
Molecular Simulation of Crystallisation in External Electric Fields: A Review


School of Chemical and Bioprocess Engineering, University College Dublin, Belfield, 4 Dublin, Ireland
Crystals 2021, 11(3), 316; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030316
Submission received: 27 February 2021
/
Revised: 17 March 2021
/
Accepted: 17 March 2021
/
Published: 22 March 2021
(This article belongs to the Special Issue Feature Papers on "Hybrid and Composite Crystalline Materials" 2021-2022)
{kind=link}
{kind=link}
Abstract
:Elucidating the underlying mechanisms of molecular solidification in both homo- and hetero-geneous systems is of paramount importance for a large swathe of natural phenomena (whether on Earth or throughout the Universe), as well as a whole litany of industrial processes. One lesser-studied aspect of these disorder-order transitions is the effect of external applied fields, shifting both thermodynamic driving forces and underlying kinetics, and, indeed, fundamental mechanisms themselves. Perhaps this is nowhere more apparent than in the case of externally-applied electric fields, where there has been a gradually increasing number of reports in recent years of electro-manipulated crystallisation imparted by such electric fields. Drawing motivations from both natural phenomena, state-of-the-art experiments and, indeed, industrial applications, this review focusses on how non-equilibrium molecular simulation has helped to elucidate crystallisation phenomena from a microscopic perspective, as well as offering an important, predictive molecular-design approach with which to further refine in-field-crystallisation operations.
1. Introduction
In recent decades, the effect of externally-applied fields of various types (e.g., sonic, acoustic, electric, and electromagnetic) on solidification and wider disorder-order transitions has become more important, in both homo- and hetero-geneous systems. In part, this has arisen from realisation of the importance of these external fields in shifting thermodynamic and kinetic crystallisation phenomena, as well as their underlying mechanisms; indeed, this greater awareness has both motivated and facilitated their increasing study in terms of both natural phenomena (whether on Earth itself, or farther afield in the Universe) and industrial processes.
Although various types of external fields have been applied to manipulate and control crystallisation, this is, in general, an under-studied and less-well-understood area. Despite their ubiquity and importance, perhaps this is nowhere more apparent than in the case of externally-applied electric fields, where there has been a gradually increasing number of reports in recent years of electro-manipulated crystallisation and intra-crystal phenomena imparted by such electric fields, which we shall discuss in the present review in due course.
In addition, non-equilibrium molecular simulation, carried out in explicit externally-applied fields, has, in recent years, contributed a great deal towards our understanding of molecular non-equilibrium phenomena, in terms of qualitative mechanisms and on how energy landscapes and reaction-energy barriers, as well as kinetic and fluctuation phenomena themselves, are altered [1]. The ability of in-field, non-equilibrium molecular simulation, if carried out expertly, to enhance and elucidate our mechanistic understanding of molecular processes (such as crystallisation) is very attractive. From a more pragmatic standpoint, molecular simulation itself can serve as a predictive “molecular-design” prototyping tool, which can predict a material’s response to external fields, and, indeed, redesign and optimise the material’s field-response characteristics and performance (e.g., for polymorph selection, control, and realisation in crystallisation operations). This heady vision shall be discussed in the present review, and implications discussed in terms of a final outlook of how this field (pardoning the pun!) of endeavour can, and perhaps should, evolve in the coming years and decades.
Prior to dedicating the rest of this review towards how electric fields manipulate crystallisation processes through the lens of molecular simulation, it is useful and instructive to reflect briefly on how other types of externally-applied fields can affect crystallisation. An important review of acoustically-driven crystallisation (or sono-crystallisation) has been published by Zhang et al. [2], which has probed, inter alia, how power-ultrasound mechanisms can couple to phonon modes and alter kinetic pathways; other important reviews have also discussed acoustic cavitation in more detail as regards possible underlying mechanisms of sono-crystallisation, abutted by pressure waves [3,4]. Indeed, in terms of shockwave-induced crystallisation, Mirsaleh-Kohan et al. have reported laser-induced acoustic shockwaves, with various experimental imaging characterisation [5]. Here, this new method allows for greatly enhanced nucleation, with the near-instantaneous formation of many “seed” crystals, which are then available for further impurity-free crystal growth. Similar features were seen in laser-ablation-based shockwaves in the transition of water to ice VII at high-pressure conditions [6], often mimicking those present in other water-containing planetary bodies, such as Europa and Enceladus.
2. Electro-Crystallisation
Understanding crystal growth and nucleation mechanisms in externally-applied electric fields (termed electro-crystallisation), is essential towards inducing crystallisation and controlling the crystal polymorphic outcome [7,8,9,10,11,12], enhancing protein-crystal quality [13,14,15,16,17], and, excitingly, as an agent for segregating components from suspension in multicomponent crystallite systems [18]. For instance, Aber et al. applied a strong static electric field to supersaturated aqueous glycine solutions, giving rise to γ-polymorph nucleation. This was a first for a strong DC field inducing the nucleation of a neutral solute in a supersaturated solution; it stemmed from fields mediating alignment of very polar glycine molecules—facilitating the adoption of more polar, field-oriented crystalline structures [7].
During electro-crystallisation, exposure to electric fields during freezing is thought to promote ice nucleation at temperatures above the spontaneous nucleation temperature—resulting in a lower degree of supercooling. This serves to reduce the induction (nucleation) time, to elongate the phase-transition time by itself, and, ultimately, to interfere with the growth mechanics of ice crystals [19,20]. Moreover, usage of electro-crystallisation can often induce ice nucleation at the desired degree of supercooling [19], although it must be noted that the probability of ice “electro-nucleation” in this case does depend on the strength of the electric field and the nucleation temperature. It is important to note that the probability of nucleation increases as the nucleation temperature approaches the spontaneous nucleation temperature, and at a greater electric-field strength [19], as one might expect. As an industrial application, electro-crystallisation has enjoyed success and indeed, increasing prominence in the food industry: for instance, freezing under static electric fields produces numerous small-sized ice crystals in frozen matrices, and thus, minimises cell disruption, serves to reduce inevitable drip loss, lessens the level of protein denaturation, and preserves the texture of fresh food to a greater extent after defrosting [20,21,22].
Naturally, electro-crystallisation itself has important general applications for manipulating crystallisation in food systems [23]. Indeed, the handling of water electro-freezing itself is pivotal in food-, and wider cryo-, preservation. In this context, it is important to note that traditional freezing methods, such as air-blast or immersion freezing, could cause inferior quality of frozen products; thus, novel freezing methods, such as ultrasound-assisted freezing or electro-freezing, have been developed [24,25,26].
As we shall discuss, (non-equilibrium) molecular simulation has much to offer in terms of fundamental insights into field-driven crystallisation.
3. Crystallisation Theory in External Electric Fields
Prior to discussing non-equilibrium molecular-simulation per se, and how this has been used to instigate electro-crystallisation, and explore its underlying field-induced mechanisms, we put this into the general context of molecular and microscopic crystallisation theory—in terms of both kinetics and thermodynamic driving forces.
At low temperatures, almost all materials order into a crystalline form, in which molecules are arranged in a regular lattice. This corresponds to the configuration with the minimum potential energy U. At higher temperatures, in irregular arrangements such as liquids and gases, molecules move more freely and thus gain extra entropy in spite of the potential energy cost. For a given temperature and pressure, the equilibrium state is determined by the second law of thermodynamics such that the Gibbs free energy is minimised, where the enthalpy H is defined as . At low temperatures, the entropy S contributes less than the enthalpy and the crystal with a low enthalpy is realised. At high temperatures, however, the entropy S constitutes the dominant contribution and the configuration with a large entropy has the low free energy G: the liquid or the gas phase is realised. The melting point is the temperature at which the free energies of the solid and liquid phases, and , respectively, are equal, i.e., . The liquid becomes more stable for .
