
Dicarboxylic Acid-Based Co-Crystals of Pyridine Derivatives Involving Structure Guiding Unconventional Synthons: Experimental and Theoretical Studies
 , ,
, ,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Methods
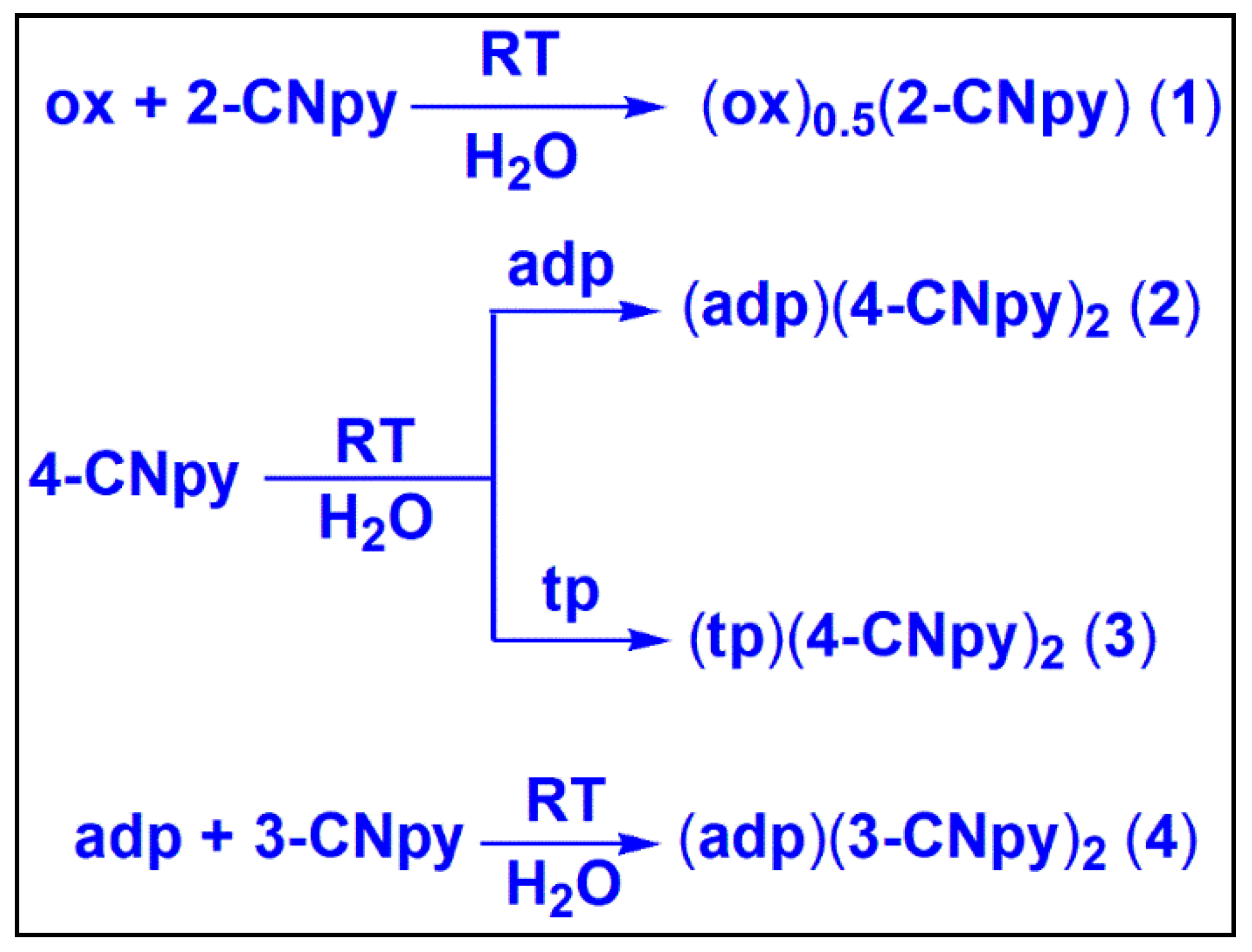
2.2. Syntheses of the Co-Crystals
2.3. Crystallographic Data Collection and Refinement
2.4. Computational Methods
3. Results
3.1. Syntheses and General Aspects
3.2. pKa-Rule
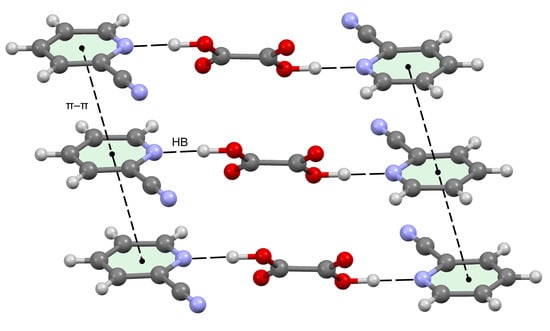
3.3. Crystal Structure Analysis
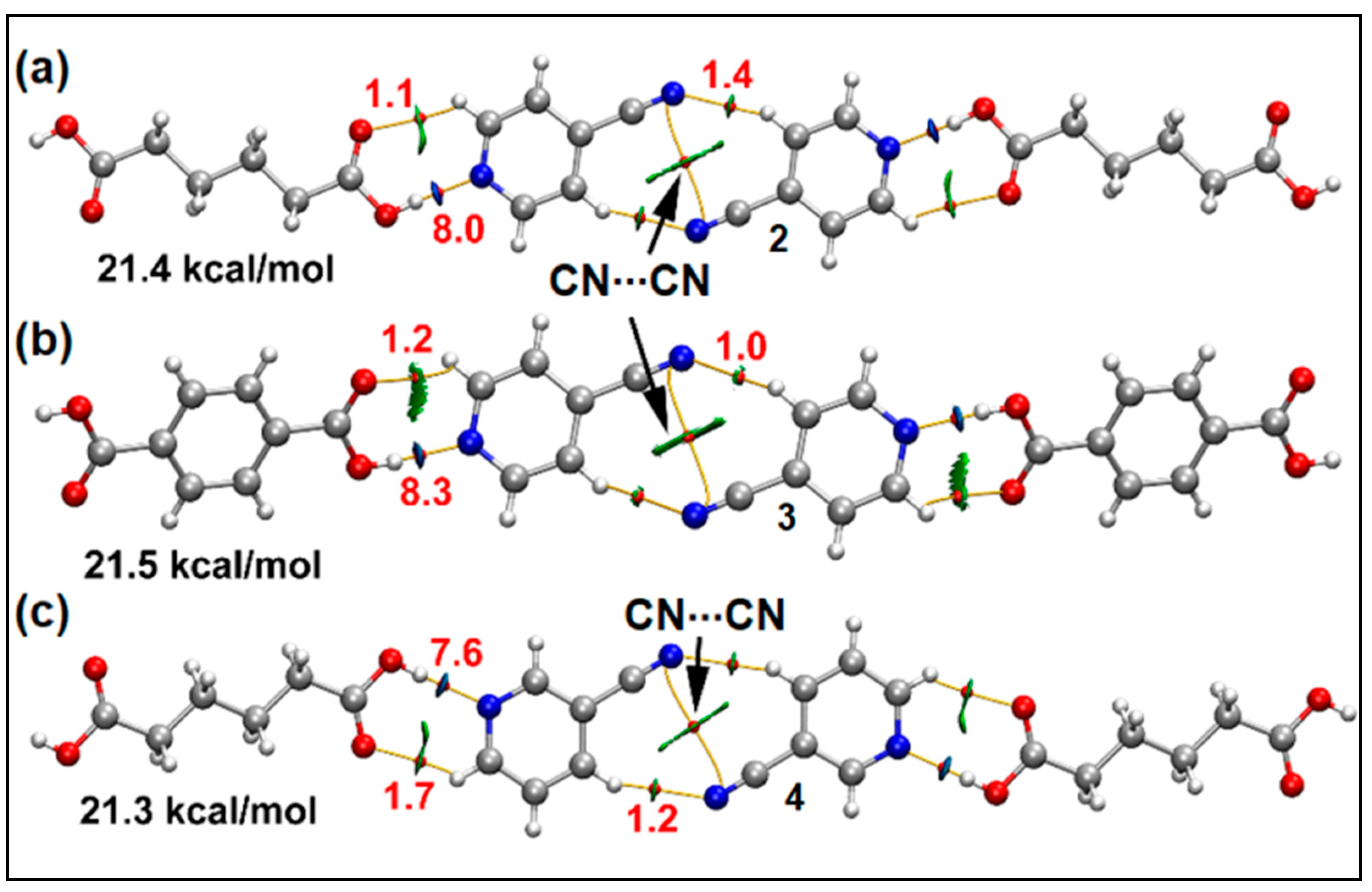
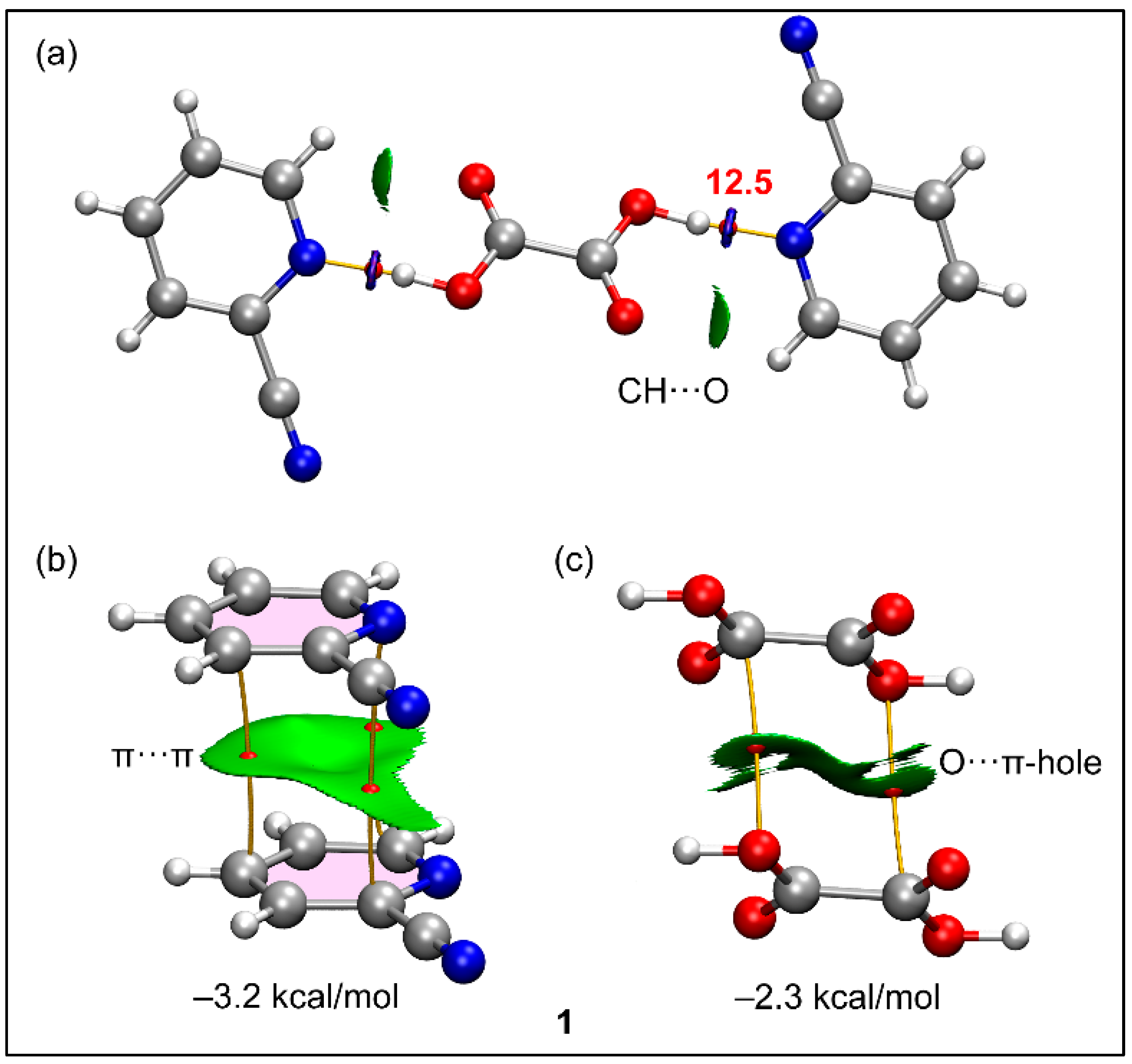
3.4. Theoretical Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chameli, D.; Bhavna, J.; Manish, K. Co-crystals of valsartan: Preparation and characterization. Res. J. Pharm. Dosage Forms Technol. 2019, 11, 143–146. [Google Scholar]
- Jayram, P.; Sudheer, P. Pharmaceutical co-crystals: A systematic review. Int. J. Pharm. Investig. 2020, 10, 246–256. [Google Scholar] [CrossRef]
- Wen, X.; Lu, Y.; Jin, S.; Zhu, Y.; Liu, B.; Wang, D.; Chen, B.; Wang, P. Crystal structures of six salts from nicotinamide and organic acids by classical H-bonds and other non-covalent forces. J. Mol. Struct. 2022, 1254, 132332–132345. [Google Scholar] [CrossRef]
- Yadav, A.V.; Shete, A.S.; Dabke, A.P.; Kulkarni, P.V.; Sakhare, S.S. Co-crystals: A novel approach to modify physicochemical properties of active pharmaceutical ingredients. Indian J. Pharm. Sci. 2009, 71, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishweshwar, P.; McMahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, J.; Mei, X. Enhancing the stability of active pharmaceutical ingredients by the cocrystal strategy. Cryst. Eng. Comm. 2022, 22, 2015–2032. [Google Scholar] [CrossRef]
- O’Sullivan, A.; Long, B.; Verma, V.; Ryan, K.M.; Padrela, L. Solid-state and particle size control of pharmaceutical co-crystals using atomization-based techniques. Int. J. Pharm. 2022, 1, 121798–121821. [Google Scholar] [CrossRef] [PubMed]
- Nikhar, R.; Szalewicz, K. Reliable crystal structure predictions from first principles. Nat. Commun. 2022, 13, 3095–3107. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Fasulo, E.; Desper, J. Cocrystal or salt: Does it really matter? Mol. Pharm. 2007, 7, 317–322. [Google Scholar] [CrossRef]
- Thayyil, A.R.; Juturu, T.; Nayak, S.; Kamath, S. Pharmaceutical co-crystallization: Regulatory aspects, design, characterization, and applications. Adv. Pharm. Bull. 2020, 10, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Samineni, R.; Chimakurthy, J.; Sumalatha, K.; Dharani, G.; Rachana, J.; Manasa, K.; Anitha, P. Co-crystals: A review of recent trends in co crystallization of BCS class II drugs. Res. J. Pharm. Technol. 2019, 12, 3117–3124. [Google Scholar] [CrossRef]
- Ngilirabanga, J.B.; Samsodien, H. Pharmaceutical co-crystal: An alternative strategy for enhanced physicochemical properties and drug synergy. NanoSelect 2021, 2, 512–526. [Google Scholar] [CrossRef]
