.; Martínez-Lillo, J.; Lloret, F.; Julve, M.; Canadell, E.; Avarvari, N. Enantiopure Conducting Salts of Dimethylbis(ethylenedithio)tetrathiafulvalene (DM-BEDTTTF) with the Hexachlororhenate(IV) Anion. Eur. J. Inorg. Chem. 2014, 2014, 3855–3862. [Google Scholar] [CrossRef]
- Karrer, A.; Wallis, J.D.; Dunitz, J.D.; Hilti, B.; Mayer, C.W.; Bürkle, M.; Pfeiffer, J. Structures and Electrical Properties of Some New Organic Conductors Derived from the Donor Molecule TMET (S,S,S,S-Bis(dimethylethylenedithio) tetrathiafulvalene). Helv. Chim. Acta 1987, 70, 942–953. [Google Scholar] [CrossRef]
- Pop, F.; Laroussi, S.; Cauchy, T.; Gómez-García, C.J.; Wallis, J.D.; Avarvari, N. Tetramethyl-Bis(ethylenedithio)-Tetrathiafulvalene (TM-BEDT-TTF) Revisited: Crystal Structures, Chiroptical Properties, Theoretical Calculations, and a Complete Series of Conducting Radical Cation Salts. Chirality 2013, 25, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Pop, F.; Melan, C.; Brooks, A.C.; Martin, L.; Horton, P.; Auban-Senzier, P.; Rikken, G.L.J.A.; Avarvari, N.; Wallis, J.D. Charge transfer complexes and radical cation salts of chiral methylated organosulfur donors. CrystEngComm 2014, 16, 3906–3916. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Batail, P.; Avarvari, N. Enantiopure Radical Cation Salt Based on Tetramethyl-bis(ethylenedithio)-tetrathiafulvalene and Hexanuclear Rhenium Cluster. Crystals 2016, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Rikken, G.L.J.A.; Avarvari, N. Electrical magneto-chiral anisotropy in a bulk chiral molecular conductor. Nat. Commun. 2014, 5, 3757. [Google Scholar] [CrossRef] [Green Version]
- Kini, A.M.; Parakka, J.P.; Geiser, U.; Wang, H.-H.; Rivas, F.; DiNino, E.; Thomas, S.; Dudek, J.D.; Williams, J.M. Tetraalkyl- and dialkyl-substituted BEDT-TTF derivatives and their cation-radical salts: Synthesis, structure, and properties. J. Mater. Chem. 1999, 9, 883–892. [Google Scholar] [CrossRef]
- Zambounis, J.S.; Mayer, C.W.; Hauenstein, K.; Hilti, B.; Hofherr, W.; Pfeiffer, J.; Bürkle, M.; Rihs, G. Crystal Structure and Electrical Properties of κ-((S,S)-DMBEDT-TTF)2ClO4. Adv. Mater. 1992, 4, 33–35. [Google Scholar] [CrossRef]
- Mroweh, N.; Mézière, C.; Pop, F.; Auban-Senzier, P.; Alemany, P.; Canadell, E.; Avarvari, N. In Search of Chiral Molecular Superconductors: κ-[(S,S)-DM-BEDT-TTF]2ClO4 Revisited. Adv. Mater. 2020, 32, 2002811. [Google Scholar] [CrossRef]
- Chaikin, P.M.; Kwak, J.F. Apparatus for thermopower measurements on organic conductors. Rev. Sci. Instrum. 1975, 46, 218–220. [Google Scholar] [CrossRef]
- Almeida, M.; Alcácer, L.; Oostra, S. Anisotropy of thermopower in N-methyl-N-ethylmorpholinium bistetracyanoquinodimethane, MEM(TCNQ)2, in the region of the high-temperature phase transitions. Phys. Rev. B 1984, 30, 2839–2844. [Google Scholar] [CrossRef]
- Lopes, E.B. INETI-Sacavém; Internal Report; INETI Press: Sacavém, Portugal, 1991. [Google Scholar]