Since the Gibbs free energies of both phases are equal at the melting point, , then the latent heat is proportional to the entropy difference :
Now, in the presence of external electric fields, in terms of perturbation theory of thermodynamic properties (such as Gibbs energies and enthalpies of the liquid and crystal states) relevant to simulation, Milchev [27] has offered a very lucid and detailed description of the alteration of thermodynamic quantities and landscapes vis-à-vis crystal nucleation. Equally, Makogon [28] and Kaschiev [29] have devised expressions for shifts in thermodynamic-equilibrium conditions using clathrate hydrates and ice, which affects Equation (1)′s latent-heat value, and overall thermodynamic driving forces and quantities of the separate phases giving rise to this fundamental thermodynamic backdrop, as explained above. The external-field strength would have to be of the order of 0.001 V/Å to have any significant effect—which was confirmed, more or less, by EMF-NEMD work of English and MacElroy [30]. Amadei et al. developed a quasi-Gaussian entropy (QGE) theory, in which free-energy changes are formulated in terms of moment-generating functions related to properties’ fluctuations [31]—allowing equations of states as a function of temperature and external-field intensity. Importantly, Aragones et al. used a Monte-Carlo simulation and Gibbs–Duhem approach to build a phase diagram for TIP4P/2005 water in a 0.03 V/Å static external fields [32]. All of these advances, both in non-equilibrium (in-field) thermodynamic phase-shift theory and associated molecular simulation, have allowed for a more thorough understanding of how the thermodynamic landscape itself shifts for in-field electro-crystallisation.
In any event, by general definition, ; of course, working from the field-perturbed Gibbs energy for the respective phases [28,29,30,31,32], we can derive in-field entropies. Thus, the phase transition is associated with a discontinuity in the slope of the in-field Gibbs free energy:
As such, the phase transition may be classified as a first-order one (i.e., one for which there is a discontinuity in the first derivative of G), with a driving force of .
If one considers the growth of a rough plane crystal surface from the melt at some temperature T < TM, one may formulate a basic model for crystal growth in external fields. At a rough face, kink sites are present, due to surface imperfections (such as screw dislocations), and the growth is classified as normal. The surface may be rough due to the crystallographic orientation of the face or due to entropy effects at sufficiently high temperatures. If one assumes that the molecules have to overcome a field-adjusted diffusional barrier Ediff in order to make the transition from the liquid to the solid phase (due to hindrance of the path of the molecule by its surrounding neighbours), then the rate of incorporation into the lattice is given by:
where a is some characteristic length and is the vibrational frequency about its average position, which is of the order of the lattice vibration. The distance a may be thought of as a molecular length by which the solidification front increases its height upon the crystallisation of one molecule. English and MacElroy have discussed how the diffusional quantities themselves, as opposed to thermodynamic properties, are affected by external fields [33], with important differences between static and oscillating electric fields [1,34,35].
This process is counteracted by molecules which move from the crystal to the liquid. Since the (field-adjusted) Gibbs free energy per molecule, the chemical potential , is higher in the liquid than in the crystal, the rate of melting shall be smaller than the rate of crystal formation by a factor of :
Therefore, the net rate of growth is given by:
Furthermore, at (in-field, shifted) equilibrium, we have the Wilson–Frenkel expression , and hence . Note that it is assumed, not unreasonably, that the vibrational frequencies of both processes are essentially equal.
The activation energy for diffusion from the melt may be related via Stokes–Einstein theory to the melt viscosity:
The self-diffusion coefficient refers to the motion of a ‘tracer’ particle surrounded by neighbouring molecules, and not to net material transport (and ought not to be confused with ‘diffusion-limited growth’, for which the collective diffusion coefficient of the liquid is so small that material transport to the crystal interface becomes rate-limiting).
If one expands the in-field up to first order in the temperature about the equilibrium , one has:
where the last equality follows from and the definition of . Using Equations (4)–(7) allows the Wilson–Frenkel growth law to be written as:
where d is the spacing between crystal layers, l is the diffusional mean free path, and f is an effectiveness factor, which is incorporated to account for the fact that not all molecules which cross the boundary shall adhere to the crystal surface and/or not all sites on the surface might be active growth sites. Equation (8) is an approximation and is therefore limited to temperatures near the equilibrium value . In any event, Vekilov has detailed additional formulations of these general equations and the underlying molecular framework, as well as discussing the underpinning assumptions with acuity [36], and the refer is referred thereto for a broader discussion about factors affecting crystal growth.
Jackson [37] has modified Equation (8) so that it contains an extra entropy factor, and this adjustment has been found to be a useful modification to the Wilson–Frenkel theory:
In the case of normal growth of small spherical and rounded crystallites from melts, the growth theory has been extended [38]. The result for a nucleus of radius is:
where is the critical radius of the nucleus and is the rate for growth of a rough plane face, given by Equation (9). The equation is valid at the initial stages of the crystallisation process when the radius of the growing crystal grain is comparable with that of the critical nucleus. It is important to note, however, that according to the above equation, smaller crystallites grow more slowly than larger crystallites. The equation states that the rate of growth of a crystallite whose size is equal to that of the critical nucleus is equal to zero, i.e., such a crystallite is in equilibrium with the parent phase.
The issue of diffusion of molecules from the surrounding liquid medium to the crystal face is an important one in determining the overall growth rate and may become rate-limiting. Therefore, one ought to modify Equation (10) to take this into account. In the growth equations presented thus far, the temperature T has been that at the interface of the crystal surface, say Ti. For small undercoolings, , of Equation (9) is proportional to , i.e., . The temperature is higher than the far-field value of the melt, . The issue of latent heat removal from the crystal interface by conduction to the surrounding liquid layer is an important one and is described by the heat conduction equation:
where k is the thermal conductivity and is the specific heat per unit volume of the melt. If one defines a dimensionless diffusion field in the melt as , then:
For a spherical crystal, the diffusion equation is given by:
In the above, it is assumed that the relaxation of the diffusion field is very quick compared to the shape of the crystal, i.e., the stationary approximation is valid. D refers to the bulk diffusion coefficient in the surrounding melt, as opposed to the self-diffusion coefficient Dself of a ‘tracer’ molecule. The stationary distribution of the diffusion field takes the form with an integration constant A. The far-field condition at is satisfied by this form for . Following the solution of the diffusion and heat conduction equations [39], the growth velocity of the nucleus of radius ρ is found to be:
and the constant of integration, A, is:
so that the growth velocity is:
where is related to K, the parameter in the expression :
and is the dimensionless undercooling (i.e., the undercooling normalised by the temperature increase caused by the latent heat production):
and is the capillary length proportional to the stiffness of the surface:
It may be seen that Equation (16) is an important modification of Equation (10) for the growth of a rough crystal nucleus (i.e., the bulk diffusion effects through the melt are now accounted for). However, as is the case for Equation (10), there is no crystal growth for sub-critical radii. In the case of fast bulk diffusion, D is large and Equation (16) approaches Equation (10). In the case of slow bulk diffusion, the term in the denominator of Equation (17) may become dominant and growth is retarded.