- Nugrahani, I.; Parwati, R.D. Challenges and progress in nonsteroidal anti-inflammatorydrugs co-crystal development. Molecules 2021, 26, 4185. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.I.; Boese, R.; Schauerte, C.; Merz, K. An experimental and theoretical approach to control salt vs cocrystal vs hybrid formation-crystal engineering of an E/Z-butenedioic acid/phthalazine system. Cryst. Growth Des. 2019, 19, 1616–1620. [Google Scholar] [CrossRef]
- Dai, X.; Chen, J.; Lu, T. Pharmaceutical cocrystallization: An effective approach to modulate the physicochemical properties of solid-state drugs. CrystEngComm 2018, 20, 5292–5316. [Google Scholar] [CrossRef]
- McNeil, S.K.; Kelley, S.P.; Beg, C.; Cook, H.; Rogers, R.D.; Nikles, D.E. Co-crystals of 10-methylphenthiazine and 1,3-dinitrobenzene: Implications for the optical sensing of TNT-based explosives. ACS Appl. Mater. Interfaces 2013, 5, 7647–7653. [Google Scholar] [CrossRef]
- Bucar, D.K.; MacGillivray, L.R. Preparation and reactivity of nano-crystalline co-crystals formed via sono-crystallization. J. Am. Chem. Soc. 2007, 129, 32–33. [Google Scholar] [CrossRef]
- Stoler, E.; Warner, J.C. Non-covalent derivatives: Co-crystals and eutectics. Molecules 2015, 20, 14833–14848. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Nanda, A. Pharmaceutical co-crystals: An overview. Indian J. Pharm. Sci. 2018, 20, 858–870. [Google Scholar]
- Dutta, D.; Sharma, P.; Gomila, R.; Frontera, A.; Barcelo-Oliver, M.; Verma, A.K.; Baruwa, B.; Bhattacharyya, M.K. Solvent-driven structural topologies in phenanthroline-based co-crystals of Zn(II) involving fascinating infinite chair-like {[(bzH)4Cl2]2−}n assemblies and unconventional layered infinite {bz-H2O-Cl}n anion-water clusters: Antiproliferative evaluation and theoretical studies. New J. Chem. 2022, 46, 5638–5652. [Google Scholar]
- Ashfaq, M.; Ali, A.; Kuznetsov, A.; Tahir, M.N.; Khalid, M. DFT and single-crystal investigation of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium-4-methylbenzoate hydrate (1:1:1) (DEMH). J. Mol. Struct. 2021, 1228, 129445–129459. [Google Scholar] [CrossRef]
- Iizuka, M.; Nakagawa, Y.; Moriya, Y.; Satou, E.; Fujimori, A. Comparison of structure/function correlational property of three kinds of gemini-type thixotropic surfactants capable of forming crystalline nanofiber based on hydrogen bonding-solid-state structure, two-dimensional molecular film forming, and epitaxial growth behavior. Bull. Chem. Soc. Jpn. 2018, 91, 813–823. [Google Scholar]
- Zheng, Q.; Rood, S.L.; Unruh, D.K.; Hutchins, K.M. O-crystallization of anti-inflammatory pharmaceutical contaminants and rare carboxylic acid-pyridine supramolecular synthon breakdown. CrystEngComm 2018, 20, 6377–6381. [Google Scholar] [CrossRef]
