1. Introduction
Most single-component molecular crystals are semiconductors or insulators because of large HOMO-LUMO energy band gaps (HOMO = Highest Occupied Molecular Orbital, LUMO = Lowest Unoccupied Molecular Orbital). Compared to other simple organic molecular crystals, metal–dithiolene complexes have small HOMO-LUMO gaps (~0.5 eV) and are, therefore, considered to be important candidates in the search for conducting single-component molecular systems. Through the last two decades of research, it has been revealed that chemical modifications, such as increasing π-conjugation and ligand planarity, can reduce the HOMO-LUMO band gap. This method, however, reduces the solubility of source materials and makes crystal growth very difficult. So far, only three single-component molecular metals, [M(tmdt)
2] (M = Ni, Au; tmdt = trimethylenetetrathiafulvalenedithiolate) [
1,
2] and [Au(Me-thiazdt)
2] (Me-thiazdt = N-methyl-1,3-thiazoline-2-thione-4,5-dithiolate) [
3], and one single-component Dirac electron candidate [Pt(dmdt)
2] (dmdt = dimethyltetrathiafulvalenedithiolate) [
4], have been reported under ambient conditions. On the other hand, molecular crystals have soft lattices and so the bandwidths of HOMO and LUMO bands may be easily enlarged by the application of external pressure. Therefore, the application of pressure has been found to be an efficient way to discover new single-component molecular metals and superconductors. Indeed, three pressure-induced single-component molecular metals, [Ni(ptdt)
2] (ptdt = propylenedithiotetrathiafulvalenedithiolate) [
5], [Cu(dmdt)
2] [
6] and [Ni(dmit)
2] (dmit = 1,3-dithiole-2-thione-4,5-dithiolate) [
7], have been found at pressures of 19.4 GPa, 4.7 GPa, and 15.9 GPa, respectively. Furthermore, we also discovered the first pressure-induced single-component molecular superconductor, [Ni(hfdt)
2] (hfdt = bis(trifluoromethyl)tetrathiafulvalenedithiolate), at 8 GPa (
Tc = 5.5 K) [
8]. Lorcy and coworkers reported that single-component molecular crystals [Au(Et-thiazdt)
2] (Et-thiazdt = N-ethyl-1,3-thiazoline-2-thione-4,5-dithiolate) [
9] and [Au(Et-thiazds)
2] [
10] exhibit metallic conduction at the even lower pressures of 1.3 GPa and 0.6 GPa, respectively.
Because of the multi-orbital character of single-component molecular conductors, it is also expected that under certain conditions, more exotic electronic states may occur. For example, the HOMO and LUMO bands may be induced to touch at a single point to form a Dirac electron system. Indeed, we have found that a single-component molecular crystal, [Pd(dddt)
2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate), forms a Dirac electron system under high pressure. First-principles density functional theory (DFT) calculations indicated that Dirac cones emerge from the crossing between the HOMO and LUMO bands, which originate from crystallographically independent layers within the crystal [
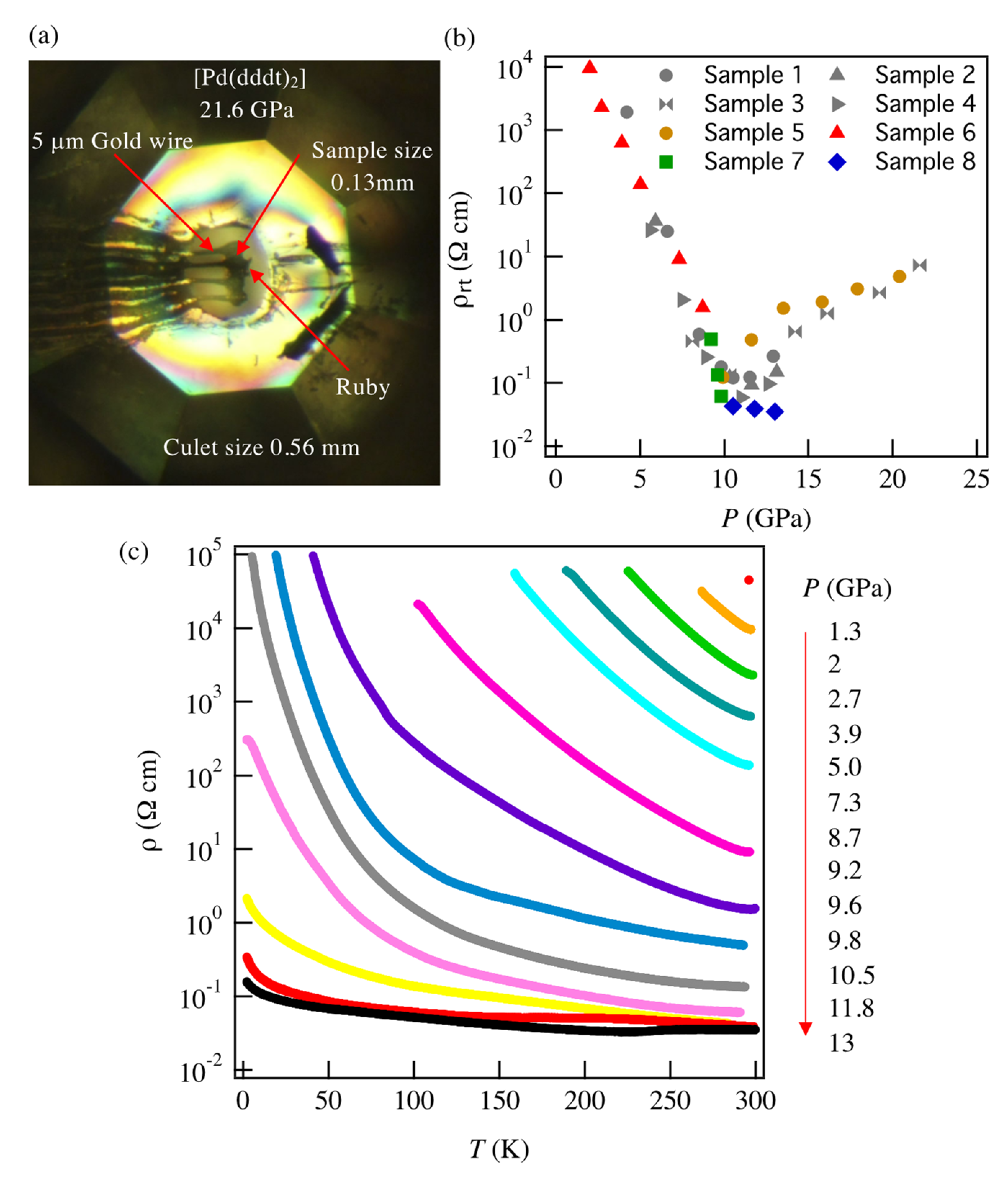
11]. In that work, the HOMO-LUMO energy gap was observed to be 0.65 eV at ambient pressure, and the room temperature resistivity was higher than the measurable range of the equipment. Pressure-dependent resistivity measurements revealed that the room temperature resistivity (
ρrt) and activation energy (
Ea) decrease sharply with increasing pressure above 4.2 GPa and increase slowly with increasing pressure above 12.6 GPa [
12]; electrical properties in the lower pressure region are still unknown.
[Pd(dddt)
2] belongs to the monoclinic crystal system with space group
P2
1/
n (no. 14) [
11]. The unit cell parameters at ambient pressure are as follows:
a = 17.8698(82)) Å,
b = 4.7281(19) Å,
c = 18.4657(85) Å,
β = 111.6313(63)°,
V = 1450.3(1) Å
3,
Z = 4.