- Huebener, R.P. Thermoelectric Power of Lattice Vacancies in Gold. Phys. Rev. 1964, 135, A1281–A1291. [Google Scholar] [CrossRef]
- Whangbo, M.-H.; Hoffmann, R. The Band Structure of the Tetracyanoplatinate Chain. J. Am. Chem. Soc. 1978, 100, 6093–6098. [Google Scholar] [CrossRef]
- Ammeter, J.H.; Bürgi, H.-B.; Thibeault, J.; Hoffmann, R. Counterintuitive Orbital Mixing in Semiempirical and ab Initio Molecular Orbital Calculations. J. Am. Chem. Soc. 1978, 100, 3686–3692. [Google Scholar] [CrossRef]
- Pénicaud, A.; Boubekeur, K.; Batail, P.; Canadell, E.; Auban-Senzier, P.; Jérome, D. Hydrogen-Bond Tuning of Macroscopic Transport Properties from the Neutral Molecular Component Site along the Series of Metallic Organic-Inorganic Solvates (BEDT-TTF)4Re6Se5C19·[guest], [guest = DMF, THF, dioxane]. J. Am. Chem. Soc. 1993, 115, 4101–4112. [Google Scholar] [CrossRef]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors I. Parallel Molecules: β and β” Phases. Bull. Chem. Soc. Jpn. 1998, 71, 2509–2526. [Google Scholar] [CrossRef]
- Whangbo, M.-H.; Williams, J.M.; Leung, P.C.W.; Beno, M.A.; Emge, T.J.; Wang, H.H. Role of the Intermolecular Interactions in the Two-Dimensional Ambient-Pressure Organic Superconductors β-(ET)2I3 and β-(ET)2IBr2. Inorg. Chem. 1985, 24, 3500–3502. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | [(rac)-1]ClO4 | [(R)-1]2ClO4 | [(S)-1]2ClO4 |
|---|---|---|---|
| formula | C9H8ClO4S6 | C18H16ClO4S12 | C18H16ClO4S12 |
| M (g mol−1) | 407.96 | 716.48 | 716.48 |
| (T) | 150.00(10) | 155.00(7) | 170.58(10) |
| crystal system | Triclinic | Monoclinic | Monoclinic |
| space group | P-1 | C2 | C2 |
| a (Å) | 6.4126(4) | 15.1135(5) | 15.1561 (5) |
| b (Å) | 9.4439(6) | 6.5681(2) | 6.5791(2) |
| c (Å) | 12.2713(8) | 26.6686(9) | 26.6154(9) |
| α (°) | 97.293(5) | 90 | 90 |
| β (°) | 94.927(5) | 091.898(3) | 91.828(3) |
| γ (°) | 96.828(5) | 90 | 90 |
| V (Å3) | 728.05(8) | 2645.86(15) | 2652.56(15) |
| Z | 2 | 4 | 4 |
| ρcalcd (g cm−3) | 1.861 | 1.799 | 1.794 |
| μ (mm−1) | 10.464 | 10.390 | 10.364 |
| Flack parameter | - | 0.06(2) | 0.04(4) |
| Completeness (%) | 99.1% | 99.5% | 99.8% |
| goodness-of-fit on F2 | 1.061 | 1.081 | 1.040 |
| final R1/wR2 [I > 2σ(I)] | R1 = 0.0676, wR2 = 0.1781 | R1 = 0.0611, wR2 = 0.1583 | R1 = 0.0515, wR2 = 0.1331 |
| R1/wR2 (all data) | R1 = 0.0809, wR2 = 0.1903 | R1 = 0.0629, wR2 = 0.1597 | R1 = 0.0560, wR2 = 0.1361 |
| Compound | [(rac)-2]ClO4 | [(R)-2]ClO4 |
|---|---|---|
| formula | C10H10ClO4S6 | C10H10ClO4S6 |
| M (g mol−1) | 421.99 | 421.99 |
| T (K) | 150.00(10) | 150.00(10) |
| crystal system | Triclinic | Triclinic |
| space group | P-1 | P1 |
| a (Å) | 7.5501(9) | 7.5729(5) |
| b (Å) | 9.3157(10) | 9.1781(6) |
| c (Å) | 11.7417(11) | 11.8048(9) |
| α (°) | 80.593(9) | 80.392(6) |
| β (°) | 75.196(11) | 76.155(6) |
| γ (°) | 79.910(9) | 81.022(6) |
| V (Å3) | 779.95(14) | 779.68(9) |
| Z | 2 | 2 |
| ρcalcd (g cm−3) | 1.797 | 1.797 |
| μ (mm−1) | 9.791 | 9.794 |