- Ilyukhin, A.B.; Koroteev, P.S.; Novotortsev, V.M. Supramolecular interactions and self-assembling in adducts of cymantrenecarboxylic acid with amino derivatives of five- and six-membered heterocyclic N-bases. J. Mol. Struct. 2019, 1187, 38–49. [Google Scholar] [CrossRef]
- Sangeetha, M.; Mathammal, R. Establishment of the structural and enhanced physicochemical properties of the cocrystal-2-benzyl amino pyridine with oxalic acid. J. Mol. Struct. 2017, 1143, 192–203. [Google Scholar] [CrossRef]
- Wheeler, S.E. Local nature of substituent effects in stacking interactions. J. Am. Chem. Soc. 2011, 133, 10262–10274. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Substituent effects in the benzene dimer are due to direct interactions of the substituents with the unsubstituted benzene. J. Am. Chem. Soc. 2008, 130, 10854–10855. [Google Scholar] [CrossRef] [Green Version]
- Sikorski, A.; Trzybinski, D. Networks of intermolecular interactions involving nitro groups in the crystals of three polymorphs of 9-aminoacridinium 2,4-dinitrobenzoate-2,4-dinitrobenzoic acid. J. Mol. Struct. 2013, 1049, 90–98. [Google Scholar] [CrossRef]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting anion-π interactions into perspective. Angew. Chem. Int. Ed. 2011, 50, 9564–9583. [Google Scholar] [CrossRef]
- Schottel, B.L.; Chifotides, H.T.; Dunbar, K.R. Anion-π interactions. Chem. Soc. Rev. 2008, 37, 68–83. [Google Scholar] [CrossRef]
- Caltagirone, C.; Gale, P.A. Anion receptor chemistry: Highlights from 2007. Chem. Soc. Rev. 2009, 38, 520–563. [Google Scholar] [CrossRef] [PubMed]
- Ojala, C.R.; Ojala, W.H.; Britton, D. Solid-State intermolecular contacts involving the nitrile group in p-cyano-N-(p-cyanobenzylidene) aniline and 4, 4′-(azinodimethylidyne) bis-benzonitrile. J. Chem. Crystallogr. 2011, 41, 464–469. [Google Scholar] [CrossRef]
- Corey, E.J. General methods for the construction of complex molecules. Pure Appl. Chem. 1967, 14, 30–49. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Nanda, A. A review about regulatory status and recent patents of pharmaceutical co-crystals. Adv. Pharm. Bull. 2018, 8, 355–363. [Google Scholar] [CrossRef]
- Batisai, E. Multi-component crystals of anti-tuberculosis drugs: A mini-review. RSC Adv. 2020, 10, 37134–37141. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering-A new organic synthesis. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Ganie, A.A.; Rashid, S.; Ahangar, A.A.; Ismail, T.M.; Sajith, P.A.; Dar, A.A. Expanding the scope of hydroxyl-pyridine supramolecular synthon to design molecular solids. Cryst. Growth Des. 2022, 22, 1972–1983. [Google Scholar] [CrossRef]
- Denisov, G.L.; Nelyubina, Y.V. New co-crystals/salts of gallic acid and substituted pyridines: An effect of ortho-substituents on the formation of an acid–pyridine heterosynthon. Crystals 2022, 12, 497. [Google Scholar] [CrossRef]