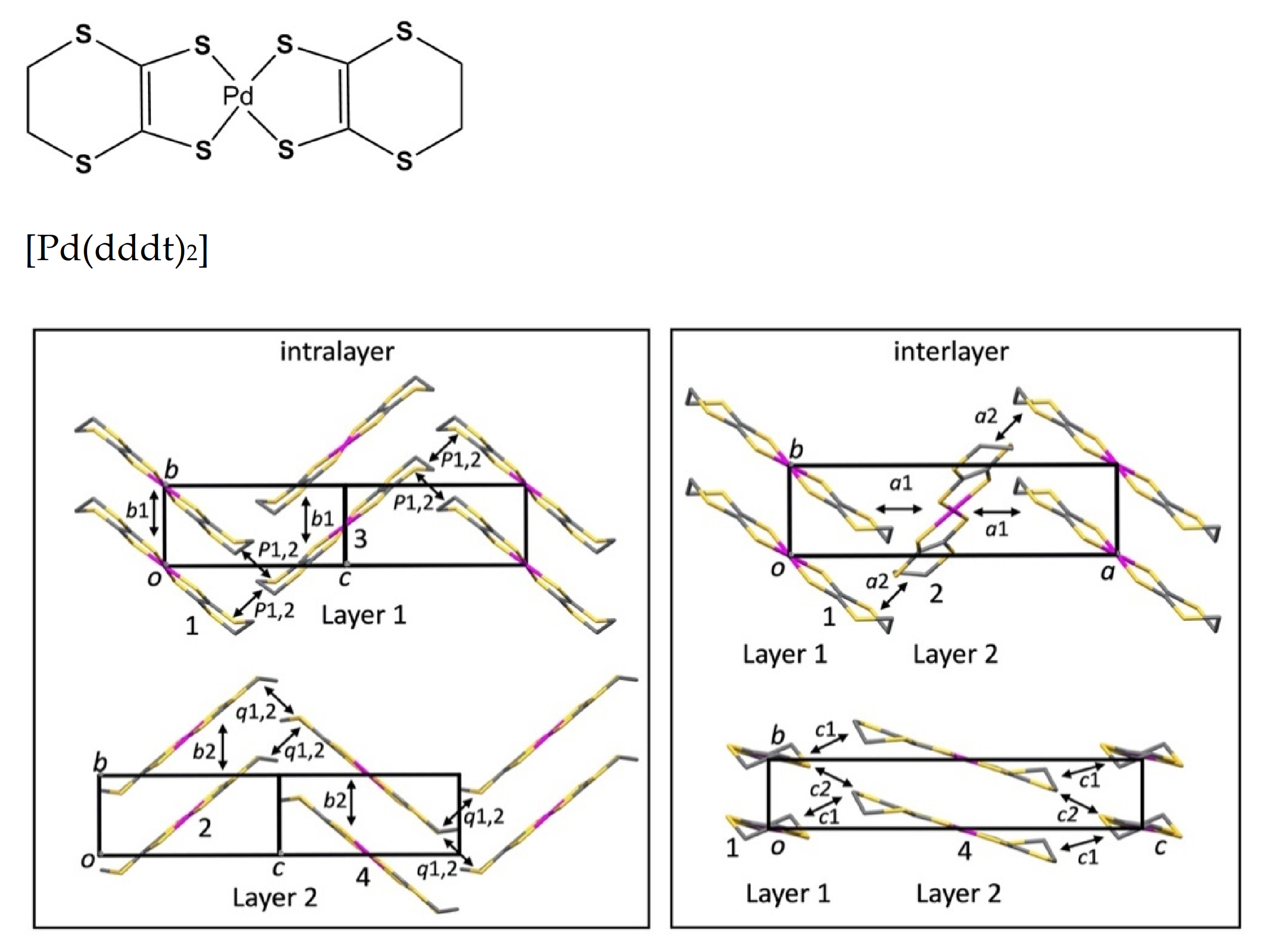
Figure 1 shows the molecular structure and crystal structure of [Pd(dddt)
2]. [Pd(dddt)
2] molecules are uniformly stacked along the
b axis, where Pd atoms are located at inversion centers. The Pd···Pd distance is 4.728 Å, which is identical to the magnitude of the
b axis. The structure may further be thought of as crystallographically independent layers stacked in the
a–
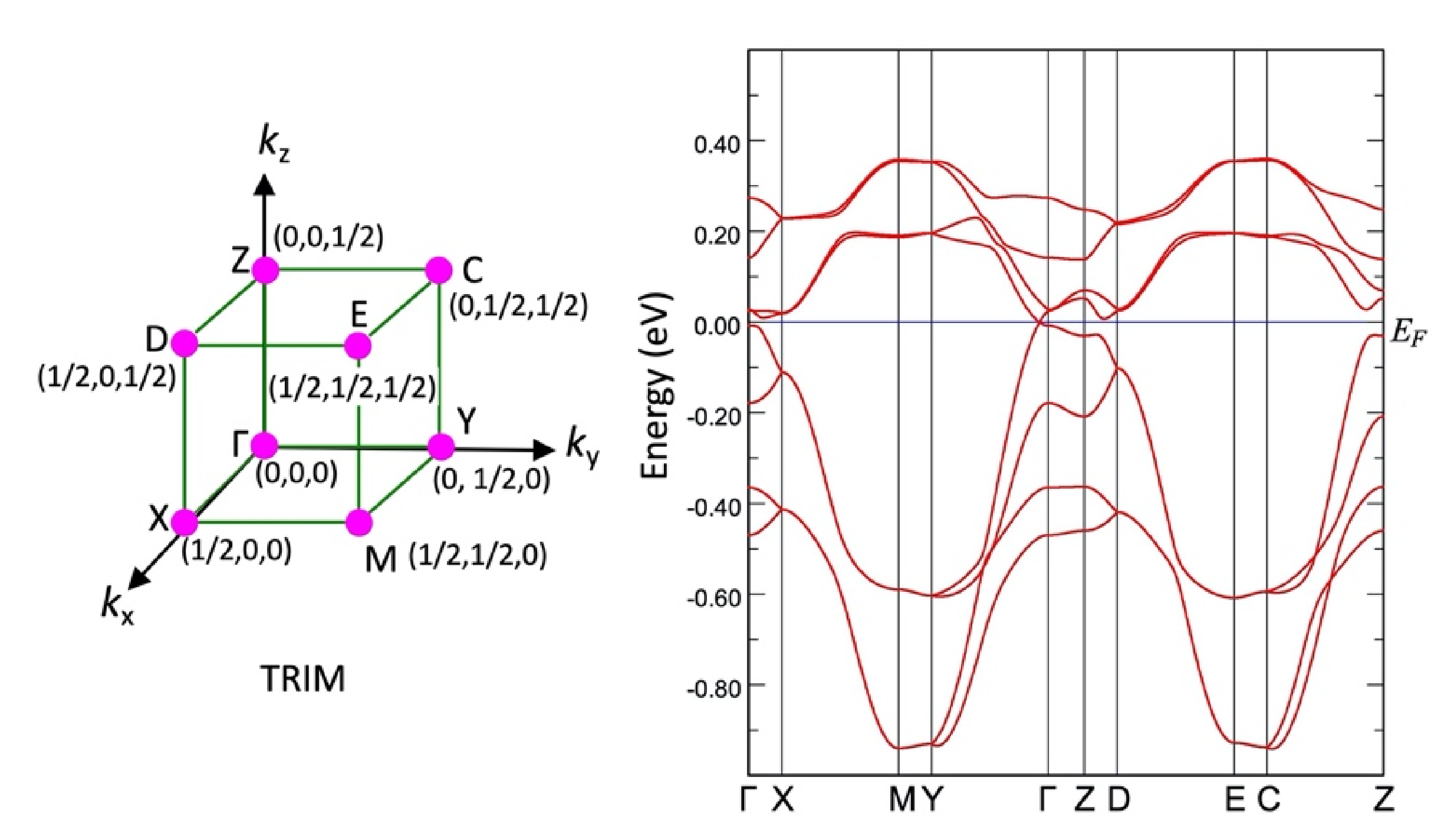
c direction: Layer 1 contains molecules in which the Pd atoms are located on the 2b Wyckoff sites at (0, 0, 1/2) and (1/2, 1/2, 0), while Layer 2 contains molecules in which the Pd atoms are located on the 2a Wyckoff sites at (0, 0, 0) and (1/2, 1/2, 1/2). It is this layered structure that gives rise to the Dirac electron state at high pressure.
In other materials, the Dirac electron system shows interesting quantum magnetotransport phenomena, such as quantum Hall effects (QHEs) reported in graphene [
13] and topological insulators [
14], and linear relationships of magnetoresistance (MR = [
ρ(
B) −
ρ(0)]/
ρ(0)) with magnetic field in iron pnictide, Ba(FeAs)
2, single crystals, when the magnetic field is perpendicular to the current direction at low temperature [
15]. Magnetoresistance (MR) measurements in a bulk two-dimensional Dirac electron system,
α-(BEDT-TTF)
2I
3, showed an unusual negative interlayer MR in the low magnetic field region [
16]. In the case of [Pd(dddt)
2], no MR measurements have been reported to date. Theoretical calculations have shown that it is a nodal line semimetal, in which the Dirac points form a loop in three-dimensional reciprocal space [
17,
18], which may give rise to further interesting MR behavior.