So far, only the in-field growth of single species crystals from their melts has been considered. The case of rough growth from solutions shall be examined presently (as would apply in the case of crystallisation of clathrate hydrates to form solid solution crystals). For solution growth, there should be a solute molecule in front of an active kink site on the interface to crystallise, in the appropriate geometric positioning with the surrounding solvent molecules at the interface. The probability of finding a solute molecule at a certain crystallisation point with a unit volume is for a solution with concentration C. This molecule oscillates about the average position with a frequency , and attempts crystallisation. However, in the solution, the solute molecules are involved in bonding with the surrounding solvent molecules, and, therefore, desolvation of solute molecules must take place prior to any incorporation into the crystal lattice (along with solvent molecules in the appropriate crystal spatial arrangement). There is an energy barrier for this desolvation process. Among trials of attachment, the rate which overcomes the desolvation energy barrier is given by the Boltzmann weight . The net velocity of crystal growth is then given by:
where is the equilibrium concentration of solute in the solution. As expected, when , there is no driving force for crystal growth and the net growth rate is zero. If one relates the solution chemical potential to C, then . This allows Equation (20) to be written as:
where .
Equation (21) is valid only for a rough plane interface in which the whole surface is contributing to the growth. It is possible to recast the definition above for the growth of spherical clusters, similar to that for growth of single species crystals from the melt [38,40]. Moreover, it is possible to account for the effect of bulk diffusion effects of the solute molecules in the solution to the crystal interface on the crystal growth rate [39].
In the case of gas also being present for the solid-liquid system undergoing (electro-)solidification, such as in clathrate-hydrate (electro-)crystallisation, there may be local variations in solute (gas) concentration near the crystal boundary away from Henry’s law [41], and the whole surface may not be active, due to less partial cavities of solvent (water) molecules ready to accommodate the solute molecules. Consequently, hydrate crystal growth would appear to be substantially less deterministic than Equation (21) might suggest.
4. Molecular Simulation in External Electric Fields
Having described basic crystal-growth theory from a molecular vantage, and how the underlying thermodynamic and dynamical quantities underpinning this may be shifted and perturbed by external electric fields, we now turn to a mechanistic description of how we may apply external electric fields in non-equilibrium molecular simulation (such as non-equilibrium molecular dynamics, NEMD), prior to then reviewing how this advanced tool has been used to shed light on crystallisation phenomena in recent decades.
In NEMD, the system’s Hamiltonian, H, is perturbed by adding a term HE to represent the external field, e.g., for dipolar particles with a field E acting along the z-axis, this is [1]:
where μtot is the total, collective dipole vector. E(t), may be time-independent (i.e., static) or time-dependent (e.g., an oscillating field, with an electromagnetic (e/m) field being a good example). Naturally, the specifics of the field perturbation itself hinge on precisely how the system’s computational handling and on how forces and energy are computed (e.g., possibly as a collection of point charges behaving classically or using electronic-structure methods, etc.).
In the (spatial-) derivative framework of how fields perturb the system Hamiltonian of Equation (22), we present Equation (23) below, expressed for the example of an e/m field (i.e., with a magnetic component, as well). If an e/m-, or electric-, field force, fi,elec = qiE, is applied to each partial-charge site i, featuring charge qi (static or fluctuating), Newton’s second law becomes:
where the intrinsic force fi is due to interactions with all other particles via the potential V, from standard vector differentiation [42]. Using the e/m field in Equation (23) as an example, the electric and magnetic components are understood to act along the laboratory x and y directions, respectively, so that the plane of polarisation is x-y, with propagation along the z-axis, from Maxwell’s equations. Typically, as nanometre dimensions of simulation boxes are many orders of magnitude smaller than e/m-field wavelengths of practical interest, field strengths can be regarded as uniform throughout the system. Moreover, the time-varying e/m-field electric-component vector may be expressed as:
in terms of the maximum of the electric- and magnetic-component intensities, Emax and Bmax. Here, Emax/Bmax = c/n and the root-mean-square (r.m.s.) electric-field intensity for an e/m field is given by Erms = Emax/, where n is the system’s refractive index and c is the speed of light.
For time-varying, cyclic fields (e.g., e/m fields), an important point in NEMD is to simulate a sufficient number of cycles in any NEMD simulation to ensure adequate statistical sampling of field effects—which is especially the case in lower-frequency alternating fields, as the oscillating-field period is long vis-à-vis the system’s underlying (translational) frequency modes [1]. Another important consideration for such NEMD is that the fields strengths required to witness clear-cut external-field effects are typically much higher, ipso facto, than underpinning local, or intrinsic, fields arising in the system per se (typically of the order of 1–3 V/Å [1,36]); this fundamental disparity is motivated by the requirement to bridge the gulf between experimental and simulation timescales. In any case, a general linear-response régime prevails (for dipolar and other, e.g., dynamical, properties) in the range of external-field strengths up to ~0.05 V/Å [35], for which the external-field forces or torques are no greater than a few percent of local fields (e.g., in Equation (23)).
Thermostatting in NEMD is, of course, of foundational importance in crystallisation applications (whether NEMD itself is propagated using empirical potentials or electronic-structure approaches). For example, in ‘real-world’ systems which generally approximate—albeit imperfectly—‘sluggish’ NPT or NVT conditions, there is (slow) leakage of field-mediated thermal energy, itself generated by friction of molecules or atoms rotating or translating [1]. It is only under strictly adiabatic conditions extending to the molecular scale would the non-use of any thermostatting in NEMD be appropriate (e.g., for study of field-induced heating). Equally, however, with the dissipation of latent heat (cf. Equation (1)) being of paramount importance in crystallisation, excessive thermostatting, even in the absence of applied fields can become very problematic in terms of realistic representation of such heat transfer. To this end, in recent years, there has been important progress in developing thermostatting protocols to allow for a steady-state latent-heat exchange with the reservoir in crystallisation/melting events by means of a spatially varying thermostat-coupling strength of atoms and molecules, allowing for canny ‘thermostat engineering’ [30,42].
In NEMD, it is essential to gauge the appropriateness of force-fields with a critical eye. In particular, in external electric fields, the treatment of atomic and molecular polarisability becomes important [1,33], ipso facto, and [30] uses the TIP4P-FQ water model in external electromagnetic fields to gauge e/m-field perturbation of methane-hydrate crystallisation, and it was found that TIP4P-FQ is more accurate for capturing water’s fundamental behaviour in e/m fields [1,30,33].
The underpinning principles of ab-initio molecular dynamics (AIMD) generally involve the system’s Hamiltonian to be time-propagated classically, although now the configurational energy itself is computed via electronic-structure techniques, alongside (Hellman–Feynman) nuclear forces. Umari and Pasquarello devised the underlying modern theory for (ground-state) density-functional theory in external fields under periodic boundary conditions (PBC), using a ‘Berry-phase’ framework for polarisation theory [43], with developments in time-varying fields and progress in time-propagation approaches honed specifically for AIMD [44]. A particular advantage of in-field DFT under PBC is that condensed-phase, periodic systems can be simulated in EEFs with fully self-consistent electron-density rearrangement, and this is of pivotal importance to allow for the accurate representation of electronically-delocalised states. However, non-equilbrium AIMD, as we shall see, has been applied much less to study field-manipulated crystallisation per se, although its initial usage to model field-driven intra-crystal phenomena is certainly encouraging (vide infra).
5. Non-Equilibrium Molecular Simulation of Crystallisation in Electric Fields
Applying static external electric fields in the NEMD simulation of supercooled water has resulted in striking and exciting findings of ‘electro-freezing’ [45,46,47,48,49,50,51,52,53,54,55]. These fields instigate collective alignment of dipoles, alongside accessible thermodynamic (e.g., phase-diagram and melting characteristics [32]) and kinetic pathways towards crystallisation, as well as given polymorph and morphology states. ‘Electro-frozen’ ice arising from homogenous nucleation of bulk supercooled water has not been realised experimentally [56]; for that, sustained field strengths would need to be ~0.02 V/Å to bring about this incipient homogeneous nucleation even as a viable thermodynamic possibility [29,56], which is very challenging to achieve experimentally. This field intensity is of the same order as suggested by Makogon by way of noticeable phase-equilibrium movement in the case of gas hydrates [28], which was also concluded of electric-field conditions by NEMD for methane hydrate [30]. In any case, field intensities needed for uniform bulk ice formation, at ~0.05 V/Å (i.e., close to dielectric saturation for water) are orders of magnitude higher than what is practically achievable in anything other than micro- or nano-scale systems [1]. To help address the long timescales needed in NEMD, in using more moderate external-field strengths more representative of real-world and experimental conditions, a number of ‘rare-event’ techniques in molecular simulations have become available in recent years to overcome long-timescale challenges [1,42]. These include, inter alia, path sampling, metadynamics and adaptive-bias dynamics [1,42].