- Pathare, P.; Tekale, S.; Shaikh, R.; Damale, M.; Sangshetti, J.; Rajani, D.; Pawar, R. Pyridine and benzoisothiazole decorated vanillin chalcones: Synthesis, antimicrobial, antioxidant, molecular docking study and ADMET properties. Curr. Org. Synth. 2020, 17, 367–381. [Google Scholar] [CrossRef]
- Amr, A.G.; Mohamed, A.M.; Mohamed, S.F.; Abdel-Hafez, N.A.; Hammam, A.G. Synthesis and anticancer activities of new pyridine, pyrane and pyrimidine derivatives fused with nitrobenzosubetrone moiety. Bioorg. Med. Chem. 2006, 14, 5481–5488. [Google Scholar] [CrossRef]
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. A review on the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem. 2015, 1, 1–11. [Google Scholar]
- Henry, M.; Kananda, C.; Rotoloac, L.; Savinoa, B.; Owensa, E.A.; Cravotto, G. Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Adv. 2020, 10, 14170–14197. [Google Scholar] [CrossRef]
- Przybyłek, M.; Jeliński, T.; Słabuszewska, J.; Ziołkowska, D.; Mroczynska, K.; Cysewski, P. Application of multivariate adaptive regression splines (MARSplines) Methodology for screening of dicarboxylic acid cocrystal using 1D and 2D molecular descriptors. Cryst. Growth Des. 2019, 19, 3876–3887. [Google Scholar] [CrossRef]
- Suzuki, N.; Kawahata, M.; Yamaguchi, K.; Suzuki, T.; Tomono, K.; Fukami, T. Comparison of the relative stability of pharmaceutical co-crystals consisting of paracetamol and dicarboxylic acids. Drug Dev. Ind. Pharm. 2018, 44, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Bhattacharyya, P.K.; Baruah, J.B. Structural studies on solvates of cyclic imide tethered carboxylic acids with pyridine and quinoline. Cryst. Growth Des. 2010, 10, 348–356. [Google Scholar] [CrossRef]
- Bedekovic, N.; Stilinovic, V.; Pitesa, T. Aromatic versus aliphatic carboxyl group as a hydrogen bond donor in salts and co-crystals of an asymmetric diacid and pyridine derivatives. Cryst. Growth Des. 2017, 17, 5732–5743. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, J.; Wu, Y.; Wang, P.; Jin, S.; Lu, Y.; Wang, D. Non-covalent bonded 2D/3D supramolecular adducts from 6-methylpyridine-3-carboxamide and carboxylic acids. J. Mol. Struct. 2022, 1264, 133256–133267. [Google Scholar] [CrossRef]
- Lakshmipathi, A.; Emmerling, F.; Bhattacharya, B.; Ghosh, S. Structure-mechanical property correlation of a series of 4-(1-Napthylvinyl) pyridine based co-crystals. J. Mol. Struct. 2022, 1268, 133670–133681. [Google Scholar] [CrossRef]
- SADABS, V2.05; Bruker AXS: Madison, WI, USA, 1999.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Hacer, M.; Horn, H.; Kömel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Gural’skiy, I.A.; Escudero, D.; Frontera, A.; Solntsev, P.V.; Rusanov, E.B.; Chernega, A.N.; Krautscheid, H.; Domasevitch, K.V. 1,2,4,5-Tetrazine: An unprecedented μ4-coordination that enhances ability for anion⋯π interactions. Dalton Trans. 2009, 38, 2856–2864. [Google Scholar] [CrossRef]