To better understand the high-pressure behavior of [Pd(dddt)2], we report a series of new measurements and calculations that verify previous theoretical calculations and provide insight into the Dirac electron state. In particular, we report more detailed high-pressure resistivity measurements, the first experimental structure determinations performed at high pressure using single-crystal synchrotron X-ray diffraction, tight binding calculations performed using these data, and the first high-pressure MR measurements performed on single crystals of [Pd(dddt)2]. We find that the new experimental crystal structures verify previous theoretical reports of the Dirac electron state and suggest that changes in resistivity may be affected by structural disorder. Tight binding calculations indicate that the Dirac electron state coexists with semiconducting behavior, also owing to the disorder. The new MR measurements are also indicative of exotic quantum transport behavior: negative MR is observed at low temperatures and the curvature of the field-dependent MR is heavily dependent on crystal orientation.
4. Discussion
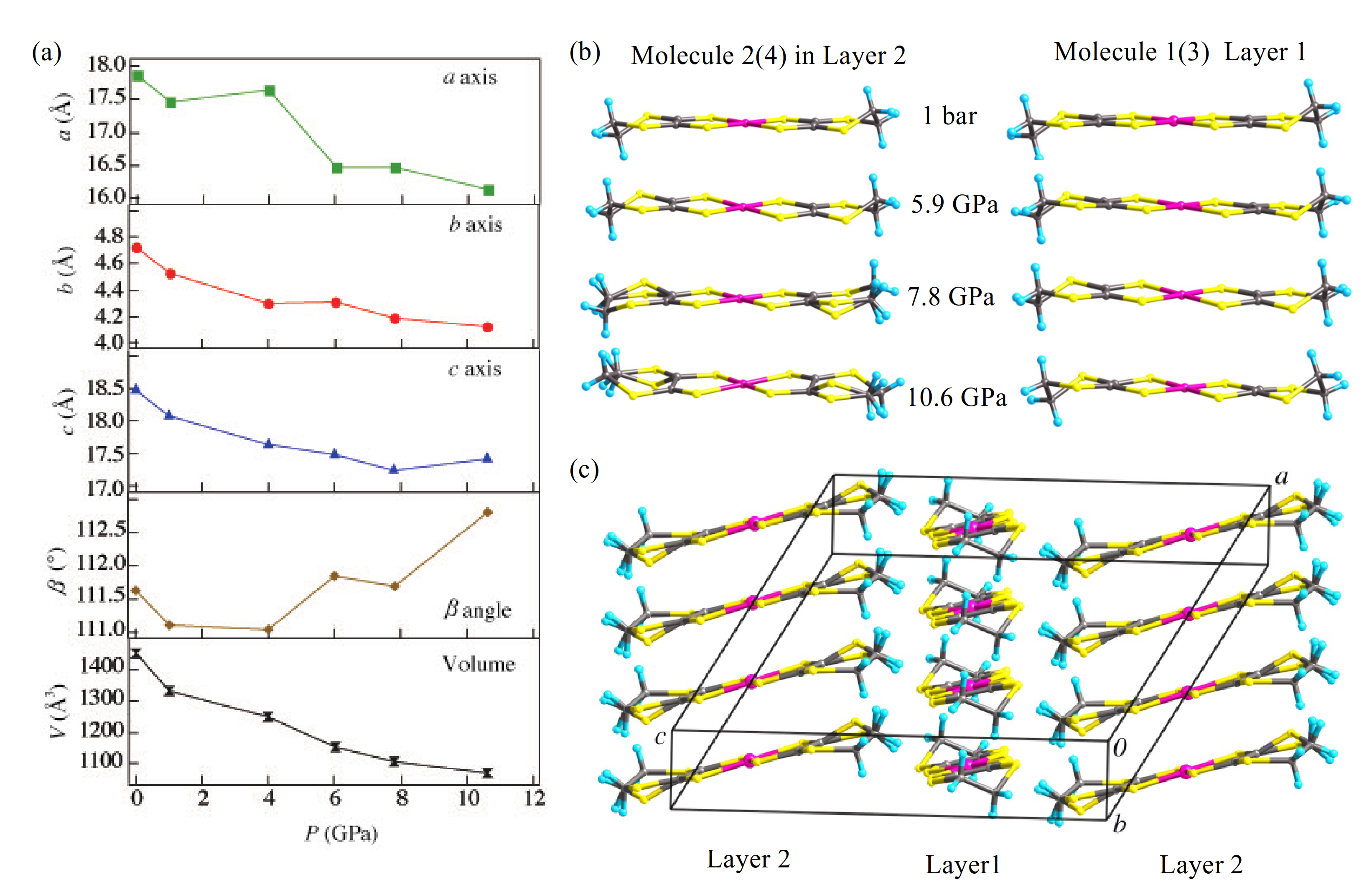
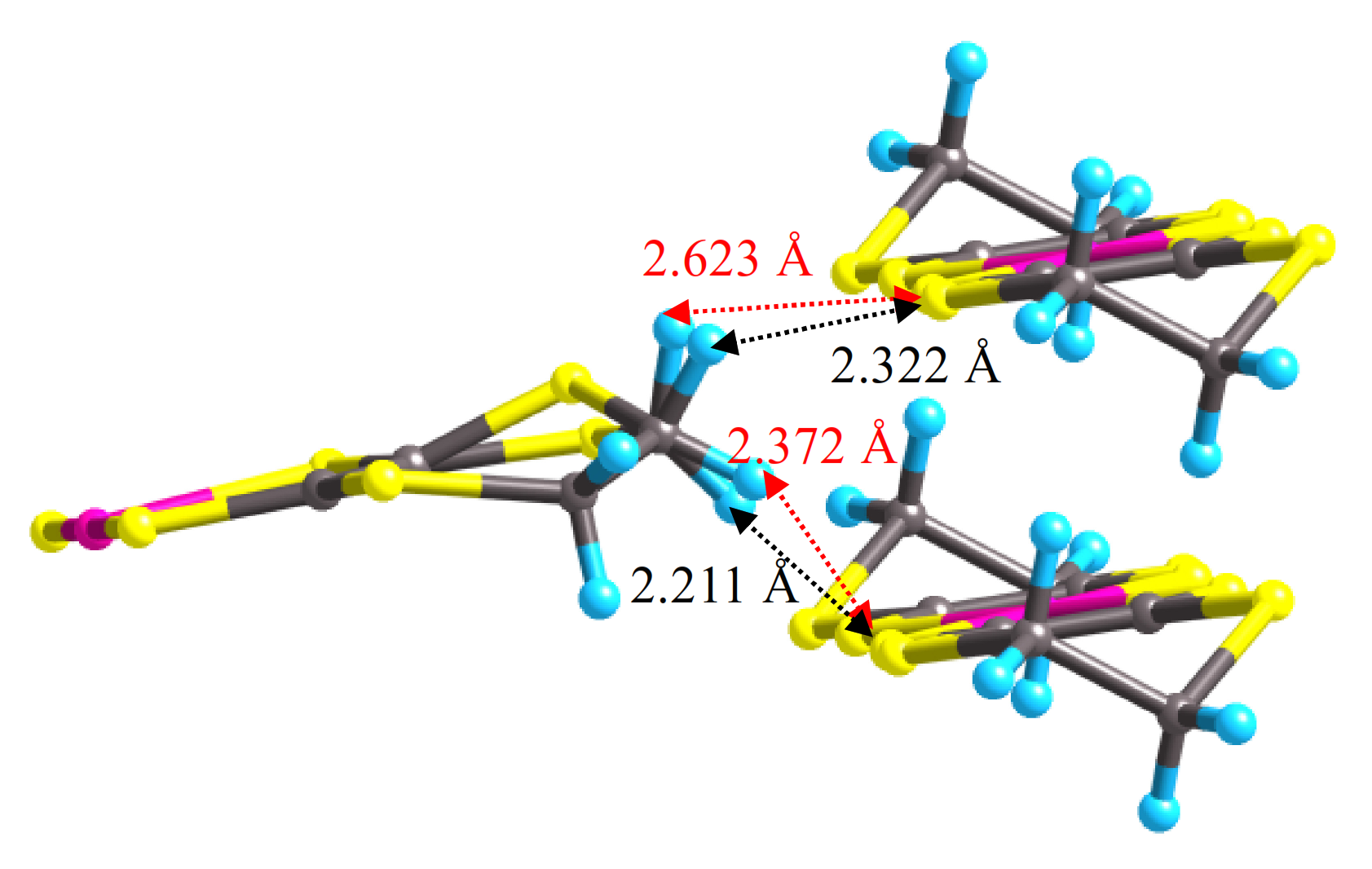
For isostructural single-component molecular conductors, the contribution of the central transition metal
d-orbitals to the LUMO is often assumed to be similar. However, our experimental results show that the central metal substitution in [M(dddt)
2] (M = Ni, Pd), directly effects the high-pressure properties and structure. In contrast to the high-pressure structure of the Ni analogue [
29], the Pd-Pd distance in [Pd(dddt)
2] decreases smoothly from 4.7281 Å at 1 bar to 4.117 Å at 10.6 GPa. Unlike [Ni(dddt)
2], which does not exhibit a structural transition up to 11.2 GPa [
29], [Pd(dddt)
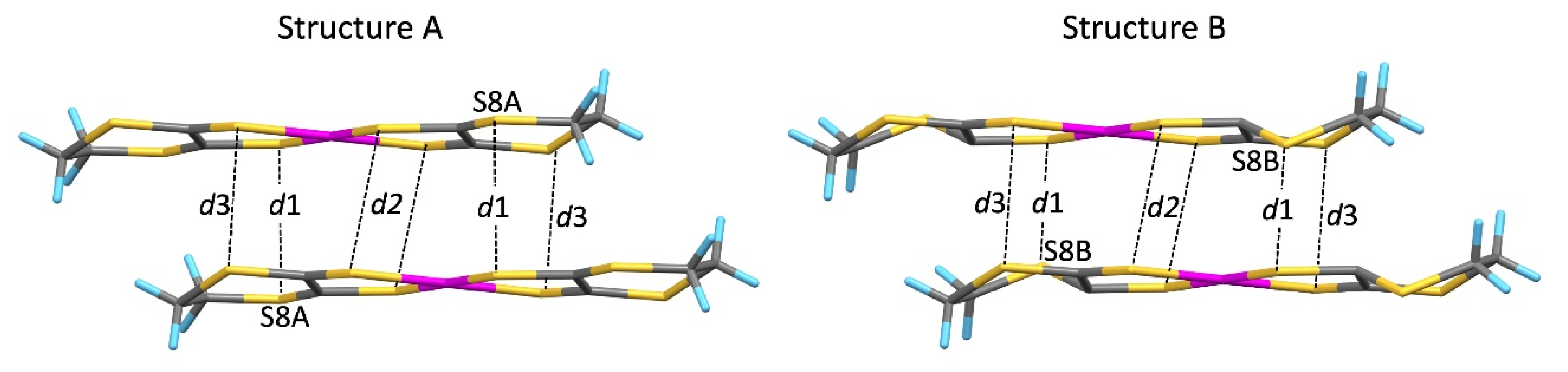
2] undergoes an order–disorder transition, which may be the cause of the increase in resistivity and coexistence of the Dirac cone state with semiconductivity in the higher-pressure region.