Svishchev and Kusalik [45,47,48] performed seminal and important NEMD simulations of water’s electro-freezing in 1994–1996. At the outset, they simulated supercooled TIP4P water in a 0.5 V/Å field, with the liquid crystallsing rapidly (within 0.2 ns) [47]. For electro-crystallisation at a threshold between 0.1 and 0.5 V/Å, non-tetrahedral interstitial coordination in liquid water dissipated, leading to neighbouring hydrogen-bonded molecules taking up distinctive local crystallographic sites in cubic ice [47]. Electro-crystallisation NEMD with TIP4P and SPC/E models in truncated octahedral and cubic simulation cells led to polar-ice formation, with a density of 0.94–0.96 g/cm3, with amorphous ices electro-freezing from initial structures of low- and high-density supercooled. In the case of TIP4P, electro-freezing at elevated pressures of 3 to 5 kbar in 0.5 V/Å static fields led to an open quartz-like structure—essentially a defective type of ice XII, within around 1 ns in truncated octahedral and cubic simulation cells [48]. Constant-volume (NVT) simulations, carried out at 1.160–1.2 g/cm3 system densities, were more favourable in achieving ice-XII structures featuring less defects.
Further NEMD explorations of ice electro-freezing starting from supercooled water have been performed by Sutmann, employing a flexible water model at 300 K, with a (very elevated) 2.5–4 V/Å threshold identified (indeed comparable in intensity to intrinsic electric fields in intensity) to give rise to a complete transformation to a polar, crystal-like structure featuring dual-layer periodicity; this was despite box-size challenges [49]. Jung et al. found that the threshold for inducing structural change at 243 K lied in the region of 0.15–0.2 V/Å: above that, a structure broadly comparable to cubic ice was realised, although diffusivities were admittedly too elevated to classify this as a solid [50]. Yan and Patey determined that for a six-site water model, the highest temperature where field-induced ice electro-nucleation occurred was at 280 K, despite the model’s freezing temperature being at 289 K; in contrast, for TIP4P/Ice, the corresponding most elevated temperature for electro-freezing was determined at its melting point (i.e., 270 K) [51]. Yan and Patey also found that surface layers supported more easily a ferroelectric cubic-ice form, whilst the remainder of the liquid took on a dipole-disordered cubic structure [52,53]. As a result, a revealing, and potent finding, from NEMD is that although field-induced electro-crystallisation may not be easily realisable in the bulk, adept and clever surface design may render this achievable at surfaces in external electric fields (particularly for those undergoing reinforcing interaction with the local electric field at the surface). This may well be pivotal in enhancing electro-nucleation and in controlling the crystal phase produced, given that some NEMD simulations showed cubic-ice formation at surfaces [51,52].
Yan and Patey carried out further NEMD simulations featuring ‘electric-field bands’, where the electric field is applied only to a restricted area of surface, as opposed to the full surface [53]. This led to ice which formed as a mixture of cubic and hexagonal phases rather than an ‘polytype-pure’ cubic ice, as found in previous simulations (and experiments) [53]. Yan and Patey pointed out, not unconvincingly, that this may be a more useful reflection of surface-based electro-crystallisation, as cubic ice may have formed in previous ‘complete-surface’ electric fields arising from periodic boundary conditions (PBCs) favouring cubic ice; in ‘partial-field bands’, PBCs do not affect the edges of the external field, allowing for parallel Ih/Ic formation.
Along this general vein, recent studies suggest that ‘geometric matching’ does not influence ice nucleation greatly; rather, nucleation is more influenced by cracks, pores, steps, and other surface features of solutes, although exact mechanistic workings are unknown [53]. This has also been discussed with acuity by Sosso et al. [57,58], who studied ice formation on organic crystals and kaolinite, alongside all of the associated surface features and quirks. Still, it may well be the case that the local electric fields intrinsic to such nano-scale features exert important control over such local ice formation. NEMD simulations have emphasised cases where such geometric matching does not rationalise nucleation observations. For example, ice nucleated in a more facile manner directly underneath a free surface comparted to the bulk liquid, due to the small local electric field (as opposed to the externally-applied one) arising from ordered arrangements of water molecules at the water-air interface [59].
Outside electric-field-based ice (electro-)crystallisation, field-induced changes in liquid-solid phase-transition kinetics in other systems have been much less reported—particularly so for the case of oscillating electric fields. Motivated in part by Rojey’s patent on e/m-field-based gas-hydrate inhibition [60], English and MacElroy performed NEMD of methane-hydrate nanocrystals in e/m fields [30]. It was found that the constantly-alternating dipole alignment inside the crystallite undermined the structural stability of water-lattice hydrogen bonds, with ~0.15 V/Å leading to hydrate electro-dissociation (typically corresponding to several percent of intrinsic electric fields in intensity, i.e., 1–3 V/Å); frequencies inducing the most noticeable ‘resonant’ dissociation were found to be in the 20–200 GHz range (overlapping with hydrogen-bond relaxation times) [30]. It was also found in [30] that elongation of the methane-hydrate nano-crystallites in the direction of e/m-field application occurred, which is important for field-mediated influence on both the habit and morphology of crystallites (vide infra).
In more important gas-hydrate work, Mayorga et al. investigated static fields on bulk hydrates via NEMD, speculating that very strong electric fields could initiate dissociation [61]. They found that at an external-field intensity of ~0.1 V/Å led to hydrate dissolution [61]—an electro-dissociation threshold in good agreement with e/m fields found by English and MacElroy [30]. In addition, for bulk hydrates, Waldron and English scrutinised how e/m fields adjust the bulk crystal structure’s structural and dynamical characteristics, such as vibrational and energy-transfer properties, albeit without inducing electro-dissociation per se [62]. They also found a similar electro-dissolution threshold to Mayorga et al. for static-field-induced dissociation of bulk methane hydrate, observing the birth of gas nano-bubbles in the aftermath of the hydrate’s break-up [63].
In all of these studies of external-field effects on crystallisation (and underlying molecular diffusion [64]) in water-containing systems, e.g., ice and gas hydrates, an important, universal feature is how hydrogen-bond structure and dynamics is manipulated by the external field via toque-induced dipole alignment (cf. Equations (22) and (23))—whether by static or oscillating fields [1,33,34,35,36,65]. This ‘torque control’ allows for manipulation of the field-adjusted diffusional barrier in Equation (3) on the crystallisation rate itself, apart from alteration of the thermodynamic driving force itself (i.e., the de-facto latent heat in Equation (1)).
Moving on to matters of field manipulation of gross-crystallite morphology and shape per se, as discussed in the literature [30,66,67], static fields’ intriguing induction of ‘electro-nucleation’ and subsequent crystallisation have also been noted for liquid-droplet shape transformation, experimentally and theoretically, from spherical to prolate spheroids [66], with the emergence of sharp conical regions in field-elongated structures discussed with acuity by Luedtke et al. [67] in NEMD of formamide nano-droplets. Indeed, as mentioned previously, for aqueous systems in microwave fields, English and MacElroy have also noted shape elongation and change in habit of methane-hydrate nano-crystallites via NEMD [30]. In addition, NEMD has also shown recently the elongation of gaseous nanobubbles in water, with they themselves induced by field-driven electrostriction, walking as energetic ‘tightrope’ between competing terms [68].