- Barrios, L.A.; Aromí, G.; Frontera, A.; Quiñonero, D.; Deyà, P.M.; Gamez, P.; Roubeau, O.; Shotton, E.J.; Teat, S.J. Coordination Complexes Exhibiting Anion···π Interactions: Synthesis, Structure, and Theoretical Studies. Inorg. Chem. 2008, 47, 5873–5881. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Choudhury, S.R.; Estarellas, C.; Dey, B.; Frontera, A.; Hemming, J.; Helliwell, M.; Gamez, P.; Mukhopadhyay, S. Supramolecular assemblies involving anion–π and lone pair–π interactions: Experimental observation and theoretical analysis. CrystEngComm 2011, 13, 4519–4527. [Google Scholar] [CrossRef]
- Sadhukhan, D.; Maiti, M.; Pilet, G.; Bauzá, A.; Frontera, A.; Mitra, S. Hydrogen Bond, π–π, and CH–π Interactions Governing the Supramolecular Assembly of Some Hydrazone Ligands and Their MnII Complexes—Structural and Theoretical Interpretation. Eur. J. Inorg. Chem. 2015, 2015, 1958–1972. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, J.W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kottke, T.; Stalk, D. Crystal handling at low temperatures. J. Appl. Cryst. 1993, 26, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Enkelmann, E.; Lipinski, G.; Mer, K. Cyanopyridines-suitable heterocycles for co-crystal syntheses. Eur. J. Inorg. Chem. 2021, 33, 3367–3372. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX, SHELXTL Reference Manual; Bruker-AXS, Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Wilson, A.J.C. Statistical bias in least-squares refinement. Acta Cryst. 1976, A32, 994–996. [Google Scholar] [CrossRef] [Green Version]
- Khan, E.; Khan, A.; Gul, Z.; Ullah, F.; Tahir, M.N.; Khalid, M.; Asif, H.M.; Asim, S.; Braga, A.A.C. Molecular salts of terephthalic acids with 2-aminopyridine and 2-aminothiazole derivatives as potential antioxidant agents; Base-Acid Base type architectures. J. Mol. Struct. 2020, 1200, 127126–127140. [Google Scholar] [CrossRef]
- Kumar, S.; Nanda, A. Approaches to design of pharmaceutical co-crystals: A review. Mol. Cryst. Liq. Cryst. 2018, 667, 54–77. [Google Scholar] [CrossRef]
- Stilinovic, V.; Kaitner, B. Salts and co-crystals of gentisic acid with pyridine derivatives: The effect of proton transfer on the crystal packing (and vice versa). Cryst. Growth Des. 2012, 12, 5763–5772. [Google Scholar] [CrossRef]
- Haynes, D.A.; Jones, W.; Motherwell, W.D.S. Co-crystallisation of succinic and fumaric acids with lutidines: A systematic study. CrystEngComm 2006, 8, 830–840. [Google Scholar] [CrossRef]
- Bowker, M.J.A.; Stahl, P.H.; Wermuth, C.G. Handbook of Pharmaceutical Salts; VHCA, Wiley-VCH: New York, NY, USA, 2002. [Google Scholar]