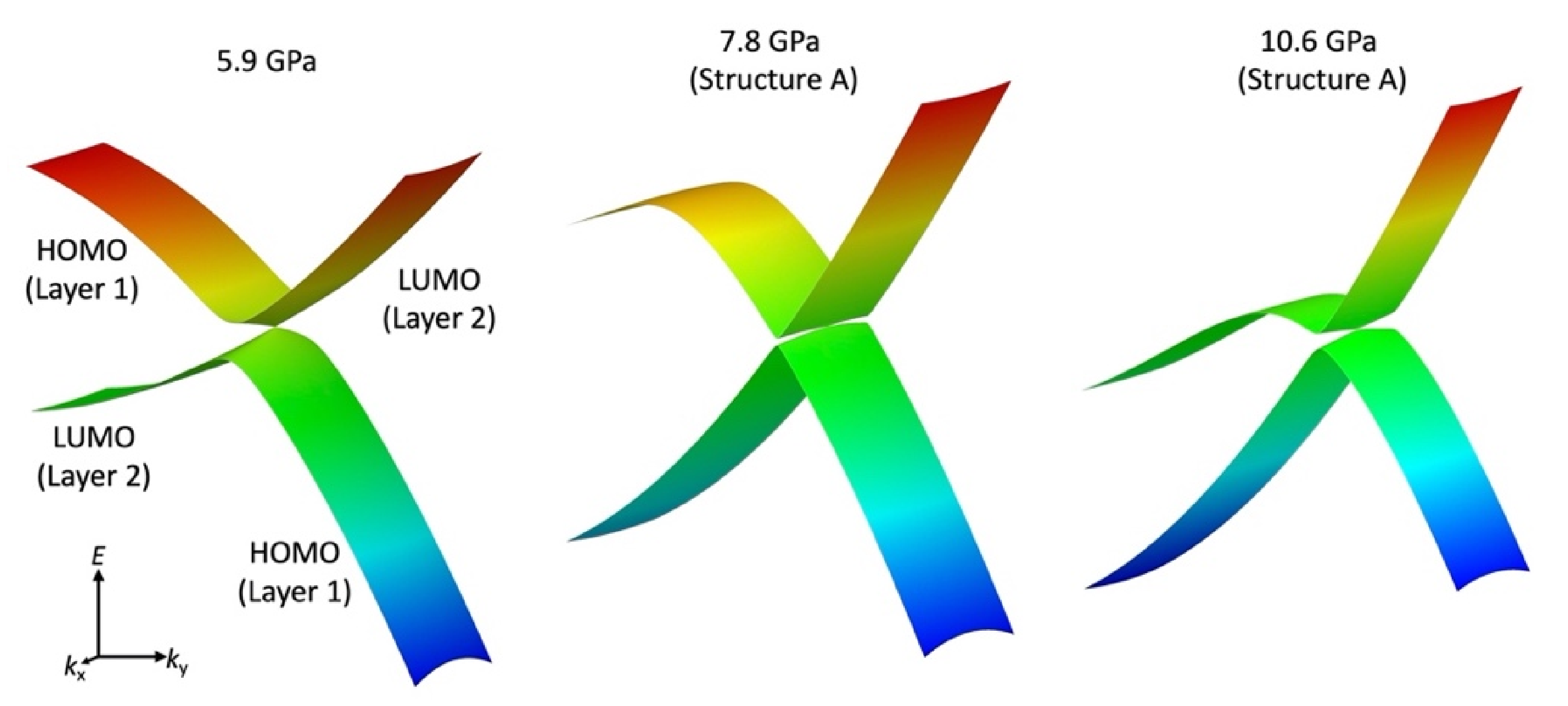
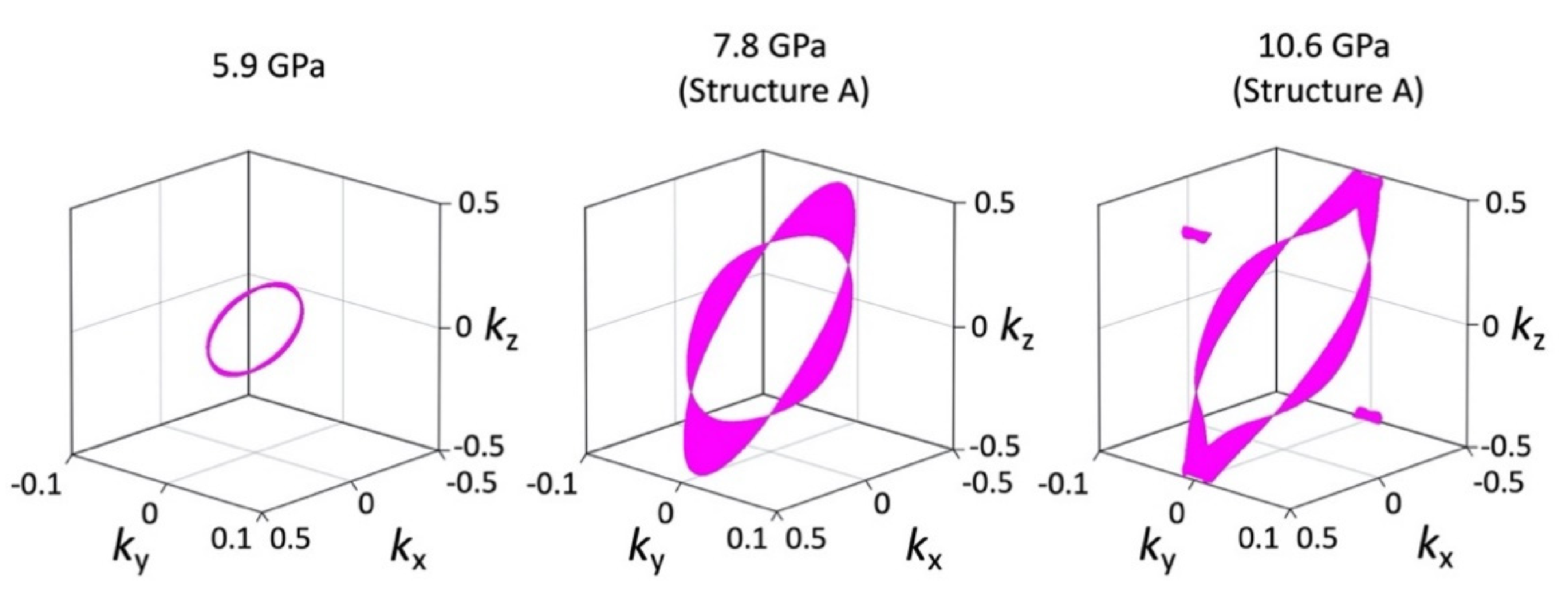
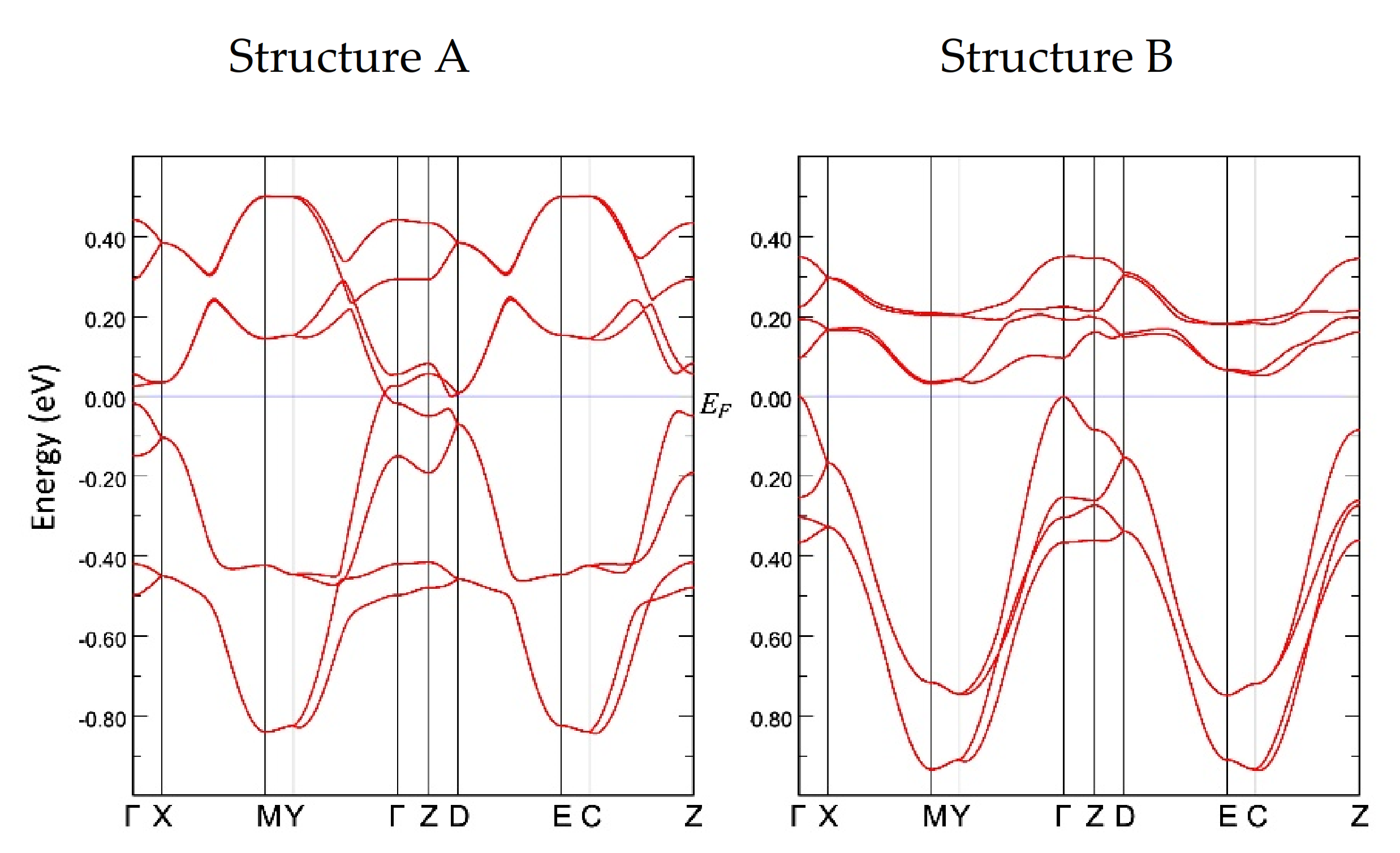
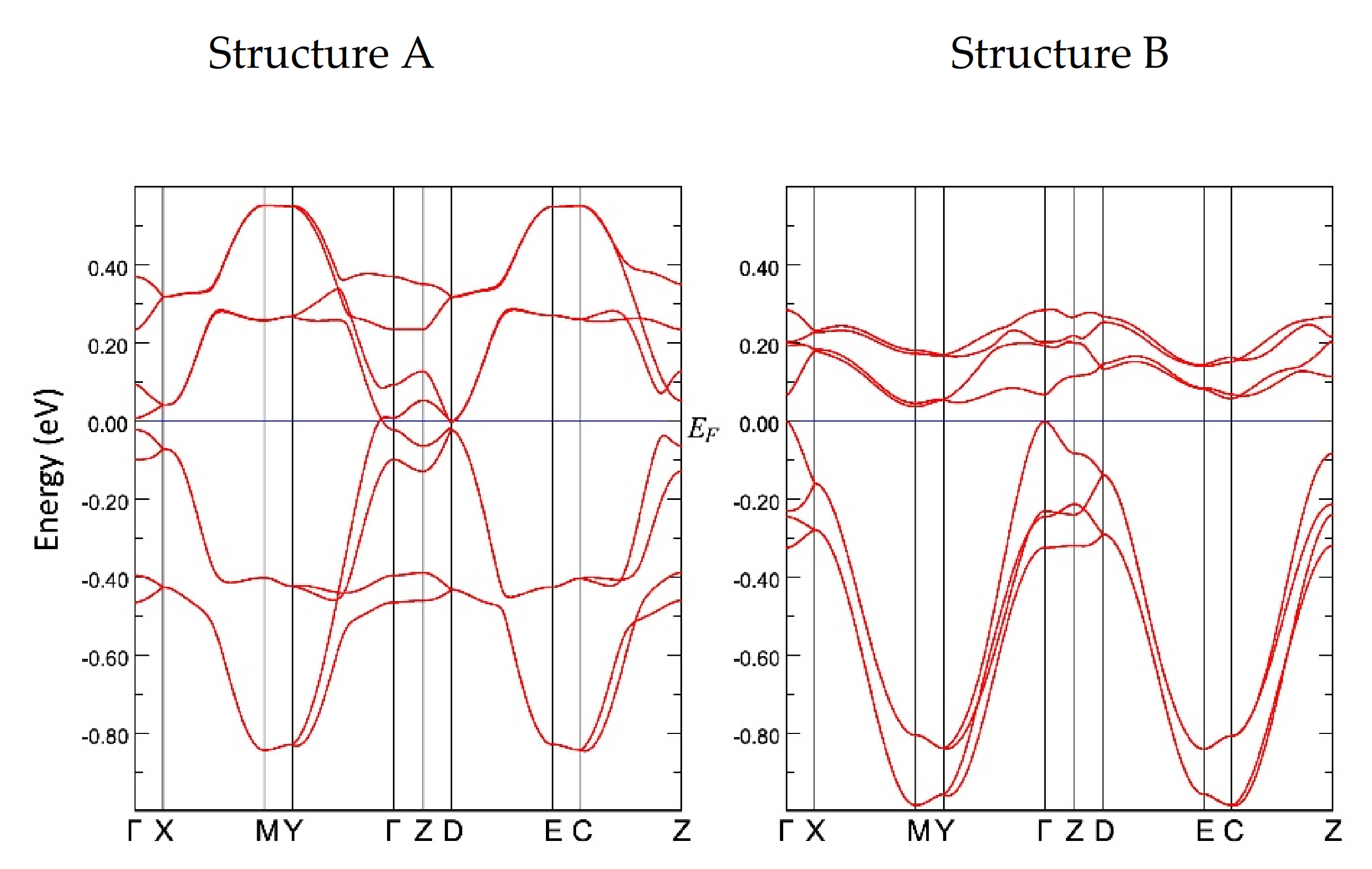
The tight-binding band structure calculations using the experimentally determined crystal structures show that the Dirac fermion state appears at least from 5.9 GPa. At 7.8 GPa and 10.6 GPa, the structural disorder causes coexistence of the Dirac cone state and a semiconducting state. This semiconductor gap state steadily appears in the resistivity data, manifesting as increasing resistivity.