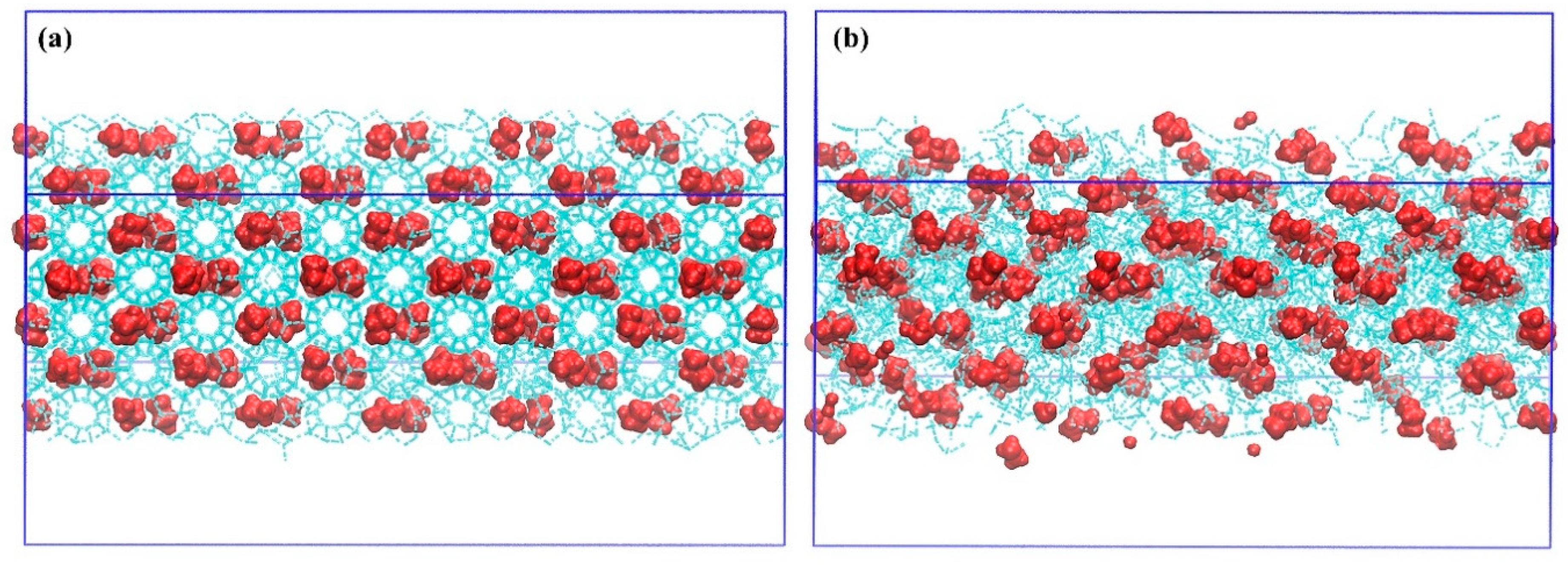
Contributing further to our understanding of field-induced phase transitions and morphology change in static electric fields for multi-phase aqueous solution, Ghaani and English studied electro-nucleation of water in heterogeneous propane-nanobubble systems to form polycrystalline ice Ic [55], building on the earlier (homogeneous) cubic-ice electro-nucleation NEMD of Nandi et al. [46]. Reprising and reproducing some simulations from [55] here, shown in Figure 1, is the incipient rearrangement of propane nanobubbles in NEMD simulations, driven by electrostriction phenomena [55,68]. As in [55], this leads to some elongation of a nanobubble and the field-assisted growth and development of cubic ice.
Turning to polymorphs, and the wider issue of manipulation of crystal habit, in the area of static fields applied to crystals composed of pharmaceutical molecules, Parks et al. made some notable, exciting NEMD progress in recent years towards external-field control over polymorph selection [11,69,70], as suggested originally by English and MacElroy [30]. Intriguingly, they have shown how static fields in NEMD alter the exploration of accessible kinetic pathways of different polymorphs of glycine (also studied originally by the seminal laser-based-field advance in [7]), allowing the adoption of otherwise-inaccessible polymorphs—which is of important industrial relevance. Parks and co-workers have also discussed external-field-control implications for crystal habit and morphology [11,69,70], echoing findings of English and MacElroy [30], as well as Luedtke et al. [67], on field-mediated manipulation of particle/crystallite shape.
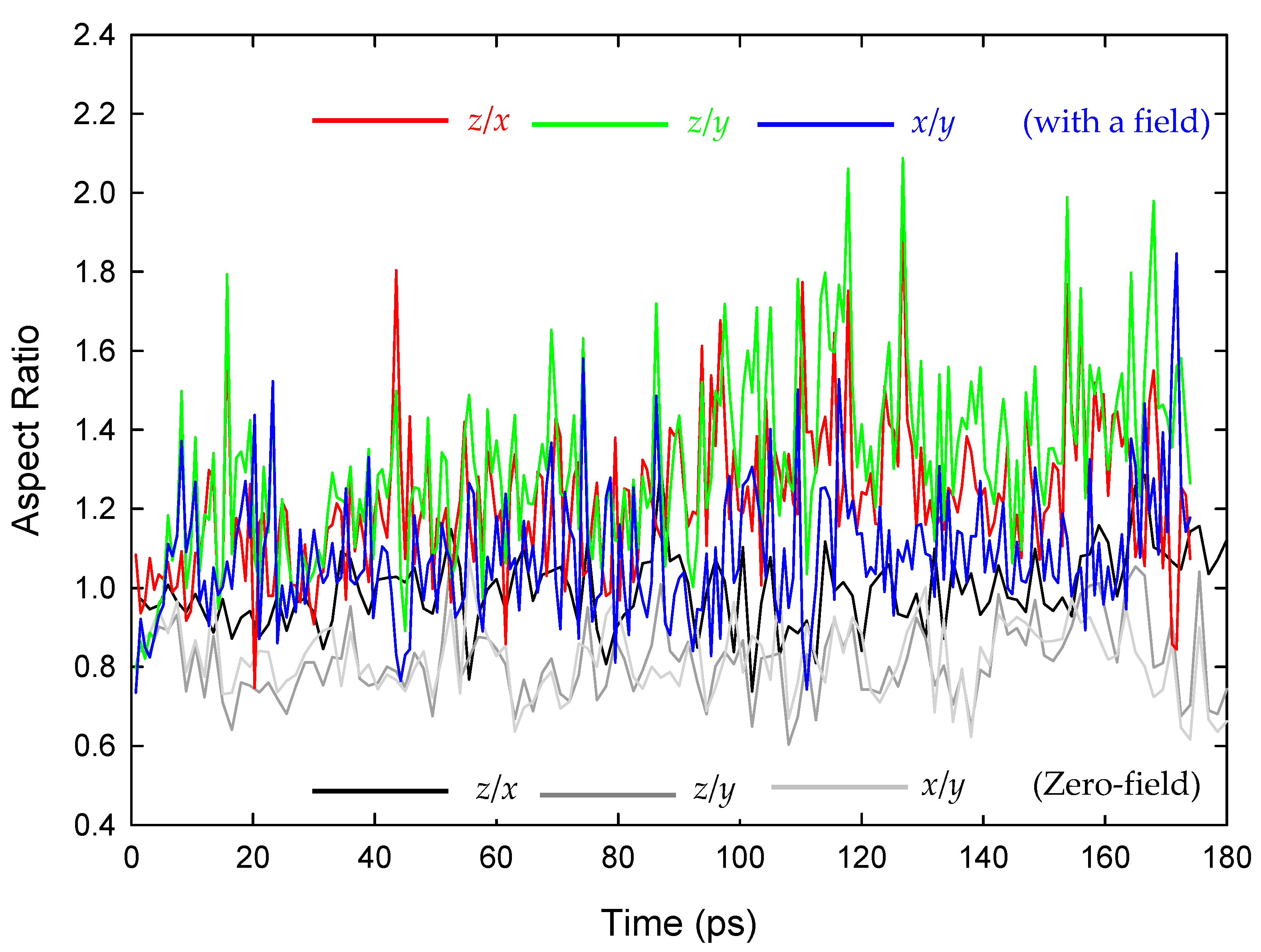
To appreciate some of the conceptual origins about field-induced crystallite shape during electro-crystallisation NEMD, we reprise herewith some of the NEMD simulations as outlined in [30] by English and MacElroy. To this end, shown in Figure 2 is the time-evolution of the aspect ratios of the initially spherical methane-hydrate crystallite, where it is clear that there is evolution in the nano-crystallite to adopt an increasingly aspherical shape as electro-crystallisation proceeds (for a 250 GHz electromagnetic field applied along the laboratory z-axis), owing to levels of collective dipole alignment. Indeed, a similar phenomenon has been witnessed in the case of water-ice nano-droplets by Nandi et al. [46], mirroring also [66,67], and laying the foundations for the technologically important and insightful work in the pharmaceutical arena of [11,69,70].