- Bhogala, R.; Basavoju, S.; Nangia, A. Tape and layer structures in co-crystals of some di- and tricarboxylic acids with 4,4′-bipyridines and isonicotinamide. From binary to ternary cocrystals. CrystEngComm 2005, 7, 551–562. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Hussain, I.; Forbes, S.; Desper, J. Exploring the hydrogen-bond preference of N–H moieties in co-crystals assembled via O–H (acid)⋯ N (py) intermolecular interactions. CrystEngComm 2007, 9, 46–54. [Google Scholar] [CrossRef]
- Schmidtmann, M.; Wilson, C.C. Hydrogen transfer in pentachlorophenol-dimethylpyridine complexes. CrystEngComm 2008, 10, 177–183. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Babu, N.J.; Nangia, A.; Mason, S.A.; Puschmann, H.; Modal, R.; Horward, A.K. Variable temperature neutron diffraction analysis of a very short O−H···O hydrogen bond in 2,3,5,6-pyrazinetetracarboxylic acid dihydrate: Synthon-assisted short Oacid−H···Owater hydrogen bonds in a multicenter array. J. Phys. Chem. A 2004, 108, 9406–9416. [Google Scholar] [CrossRef]
- Ruelas-Alvarez, G.Y.; Valenzuela, A.J.C.; Cruz-Enríquez, A.; Hopfl, H.; Campos-Gaxiola, J.J.; Rivera, M.A.R.; Rodríguez-Molina, B. Exploration of the luminescence properties of organic phosphate salts of 3-quinoline- and 5-isoquinolineboronic acid. Eur. J. Inorg. Chem. 2019, 22, 2707–2724. [Google Scholar] [CrossRef]
- Melendez, R.E.; Zaworotko, M.J. Toward crystal engineering of organic porous solids: Diammine salts of trimesic acid. Supramol. Chem. 1997, 8, 157–168. [Google Scholar] [CrossRef]
- Bis, J.A.; Vishweshwar, P.; Weyna, D.; Zaworotko, M.J. Hierarchy of supramolecular synthons: Persistent hydroxyl···pyridine hydrogen bonds in co-crystals that contain a cyano acceptor. Mol. Pharm. 2007, 4, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, B.P.; Pogoda, D.; Perry, M.L.; Vidal, L.M.T.; Zaworotko, M.J.; Ayala, A.P. Co-crystal polymorphs and solvates of the anti-trypanosomacruzi drug benznidazole with improved dissolution performance. Cryst. Growth Des. 2020, 20, 4707–4718. [Google Scholar] [CrossRef]
- Cheney, M.L.; Weyna, D.R.; Shan, N.; Hanna, H.; Wojtas, L.; Zaworotko, M.J. Coformer selection in pharmaceutical co-crystal development: A case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics. J. Pharm. Sci. 2011, 100, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Dutta, D.; Nath, H.; Frontera, A.; Bhattacharyya, M.K. A novel oxalato bridged supramolecular ternary complex of Cu(II) involving energetically significant π-hole interaction: Experimental and theoretical studies. Inorg. Chim. Acta 2019, 487, 354–361. [Google Scholar] [CrossRef]
- Dutta, D.; Islam, S.M.N.; Saha, U.; Frontera, A.; Bhattacharyya, M.K. Cu(II) and Co(II) coordination solids involving unconventional parallel nitrile(π)–nitrile(π) and energetically significant cooperative hydrogen bonding interactions: Experimental and theoretical studies. J. Mol. Struct. 2019, 1195, 733–743. [Google Scholar] [CrossRef]