| Flack parameter | - | 0.10(6) |
| Completeness (%) | 99.5% | 97.1% |
| goodness-of-fit on F2 | 1.047 | 1.049 |
| final R1/wR2 [I > 2σ(I)] | R1 = 0.0662, wR2 = 0.1751 | R1 = 0.0531, wR2 = 0.1423 |
| R1/wR2 (all data) | R1 = 0.0795, wR2 = 0.1981 | R1 = 0.0574, wR2 = 0.1487 |
| Bond Lengths (Å) | |||||
|---|---|---|---|---|---|
| A | compound | [(R)-1]2ClO4 | [(S)-1]2ClO4 | [(rac)-1]ClO4 | |
| C3A—C4A | 1.385(13) | 1.372(8) | C3—C4 | 1.390(8) | |
| S1A—C3A | 1.738(10) | 1.746(6) | S1—C3 | 1.721(6) | |
| S2A—C3A | 1.731(10) | 1.729(6) | S2—C3 | 1.715(6) | |
| S3A—C4A | 1.720(9) | 1.749(6) | S3—C4 | 1.710(6) | |
| S4A—C4A | 1.730(10) | 1.730(6) | S4—C4 | 1.722(5) | |
| B | C3B—C4B | 1.368(13) | 1.392(7) | -- | - |
| S1B—C3B | 1.744(9) | 1.734(6) | -- | - | |
| S2B—C3B | 1.736(10) | 1.733(6) | - | - | |
| S3B—C4B | 1.750(10) | 1.711(5) | -- | - | |
| S4B—C4B | 1.733(10) | 1.726(6) | - | - | |
| Interaction | S···S (Å) | |βHOMO-HOMO| (eV) |
|---|---|---|
| I | 3.385 (×2), 3.466 (×2) | 0.9401 |
| II | 3.518 (×2), 3.793 | 0.0207 |
| II | 3.394, 3.497, 3.839, 3.849, 3.877 | 0.0401 |
| Interaction | [(R)-1]2ClO4 | [(S)-1]2ClO4 |
|---|---|---|
| I (A-B) | 0.7880 | 0.7865 |
| II (A-B) | 0.6366 | 0.6285 |
| III (A-A) | 0.0303 | 0.0290 |
| IV (B-B) | 0.0370 | 0.0375 |
| V (A-B) | 0.0774 | 0.0765 |
| VI (A-B) | 0.1012 | 0.0945 |
| Radical Cation Salt | Stoichiometry (Donor:Anion) | Crystal System | Conductivity (High Temperature) |
|---|---|---|---|
| [(rac)-1]ClO4 | 1:1 | Triclinic | Poor semiconductor |
| [(R)-1]2ClO4 | 2:1 | Monoclinic | Metal |
| [(S)-1]2ClO4 | 2:1 | Monoclinic | Metal |
| [(rac)-1]2PF6 | 2:1 | Triclinic | Metal |
| [(R)-1]2PF6 | 2:1 | Triclinic | Metal |
| [(S)-1]2PF6 | 2:1 | Triclinic | Metal |
| [(rac)-2]ClO4 | 1:1 | Triclinic | Poor semiconductor |
| [(R)-2]ClO4 | 1:1 | Triclinic | Poor semiconductor |
| [(rac)-2]PF6•(C4H8O) | 1:1 | Triclinic | Poor semiconductor |
| [(R)-2]2PF6 | 2:1 | Triclinic | Semiconductor |
| [(S)-2]2PF6 | 2:1 | Triclinic | Semiconductor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroweh, N.; Auban-Senzier, P.; Vanthuyne, N.; Lopes, E.B.; Almeida, M.; Canadell, E.; Avarvari, N. Chiral Conducting Me-EDT-TTF and Et-EDT-TTF-Based Radical Cation Salts with the Perchlorate Anion. Crystals 2020, 10, 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10111069
Mroweh N, Auban-Senzier P, Vanthuyne N, Lopes EB, Almeida M, Canadell E, Avarvari N. Chiral Conducting Me-EDT-TTF and Et-EDT-TTF-Based Radical Cation Salts with the Perchlorate Anion. Crystals. 2020; 10(11):1069. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10111069
Chicago/Turabian StyleMroweh, Nabil, Pascale Auban-Senzier, Nicolas Vanthuyne, Elsa B. Lopes, Manuel Almeida, Enric Canadell, and Narcis Avarvari. 2020. "Chiral Conducting Me-EDT-TTF and Et-EDT-TTF-Based Radical Cation Salts with the Perchlorate Anion" Crystals 10, no. 11: 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10111069