6. Conclusions
The present review has discussed how external electric fields alter crystallisation pathways, thermodynamics, and kinetic/diffusional barriers through the lens of a non-equilibrium molecular simulation, often via the universal mechanistic coupling of field-induced torques and forces on molecules—with perturbation of hydrogen bonding between water molecules being an important case in point.
In terms of future outlook, although the present review has mentioned (non-equilibrium) DFT and AIMD as being important for the study of (polarisable) crystal-liquid interfaces, with non-bulk properties, this is a still-unexplored area in field-manipulated crystallisation. To make progress in this direction, careful and judicious selection and optimisation of the most appropriate DFT functionals for crystal and interfacial systems, in close accord with experiment [71,72], is essential, together with careful attention to, and characterisation of, interfacial electronic properties [73,74,75], which are highly field sensitive. In this pursuit, the use of DFT-based NEMD simulation as a prototyping and molecular-design tool is most promising, e.g., with direct mechanistic field-coupling action (via Equations (21) and (22)) evident for polar and quadrupolar molecules, e.g., CO2; in this sense, for CO2 fixation [76], external electric fields offer much promise in the design of canny field-manipulated operational strategies, including for crystallisation [77].
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
N.J.E. thanks Mohammad Reza Ghaani for help with figure preparation and some technical assistance.
Conflicts of Interest
The author declares no conflict of interest.
References
- English, N.J.; Waldron, C.J. Perspectives on External Electric Fields in Molecular Simulation: Progress, Prospects and Challenges. Phys. Chem. Chem. Phys. 2015, 17, 12407. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, D.W.; Zhu, Z.; Cheng, L. Enhancement of Crystallization Processes by Power Ultrasound: Current State-of-the-Art and Research Advances. Comp. Rev. 2015, 14, 303. [Google Scholar] [CrossRef]
- Kim, H.N.; Suslick, K.S. The Effects of Ultrasound on Crystals: Sonocrystallization and Sonofragmentation. Crystals 2018, 8, 280. [Google Scholar] [CrossRef] [Green Version]
- Nalesso, S.; Bussemaker, M.J.; Sear, R.P.; Hodnett, M.; Lee, J. A review on possible mechanisms of sonocrystallisation in solution. Ultrason. Sonochem. 2019, 57, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Mirsaleh-kohan, N.; Fischer, A.; Graves, B.; Bolorizadeh, M. Laser Shock-Wave Induced Crystallization. Cryst. Growth Des. 2017, 17. [Google Scholar] [CrossRef]
- Gleason, A.; Bolme, C.; Galtier, E.; Lee, H.; Granados, E.; Dolan, D.; Seagle, C.; Ao, T.; Ali, S.; Lazicki, A.; et al. Compression Freezing Kinetics of Water to Ice VII. Phys. Rev. Lett. 2017, 119, 025701. [Google Scholar] [CrossRef]
- Aber, J.E.; Arnold, S.; Garetz, B.A.; Myerson, A.S. Myerson, Strong dc Electric Field Applied to Supersaturated Aqueous Glycine Solution Induces Nucleation of the γ Polymorph. Phys. Rev. Lett. 2005, 94, 145503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myerson, A.S.; Ginde, R. Industrial Crystallization of Melts; CRC Press: Boca Raton, FL, USA, 2004; pp. 183–240. [Google Scholar]
- Alexandera, L.F.; Radacsi, N. Application of electric fields for controlling crystallization. CrystEngComm 2019, 21, 5014–5031. [Google Scholar] [CrossRef] [Green Version]
- Adrjanowicz, K.; Paluch, M.; Richert, R. Formation of new polymorphs and control of crystallization in molecular glass-formers by electric field. Phys. Chem. Chem. Phys. 2018, 20, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.; Koswara, A.; Tung, H.H.; Nere, N.; Bordawekar, S.; Nagy, Z.K.; Ramkrishna, D. Molecular Dynamics Electric Field Crystallization Simulations of Paracetamol Produce a New Polymorph. Cryst. Growth Des. 2017, 17, 3751–3765. [Google Scholar] [CrossRef]
- di Profio, G.; Reijonen, M.T.; Caliandro, R.; Guagliardi, A.; Curcio, E.; Drioli, E. Insights into the polymorphism of glycine: Membrane crystallization in an electric field. Phys. Chem. Chem. Phys. 2013, 15, 9271. [Google Scholar] [CrossRef]
- Taleb, M.; Didierjean, C.; Jelsch, C.; Mangeot, J.P.; Aubry, A. Equilibrium kinetics of lysozyme crystallization under an external electric field. J. Cryst. Growth 2001, 232, 250–255. [Google Scholar] [CrossRef]
- Pareja-Rivera, C.; Cuéllar-Cruz, M.; Esturau-Escofet, N.; Demitri, N.; Polentarutti, M.; Stojanoff, V.; Moreno, A. Recent Advances in the Understanding of the Influence of Electric and Magnetic Fields on Protein Crystal Growth. Cryst. Growth Des. 2017, 17, 135–145. [Google Scholar] [CrossRef]
- Hou, D.; Chang, H.-C. AC field enhanced protein crystallization. Appl. Phys. Lett. 2008, 92, 223902. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, H.; Uda, S.; Fujiwara, K.; Tachibana, M.; Kojima, K.; Nozawa, J.J. Improvement of crystal quality for tetragonal hen egg white lysozyme crystals under application of an external alternating current electric field. Appl. Crystallogr. 2013, 46, 25–29. [Google Scholar] [CrossRef]
- Rubin, E.; Owen, C.; Stojanoff, V. Crystallization under an external electric field: A case study of glucose isomerase. Crystals 2017, 7, 206. [Google Scholar] [CrossRef]
- Li, W.W.; Radacsi, N.; Kramer, H.J.M.; van der Heijden, A.E.D.M.; Horst, J.H.T. Solid Separation from a Mixed Suspension through Electric-Field-Enhanced Crystallization. Angew. Chem. Int. Ed. 2016, 55, 16088–16091. [Google Scholar] [CrossRef] [Green Version]
- Orlowska, M.; Havet, M.; Le-Bail, A. Controlled ice nucleation under high voltage DC electrostatic field conditions. Food Res. Int. 2009, 42, 879–884. [Google Scholar] [CrossRef]
- Xanthakis, E.; Havet, M.; Chevallier, S.; Abadie, J.; Le-Bail, A. Effect of static electric field on ice crystal size reduction during freezing of pork meat. Innov. Food Sci. Emerg. Technol. 2013, 20, 115–120. [Google Scholar] [CrossRef]
- Jia, G.; He, X.; Nirasawa, S.; Tatsumi, E.; Liu, H.; Liu, H. Effects of high-voltage electrostatic field on the freezing behavior and quality of pork tenderloin. J. Food Eng. 2017, 204, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Dalvi-Isfahan, M.; Hamdami, N.; Le-Bail, A. Effect of freezing under electrostatic field on the quality of lamb meat. Innov. Food Sci. Emerg. Technol. 2016, 37, 68–73. [Google Scholar] [CrossRef]