- Dutta, D.; Sharma, P.; Frontera, A.; Gogoi, A.; Verma, A.K.; Dutta, D.; Sarma, B.; Bhattacharyya, M.K. Oxalato bridged coordination polymer of manganese(III) involving unconventional O⋯π-hole(nitrile) and antiparallel nitrile⋯nitrile contacts: Antiproliferative evaluation and theoretical studies. New J. Chem. 2020, 44, 20021–20038. [Google Scholar] [CrossRef]
- Das, B.K.; Bora, S.J.; Bhattacharyya, M.K.; Barman, R.K. Inverse bilayer structure of mononuclear CoII and NiII complexes of the type [M(H2O)3(SO4)(4-CNpy)]2. Acta Cryst. 2009, B65, 467–473. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Nakayama, T.; Fukuda, H.; Kamikawa, T.; Sakamoto, Y.; Sugita, A.; Kawasaki, M.; Amano, T.; Sato, H.; Sakaki, S.; Morino, I.; et al. Effective interaction energy of water dimer at room temperature: An experimental and theoretical study. J. Chem. Phys. 2007, 127, 134302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podeszwa, R.; Bukowski, R.; Szalewicz, K. Potential Energy Surface for the Benzene Dimer and Perturbational Analysis of π−π Interactions. J. Phys. Chem. A 2006, 110, 10345–10354. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Formula | C14H10N4O4 | C18H18N4O4 | C20H14N4O4 | C18H18N4O4 |
| Formula weight | 149.13 | 354.36 | 374.35 | 354.36 |
| Temp (K) | 100.0 | 100.0 | 100.0 | 294 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/c | P | P | P |
| a, (Å) | 3.664(2) | 3.8849(3) | 3.7596(4) | 4.0270(3) |
| b, (Å) | 13.887(8) | 10.5878(9) | 6.2540(7) | 10.8650(7) |
| c, (Å) | 13.455(6) | 11.7569(10) | 18.617(2) | 11.2177(7) |
| α, (°) | 90 | 63.760(4) | 92.569(4) | 67.878(2) |
| β, (°) | 91.55(2) | 86.193(5) | 95.589(4) | 86.725(2) |
| γ, (°) | 90 | 85.953(5) | 95.113(4) | 89.843(2) |
| Volume (Å3) | 684.5(6) | 432.34(6) | 433.29(8) | 453.84(5) |
| Z | 2 | 1 | 1 | 1 |
| Absorption coefficient (mm−1) | 0.927 | 0.818 | 0.857 | 0.780 |
| F(000) | 308 | 186 | 194 | 186 |
| D(calcd) (Mg/m3) | 1.447 | 1.361 | 1.435 | 1.297 |
| Index ranges | −4 ≤ h ≤ 3, −16 ≤ k ≤ 16, −16 ≤ l ≤ 16 | −3 ≤ h ≤ 4, −12 ≤ k ≤ 12, −13 ≤ l ≤ 14 | −4 ≤ h ≤ 4, −7 ≤ k ≤ 7, −22 ≤ l ≤ 22 | −4 ≤ h ≤ 4, −13 ≤ k ≤ 13, −13 ≤ l ≤ 13 |
| Crystal size, (mm3) | 0.49 × 0.20 × 0.19 | 0.45 × 0.39 × 0.33 | 0.23 × 0.15 × 0.13 | 0.28 × 0.15 × 0.12 |
| θ range (°) | 6.374 to 66.541 | 4.195 to 67.223 | 4.776 to 133.11 | 8.524 to 140.024 |
| Independent reflections | 1114 | 1541 | 4640 | 1661 |
| Reflections collected | 1166 | 5081 | 4640 | 20138 |
| Refinement method | Full-matrix least squares on F2 | Full-matrix least squares on F2 | Full-matrix least squares on F2 | Full-matrix least squares on F2 |
| Data/restraints/parameters | 1166/0/105 | 1541/0/119 | 4640/0/129 | 1661/0/119 |
| Goodness-of-fit on F2 | 1.098 | 1.063 | 1.094 | 1.089 |
| Final R indices (I > 2σ(I)) | R1 = 0.0438, wR2 = 0.1149 | R1 = 0.0479, wR2 = 0.1327 | R1 = 0.0761, wR2 = 0.2427 | R1 = 0.0453, wR2 = 0.1220 |
| R indices (all data) | R1 = 0.0452, wR2 = 0.1162 | R1 = 0.0539, wR2 = 0.1376 | R1 = 0.0785, wR2 = 0.2455 | R1 = 0.0468, wR2 = 0.1231 |