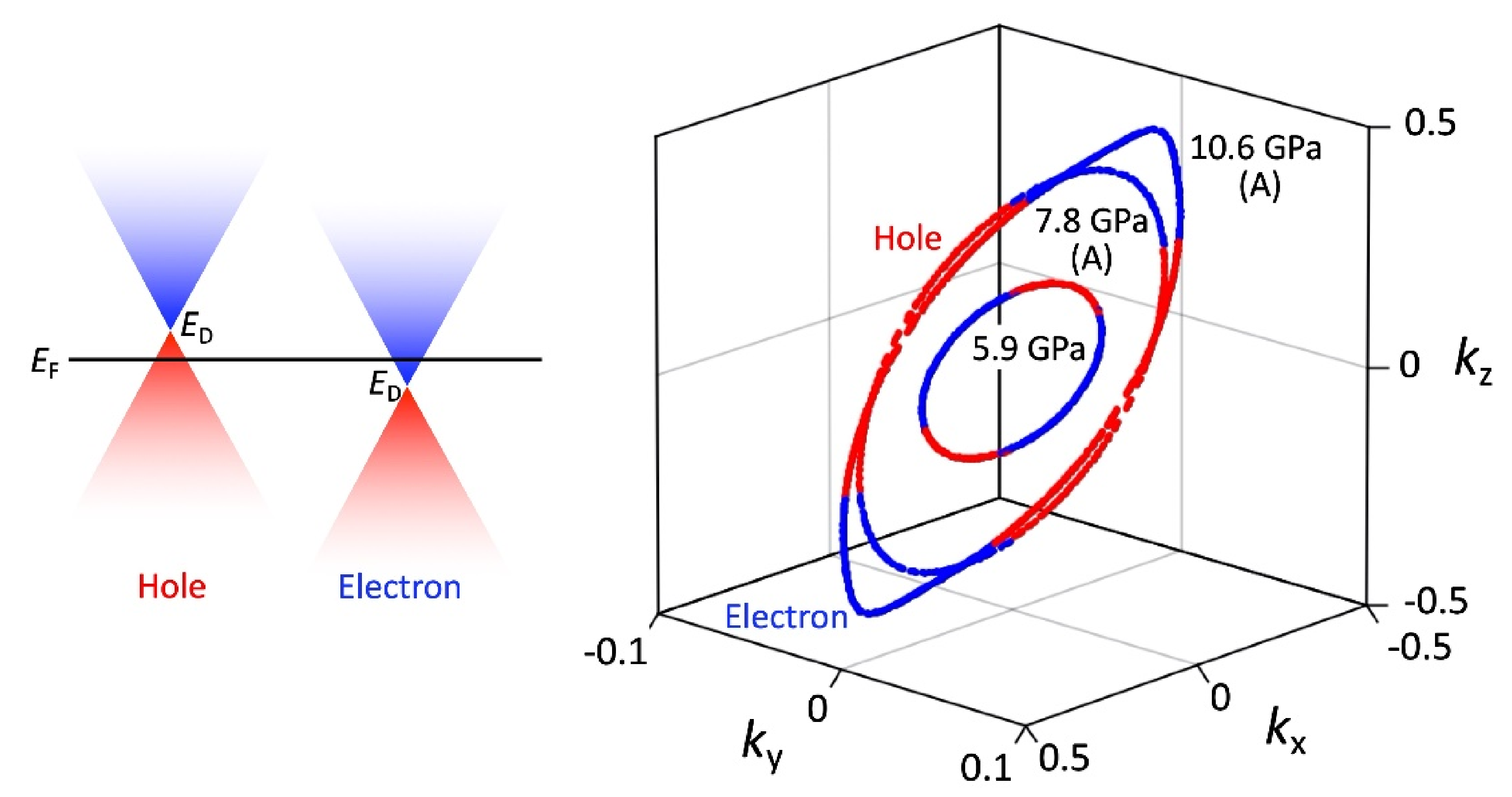
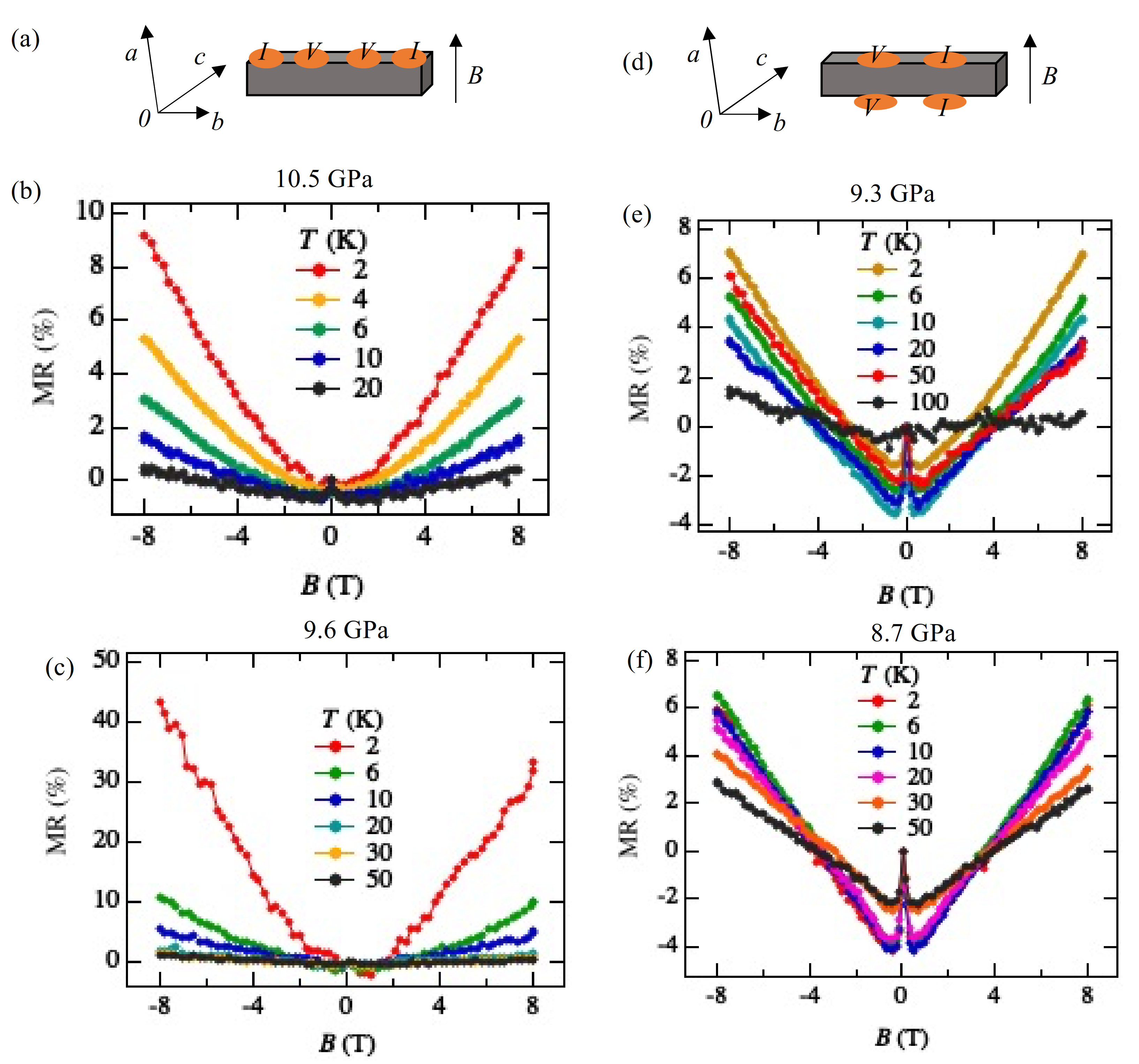
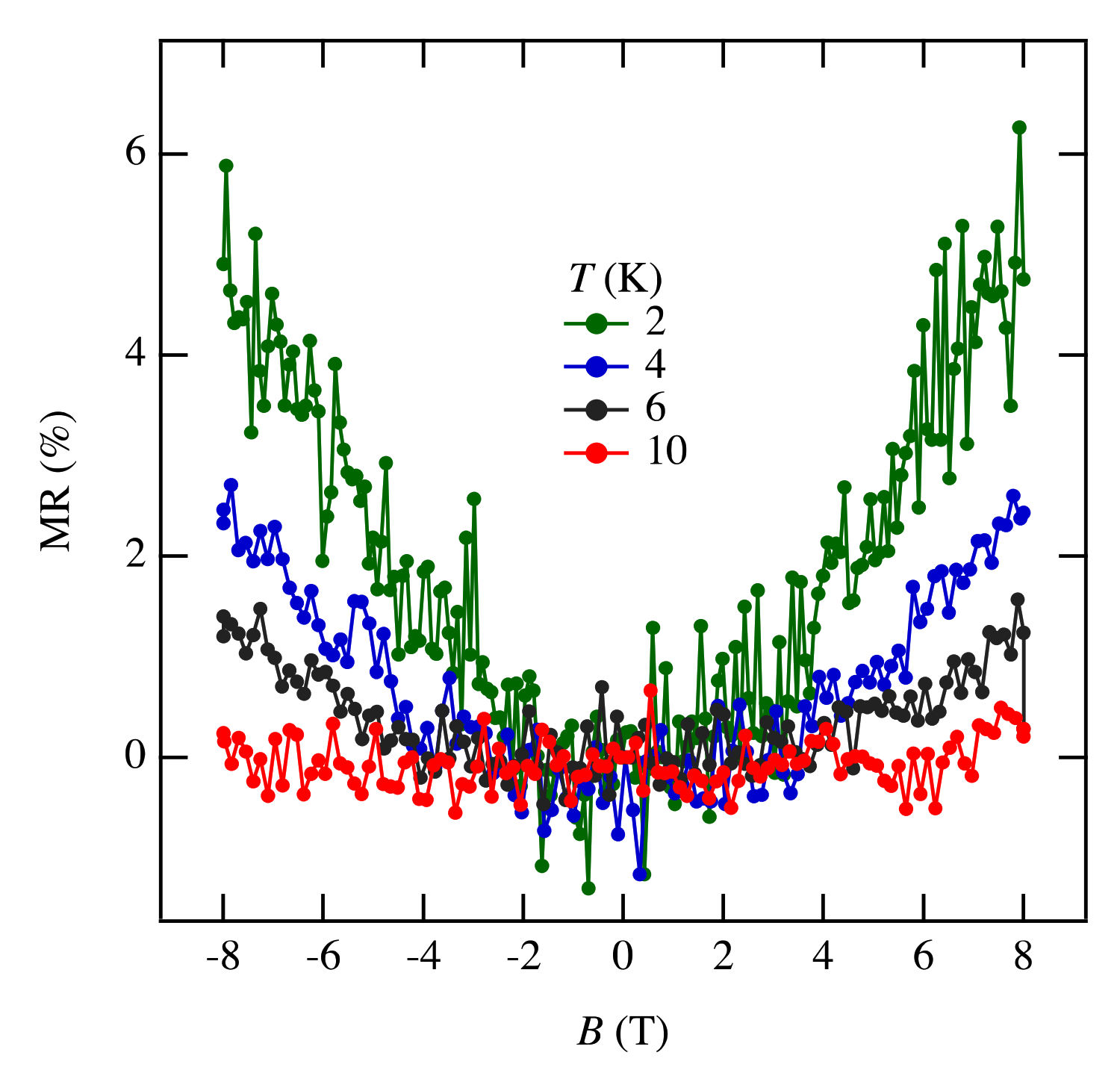
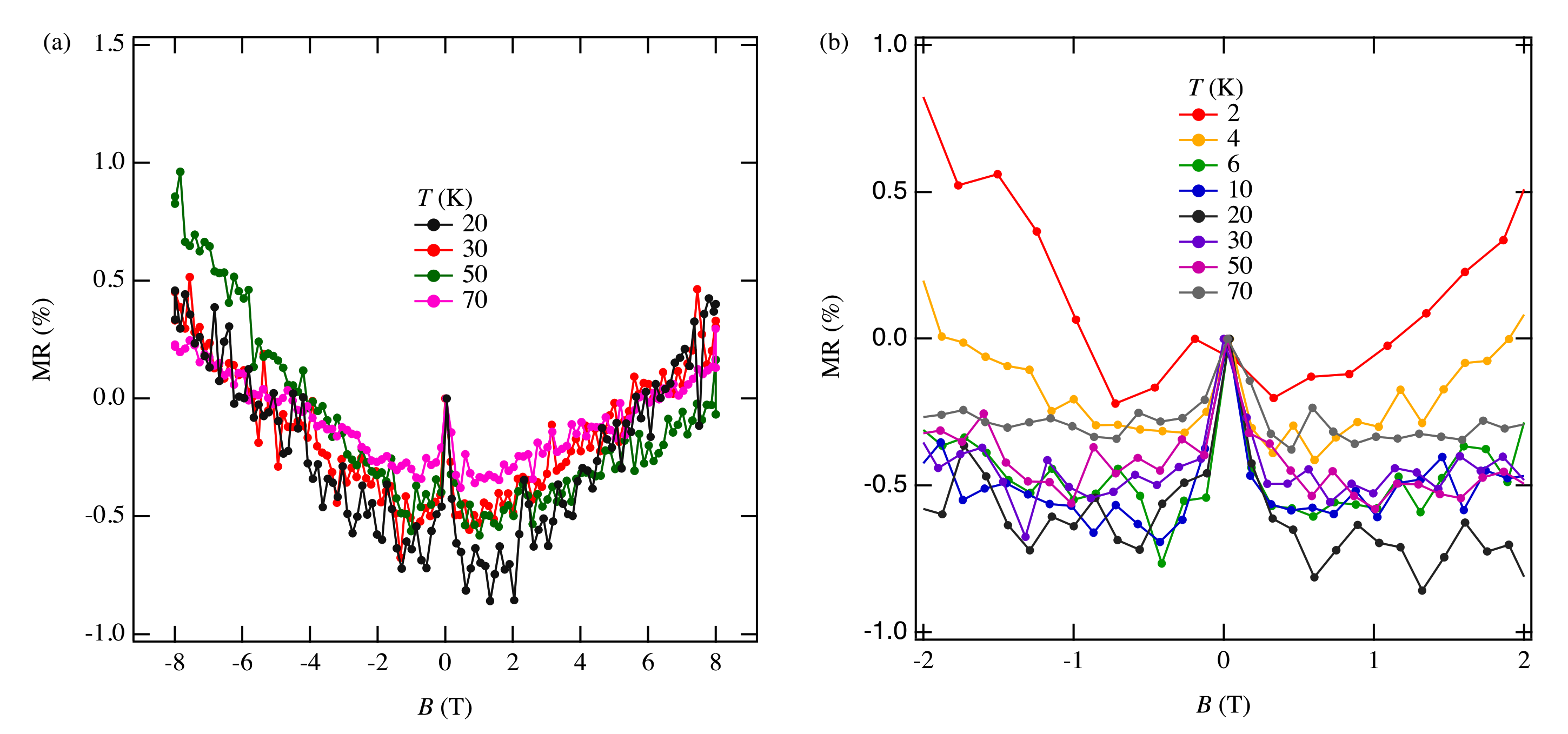
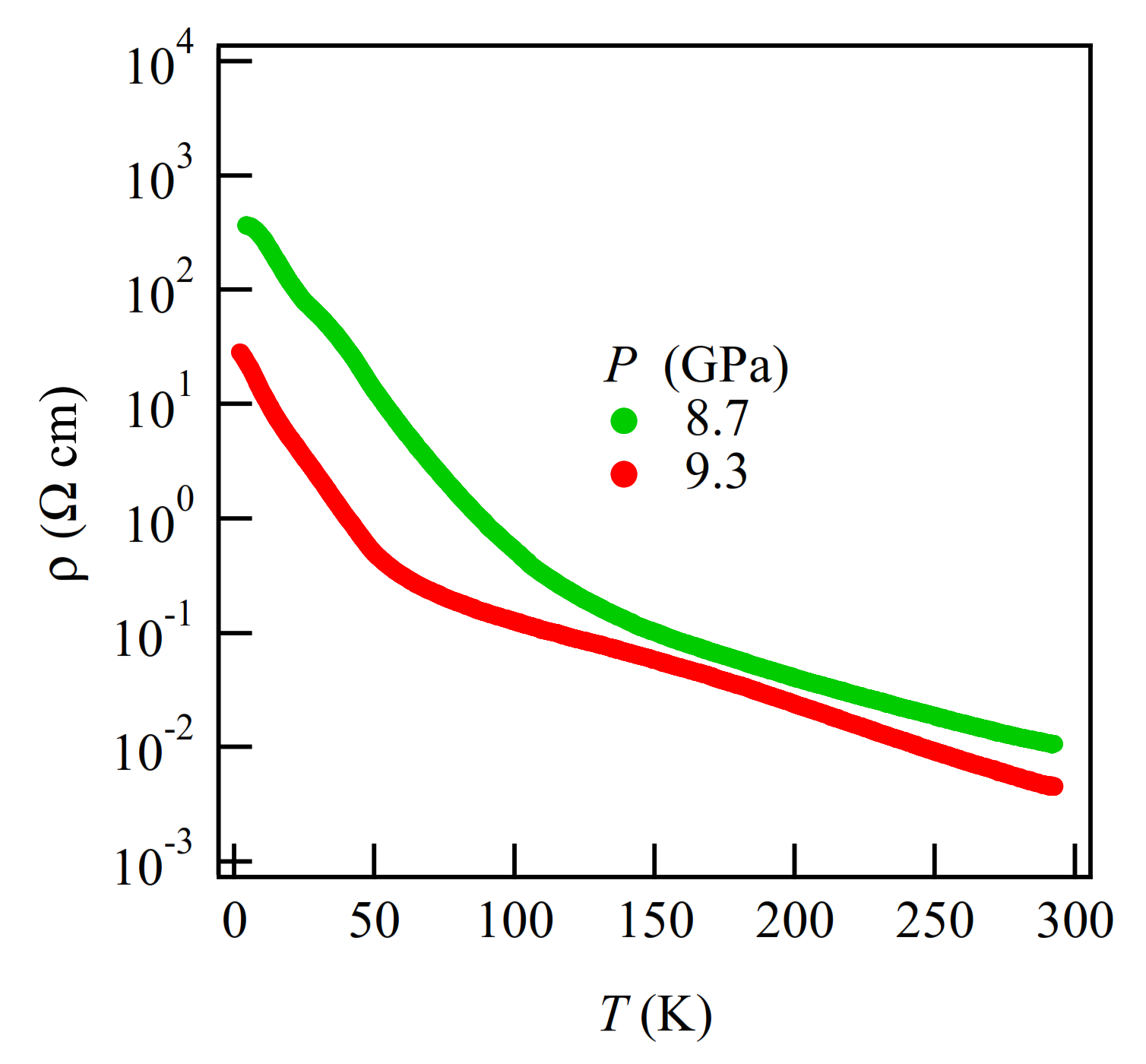
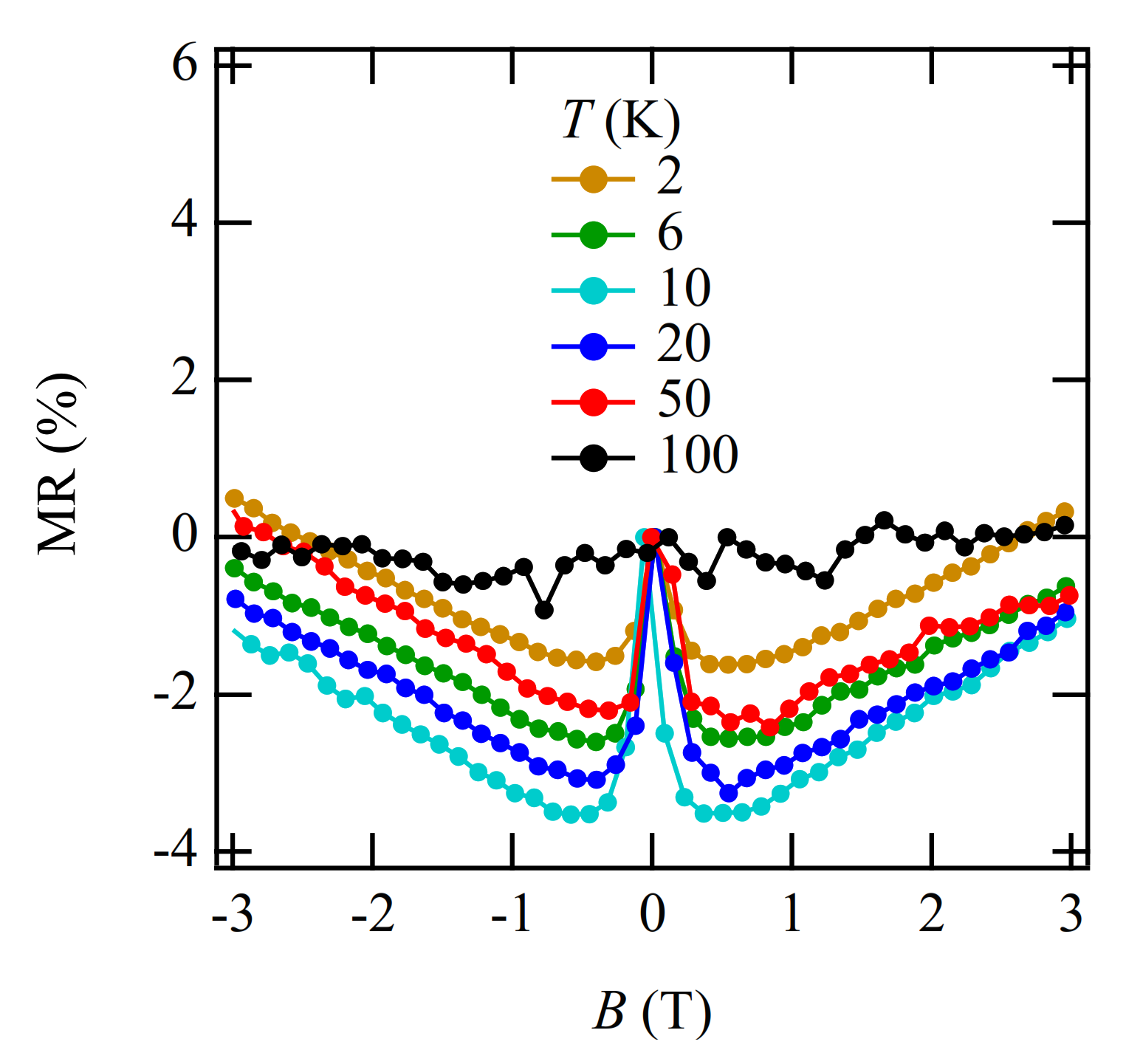
The MR measurements show a linear relationship over wide temperature and pressure ranges. If the MR linear relationship is correlated with the Dirac electron, our results indicate that Dirac cones emerged in the pressure range from 8.7 GPa to 10.5 GPa at least. This result is consistent with the theoretical calculations for resistivity under pressure that indicates a nodal line semimetal state from 7.7 GPa to 8 GPa [
34]. In this calculation, a weak metal-like behavior was also observed near the room temperature, which is in good agreement with the measured resistivity at 13 GPa. Previous studies have suggested that the temperature-independent resistivity at 12.6 GPa may be associated with the Dirac electron state [
11]. However, in the case of Sample 7 under the same sample mounting conditions as the previous measurement, no linear MR results were observed above 12.5 GPa. It is possible that the sample mounting direction was not good for MR measurements or the disordered structure may not show linear MR. To fully understand the Dirac electronic state of this material, it is necessary to measure the Hall effect by applying electric current and magnetic field in different crystal directions. The topological property of wave function given by the Berry phase [