- Jha, P.K.; Sadot, M.; Vino, S.A.; Jury, V.; Curet-Ploquin, S.; Rouaud, O.; Havet, M.; Le-Bail, A. A review on effect of DC voltage on crystallization process in food systems. Innov. Food Sci. Emerg. Technol. 2017, 42, 204–219. [Google Scholar] [CrossRef]
- Zhu, Z.; Sun, D.-W.; Zhang, Z.; Li, Y.; Cheng, L. Effects of micro-nano bubbles on the nucleation and crystal growth of sucrose and maltodextrin solutions during ultrasound-assisted freezing process. LWT 2018, 92, 404–411. [Google Scholar] [CrossRef]
- Sun, D.-W.; Zheng, L. Vacuum cooling technology for the agri-food industry: Past, present and future. J. Food Eng. 2006, 77, 203–214. [Google Scholar] [CrossRef]
- Yu, D.; Liu, B.; Wang, B. The effect of ultrasonic waves on the nucleation of pure water and degassed water. Ultrason. Sonochem. 2012, 19, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Milchev, A. Nucleation phenomena in electrochemical systems: Thermodynamic concepts. ChemTexts 2016, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Makogon, Y.F. Hydrates of Hydrocarbons; PennWell Books: Tulsa, OK, USA, 1997. [Google Scholar]
- Kaschiev, D. On the influence of the electric field on nucleation kinetics. Philos. Mag. 1972, 25, 459. [Google Scholar] [CrossRef]
- English, N.J.; MacElroy, J.M.D. Theoretical studies of the kinetics of methane hydrate crystallization in external electromagnetic fields. J. Chem. Phys. 2004, 120, 10247. [Google Scholar] [CrossRef]
- Amadei, A.; Apol, M.E.F.; Brancato, G.; di Nola, A. Theoretical equations of state for temperature and electromagnetic field dependence of fluid systems, based on the quasi-Gaussian entropy theory. J. Chem. Phys. 2002, 116, 4437. [Google Scholar] [CrossRef]
- Aragones, J.L.; MacDowell, L.G.; Siepmann, J.I.; Vega, C. Phase Diagram of Water under an Applied Electric Field. Phys. Rev. Lett. 2011, 107, 155702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, N.J.; MacElroy, J.M.D. Hydrogen bonding and molecular mobility in liquid water in external electromagnetic fields. J. Chem. Phys. 2003, 119, 11806. [Google Scholar] [CrossRef]
- Reale, R.; English, N.J.; Marracino, P.; Liberti, M.; Apollonio, F. Dipolar Response and Hydrogen-Bond Kinetics in Liquid Water in Square-Wave Time-Varying Electric Fields. Mol. Phys. 2014, 112, 1870. [Google Scholar] [CrossRef]
- Reale, R.; English, N.J.; Garate, J.-A.; Marracino, P.; Liberti, M.; Apollonio, F. Human Aquaporin 4 Gating Dynamics Under and After Nanosecond-Scale Static and Alternating Electric-Field Impulses: A Molecular Dynamics Study of Field Effects and Relaxation. J. Chem. Phys. 2013, 139, 205101. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G. What Determines the Rate of Growth of Crystals from Solution? Cryst. Growth Des. 2007, 7, 12. [Google Scholar] [CrossRef]
- Jackson, K.A. Theory of Melt Growth, in Crystal Growth and Characterization; Ueda, R., Mullin, J.B., Eds.; North-Holland: Amsterdam, The Netherlands, 1975. [Google Scholar]
- Christian, J.W. The Theory of Tranformations in Metals and Alloys, Part I: Equilibrium and General Kinetic Theory, 2nd ed.; Pergamon Press: Oxford, UK, 1981. [Google Scholar]
- Saito, Y. Statistical Physics of Crystal Growth; World Scientific: Singapore, 1996. [Google Scholar]
- Markov, I.V. Crystal Growth for Beginners; World Scientific: New York, NY, USA, 1995. [Google Scholar]
- English, N.J.; Carroll, D.G. Prediction of Henry’s Law constants by a Quantitative Structure Property Relationship and neural networks. J. Chem. Inf. Comp. Sci. 2001, 41, 1150. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Pergamon Press: Oxford, UK, 2017. [Google Scholar]
- Umari, P.; Pasquarello, A. Ab initio Molecular Dynamics in a Finite Homogeneous Electric Field. Phys. Rev. Lett. 2002, 89, 157602. [Google Scholar] [CrossRef]
- Futera, Z.; English, N.J. Communication: Influence of external static and alternating electric fields on water from long-time non-equilibrium ab initio molecular dynamics. J. Chem. Phys. 2017, 147, 031102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svishchev, I.M.; Kusalik, P.G. Electrofreezing of Liquid Water: A Microscopic Perspective. J. Am. Chem. Soc. 1996, 118, 649. [Google Scholar] [CrossRef]
- Nandi, P.K.; Burnham, C.J.; English, N.J. Electro-nucleation of water nano-droplets in No Man’s Land to fault-free ice Ic. Phys. Chem. Chem. Phys. 2018, 20, 8042. [Google Scholar] [CrossRef]
- Svishchev, I.M.; Kusalik, P.G. Crystallization of Liquid Water in a Molecular Dynamics Simulation. Phys. Rev. Lett. 1994, 73, 975–978. [Google Scholar] [CrossRef] [Green Version]
- ISvishchev, M.; Kusalik, P.G. Quartzlike polymorph of ice. Phys. Rev. B 1996, 53, R8815–R8817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutmann, G. Structure formation and dynamics of water in strong external electric fields. J. Electroanal. Chem. 1998, 450, 289. [Google Scholar] [CrossRef]
- Jung, D.H.; Yang, J.H.; Jhon, M.S. The effect of an external electric field on the structure of liquid water using molecular dynamics simulations. Chem. Phys. 1999, 244, 331. [Google Scholar] [CrossRef]
- Yan, J.Y.; Patey, G.N. Heterogeneous Ice Nucleation Induced by Electric Fields. J. Phys. Chem. Lett. 2011, 2, 2555. [Google Scholar] [CrossRef]
- Yan, J.Y.; Patey, G.N. Molecular Dynamics Simulations of Ice Nucleation by Electric Fields. J. Phys. Chem. A 2012, 116, 7057. [Google Scholar] [CrossRef]
- Yan, J.Y.; Patey, G.N. Ice nucleation by electric surface fields of varying range and geometry. J. Chem. Phys. 2013, 139, 144501. [Google Scholar] [CrossRef]
- Khusnutdinoff, R.M. Dynamics of a network of hydrogen bonds upon water electrocrystallization. Colloid J. 2013, 75, 726. [Google Scholar] [CrossRef]
- Ghaani, M.R.; English, N.J. Kinetic study on electro-nucleation of water in a heterogeneous propane nano-bubble system to form polycrystalline ice Ic. J. Chem. Phys. 2020, 153, 084501. [Google Scholar] [CrossRef]
- Stan, C.A.; Tang, S.K.Y.; Bishop, K.J.M.; Whiteside, G.M. Externally Applied Electric Fields up to 1.6 × 105 V/m Do Not Affect the Homogeneous Nucleation of Ice in Supercooled Water. J. Phys. Chem. B 2011, 115, 1089. [Google Scholar] [CrossRef] [Green Version]
- Sosso, G.C.; Whale, T.F.; Holden, M.A.; Pedevilla, P.B.; Murray, J.; Michaelides, A. Unravelling the origins of ice nucleation on organic crystals. Chem. Sci. 2018, 9, 8077–8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosso, G.C.; Tribello, G.A.; Zen, A.; Pedevilla, P.; Michaelides, A. Ice formation on kaolinite: Insights from molecular dynamics simulations. J. Chem. Phys. 2016, 145, 211927. [Google Scholar] [CrossRef] [Green Version]