| Largest peak and hole (e·Å−3) | 0.22/−0.23 | 0.22/−0.24 | 0.43/−0.37 | 0.16/−0.15 |
| Multicomponent Crystals | pKa (Base) | pKa (Acid) | pKa |
|---|---|---|---|
| (ox)0.5(2-CNpy) (1) | −0.26 | 1.27/4.28 | −1.53/−4.54 |
| (adp)(4-CNpy)2 (2) | 1.90 | 4.43/5.41 | −2.53/−3.51 |
| (tp)(4-CNpy)2 (3) | 1.90 | 3.51/4.82 | −1.61/−2.92 |
| (adp)(3-CNpy)2 (4) | 1.39 | 4.43/5.41 | −3.04/−4.02 |
| Compound | C–N–C | Bond Angle (°) |
|---|---|---|
| Compound 1 | C2–N1–C6 | 117.4 |
| Compound 2 | C2–N1–C6 | 118.4 |
| Compound 3 | C2–N1–C6 | 117.9 |
| Compound 4 | C2–N1–C6 | 117.5 |
| Compound | C–O | Bond Length (Å) |
|---|---|---|
| Compound 1 | C2’–O1´ | 1.31 |
| C2´–O2´ | 1.21 | |
| Compound 2 | C2´–O1´ | 1.33 |
| C2´–O2´ | 1.21 | |
| Compound 3 | C2´–O3´ | 1.21 |
| C2´–O1´ | 1.32 | |
| Compound 4 | C10–O11 | 1.19 |
| C10–O9 | 1.32 |
| D–H⋯A | d(D–H) | d(H⋯A) | d(D–A) | <(DHA) |
|---|---|---|---|---|
| 1 | ||||
| C3–H3⋯O2´ | 0.95 | 2.77 | 3.358(2) | 120.9 |
| O1´–H1⋯N1 | 1.02 | 1.61 | 2.633(1) | 174.9 |
| C5–H5⋯N22 | 0.95 | 2.99 | 3.458(2) | 132.7 |
| 2 | ||||
| C5–H5⋯N42 | 0.95 | 2.51 | 3.389(2) | 152.8 |
| C2–H2⋯O2´ | 0.95 | 2.66 | 3.331(1) | 127.4 |
| O1´–H1´⋯N1 | 0.84 | 1.83 | 2.668(1) | 172.6 |
| C4´–H4´B⋯N42 | 0.99 | 2.82 | 3.737(2) | 153.7 |
| 3 | ||||
| C5–H5⋯N42 | 0.94 | 2.66 | 3.517(4) | 149.2 |
| N1–H1⋯O1 | 0.84 | 1.89 | 2.728(3) | 171.2 |
| C6–H6⋯O3´ | 0.95 | 2.28 | 3.124(2) | 145.6 |
| 4 | ||||
| C4–H4⋯N8 | 0.93 | 2.58 | 3.442(2) | 154.1 |
| C12–H12B⋯N8 | 0.97 | 2.81 | 3.584(2) | 137.0 |
| C6–H6⋯O11 | 0.93 | 2.49 | 3.194(2) | 145.3 |
| C5–H5⋯O11 | 0.93 | 2.91 | 3.324(2) | 128.3 |
| C2–H2⋯O9 | 0.93 | 2.64 | 3.423(1) | 145.6 |
| O9–H9⋯N1 | 0.82 | 1.92 | 2.744(1) | 178.7 |
| C12–H12B⋯O9 | 0.95 | 2.97 | 3.654(3) | 133.6 |
| C13–H13A⋯O11 | 0.94 | 2.99 | 3.362(2) | 154.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, P.; Gomila, R.M.; Frontera, A.; Barcelo-Oliver, M.; Bhattacharyya, M.K. Dicarboxylic Acid-Based Co-Crystals of Pyridine Derivatives Involving Structure Guiding Unconventional Synthons: Experimental and Theoretical Studies. Crystals 2022, 12, 1442. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12101442
Sharma P, Gomila RM, Frontera A, Barcelo-Oliver M, Bhattacharyya MK. Dicarboxylic Acid-Based Co-Crystals of Pyridine Derivatives Involving Structure Guiding Unconventional Synthons: Experimental and Theoretical Studies. Crystals. 2022; 12(10):1442. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12101442
Chicago/Turabian StyleSharma, Pranay, Rosa M. Gomila, Antonio Frontera, Miquel Barcelo-Oliver, and Manjit K. Bhattacharyya. 2022. "Dicarboxylic Acid-Based Co-Crystals of Pyridine Derivatives Involving Structure Guiding Unconventional Synthons: Experimental and Theoretical Studies" Crystals 12, no. 10: 1442. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12101442