35] is important for understanding Dirac nodal lines; it is predicted that the Hall effect may be observed when the Berry phase component is oriented parallel to the magnetic field [
34]. To clarify these issues, additional MR measurements and Hall effect measurements are ongoing.
Regarding the negative MR below 1 T, such behavior has been previously observed in weakly localized semiconductor CrVTiAl thin films [
36] or multilayer graphene [
37]. Interestingly, in the absence of the Dirac cone, the negative MR quickly disappeared above 10 K [
36]. In the multilayer graphene measurement, the magnetic field was parallel to the current direction [
37]. MR measurements of few-layer graphene with the magnetic field perpendicular to current direction showed that increasing the number of layers enhances the weak localization effect [
38]. In both of those experiments the negative MR appears in magnetic fields below 1 T and persists up to 50 K. By comparing these results with ours, we suggest that the negative MR observed in [Pd(dddt)
2] can be understood through the localization of the Dirac electron system.
Finally, we note that the critical pressure for observation of the Dirac electron state appears to be different depending on hydrostatic conditions in different pressure cells, pressure media and in silico pressurization methods. Our resistivity measurements show the smallest band gap at 13 GPa, while first principles DFT calculations showed that the Dirac cones appear at 8 GPa. However, the result of tight-binding calculations using the structure data from high-pressure crystal structure measurements indicates that Dirac cones appear at 5.9 GPa [
31]. Several factors make the different methods challenging to compare directly. On one hand, solidification of pressure transmitting media, which occurs at different pressures depending on composition, can induce uniaxial effects in resistivity measurements. To mitigate this risk, the samples involved in both the structural and the electrical measurements were covered with a protective layer to improve the hydrostatic condition. On the other hand, the generalized gradient approximation (GGA) used in first principles calculations cannot describe long-range attractive van der Waals interactions (dispersion forces), which affects the estimation of lattice parameters in the lower pressure region. In addition, GGA calculations tend to underestimate electronic band gaps, which induces a difference between the calculated and experimental pressures [
39].




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}