- Vrbka, L.; Jungwirth, P. Homogeneous freezing of water starts in the subsurface. J. Phys. Chem. 2006, 110, 18126. [Google Scholar] [CrossRef] [PubMed]
- Rojey, A. Process and System Using an Electromagnetic Wave to Prevent the Formation of Hydrates. U.S. Patent No. 5625178, 29 April 1997. [Google Scholar]
- Mayorga, M.; López-Lemus, J.; Luis, D.P. Electrodissociation of clathrate-like structures. Mol. Simul. 2010, 36, 461. [Google Scholar]
- Waldron, C.J.; English, N.J.; Waldron, C.J.; English, N.J. Global-Density Fluctuations in Methane Clathrate Hydrates in Externally-Applied Electromagnetic Fields. J. Chem. Phys. 2017, 147, 024506. [Google Scholar] [CrossRef]
- Waldron, C.J.; English, N.J. System-Density Fluctuations and Electro-Dissociation of Methane Clathrate Hydrates in Externally-Applied Static Electric Fields. J. Chem. Thermo. 2018, 117, 68. [Google Scholar] [CrossRef]
- Cao, H.; English, N.J.; MacElroy, J.M.D. Diffusive hydrogen inter-cage migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates. J. Chem. Phys. 2013, 138, 094507. [Google Scholar] [CrossRef]
- English, N.J. Molecular dynamics simulations of liquid water using various long range electrostatics techniques. Mol. Phys. 2005, 103, 1945–1960. [Google Scholar] [CrossRef]
- Sherwood, J.D. The deformation of a fluid drop in an electric field: A slender-body analysis. J. Phys. A. Math. Gen. 1991, 24, 4047. [Google Scholar] [CrossRef]
- Luedtke, W.D.; Gao, J.; Landman, U. Dielectric nanodroplets: Structure, stability, thermodynamics, shape transitions and electrocrystallization in applied electric fields. J. Phys. Chem. C 2011, 115, 20343–20358. [Google Scholar] [CrossRef]
- Ghaani, M.R.; Kusalik, P.G.; English, N.J. Massive generation of metastable bulk nanobubbles in water by external electric fields. Sci. Adv. 2020, 6, eaaz0094. [Google Scholar] [CrossRef] [Green Version]
- Parks, C.; Koswara, A.; Tung, H.H.; Nere, N.; Bordawekar, S.; Nagy, Z.K.; Ramkrishna, D. Extending the Crystal Landscape Through Electric Field Controlled Crystallization—A Molecular Dynamics Case Study. ChemRxiv 2018. [Google Scholar] [CrossRef]
- Bulutoglu, P.S.; Parks, C.; Nere, N.K.; Bordawekar, S.; Ramkrishna, D. Exploring New Crystal Structures of Glycine via Electric Field-Induced Structural Transformations with Molecular Dynamics Simulations. Processes 2019, 7, 268. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Dev, P.; English, N.J.; Thampi, K.R.; MacElroy, J.M.D. A TD-DFT study of the effects of structural variations on the photochemistry of polyene dyes. Chem. Sci. 2012, 3, 416–424. [Google Scholar] [CrossRef]
- McDonnell, K.; Wadnerkar, N.; English, N.J.; Rahman, M.; Dowling, D. Photo-active and optical properties of bismuth ferrite (BiFeO3): An experimental and theoretical study. Chem. Phys. Lett. 2013, 572, 78–84. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Band gap engineering of double-cation-impurity-doped anatase-titania for visible-light photocatalysts: A hybrid density functional theory approach. Phys. Chem. Chem. Phys. 2011, 13, 13698–13703. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; English, N.J. Electronic structure of cation-codoped TiO2 for visible-light photocatalyst applications from hybrid density functional theory calculations. App. Phys. Lett. 2011, 98, 142103. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Density functional theory description of the mechanism of ferromagnetism in nitrogen-doped SnO2. Phys. Lett. A 2009, 374, 319–322. [Google Scholar] [CrossRef] [Green Version]
- English, N.J.; El-Hendawy, M.M.; Mooney, D.A.; MacElroy, J.M.D. Perspectives on Atmospheric CO2 Fixation in Inorganic and Biomimetic Structures. Coord. Chem. Rev. 2014, 269, 85–95. [Google Scholar] [CrossRef]
- English, N.J.; Clarke, E.T. Molecular Dynamics Study of CO2 Hydrate Dissociation: Fluctuation-Dissipation and Non-Equilibrium Analysis. J. Chem. Phys. 2013, 139, 094701. [Google Scholar] [CrossRef]
Figure 1.
Depiction of incipient rearrangements to facilitate propane-nanobubble growth in non-equilibrium molecular-dynamics (NEMD) simulation in 0.1 V/Å fields: (a) initial gas-hydrate configuration, (b) early-stage nanobubble coalescence.
Figure 1.
Depiction of incipient rearrangements to facilitate propane-nanobubble growth in non-equilibrium molecular-dynamics (NEMD) simulation in 0.1 V/Å fields: (a) initial gas-hydrate configuration, (b) early-stage nanobubble coalescence.

Figure 2.
Methane-hydrate-crystallite aspect ratios as a function of time, in the case of initially 12.5 Å-radius spheres surrounded by an aqueous phase, similar in detail and set-up to [30]. Crystallisation simulations were carried out in a similar way to [30] under both zero-field conditions (in black and grey) and in a 250 GHz/0.15 V/År.m.s. e//m field applied along the laboratory z-axis. The development of relative elongation along the laboratory z-axis (the direction of e/m-field application) vis-à-vis the x- and y-directions, is evident.
Figure 2.
Methane-hydrate-crystallite aspect ratios as a function of time, in the case of initially 12.5 Å-radius spheres surrounded by an aqueous phase, similar in detail and set-up to [30]. Crystallisation simulations were carried out in a similar way to [30] under both zero-field conditions (in black and grey) and in a 250 GHz/0.15 V/År.m.s. e//m field applied along the laboratory z-axis. The development of relative elongation along the laboratory z-axis (the direction of e/m-field application) vis-à-vis the x- and y-directions, is evident.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
English, N.J. Molecular Simulation of Crystallisation in External Electric Fields: A Review. Crystals 2021, 11, 316. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030316
AMA Style
English NJ. Molecular Simulation of Crystallisation in External Electric Fields: A Review. Crystals. 2021; 11(3):316. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030316
Chicago/Turabian StyleEnglish, Niall J. 2021. "Molecular Simulation of Crystallisation in External Electric Fields: A Review" Crystals 11, no. 3: 316. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030316
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.