
Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies
1
High Pressure and Synchrotron Radiation Physics Division, Bhabha Atomic Research Centre, Mumbai 400085, India
2
Homi Bhabha National Institute, Anushakti Nagar, Mumbai 400094, India
*
Author to whom correspondence should be addressed.
Crystals 2023, 13(2), 254; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst13020254
Submission received: 2 January 2023
/
Revised: 23 January 2023
/
Accepted: 27 January 2023
/
Published: 1 February 2023
(This article belongs to the Special Issue Pressure-Induced Phase Transformations (Volume II))
Abstract
:In this manuscript, we report the density functional theory-based first principles study of the structural and vibrational properties of technologically relevant M′ fergusonite (P2/c)-structured NdTaO4 and SmTaO4 under compression. For NdTaO4 and SmTaO4, ambient unit cell parameters, along with constituent polyhedral volume and bond lengths, have been compared with earlier reported parameters for EuTaO4 and GdTaO4 for a better understanding of the role of lanthanide radii on the primitive unit cell. For both the compounds, our calculations show the presence of first-order monoclinic to tetragonal phase transition accompanied by nearly a 1.3% volume collapse and an increase in oxygen coordination around the tantalum (Ta) cation from ambient six to eight at phase transition. A lower bulk modulus obtained in the high-pressure tetragonal phase when compared to the ambient monoclinic phase is indicative of the more compressible unit cell under pressure. Phonon modes are calculated for the ambient and high-pressure phases with compression for both the compounds along with their pressure coefficients. One particular IR mode has been observed to show red shift in the ambient monoclinic phase, possibly leading to the instability in the compounds under compression.
1. Introduction
RBO4 (R: rare earth; B: a pentavalent cation such as V, W, Mo, Nb, Ta, As, and P) compounds are the subject of extensive research due to their promising applications in areas such as proton-conducting solid oxide fuel cells [1], and as a host for nuclear radioactive waste immobilization [2,3]. It has been well established that the B cation plays a crucial role in deciding the stable structure of RBO4 compounds. Depending on the ratio of B cationic radii to lanthanide radii, RBO4 compounds are reported to crystallize in either tetragonal (zircon, scheelite type) or monoclinic (fergusonite, monazite, wulframite type) structures [4]. Rare earth orthovanadate (RVO4) are generally synthesized in a zircon (tetragonal) structure with a I41/amd space group, while rare earth orthotungstates (RWO4) and molybates (RMoO4) have been reported to crystallize in scheelite (tetragonal) structure with I41/a crystal symmetry [5,6,7]. A monoclinic M fergusonite structure with space group I2/a has been established as a stable structure for rare earth orthoniobates (RNbO4) at ambient temperature and pressure [8]. Depending upon the atomic radii of the lanthanide cation, the crystal structure of rare earth orthophosphate is either zircon (R ˂ Gd) or monazite (R ≥ Gd) [9]. All the structures are closely related to each other by group–subgroup relations. A tetragonal scheelite structure (I41/a) is a subgroup of a tetragonal zircon (I41/amd) structure and the transformation between these two structures is generally of the first-order reconstructive type. A scheelite structure is transformed to a fergusonite structure by means of another translationgleiche, which involves lowering of point group symmetry from 4/m to 2/m [10]. Among all RBO4 compounds, the RTaO4 family of compounds exhibit polymorphism at ambient pressure and temperature conditions, which makes them of great interest from theoretical and technological point of views [11,12]. In RTaO4 compounds, the final stable structure not only depends on the lanthanide radii but also depends on the heating temperature. Most of the compounds belonging to the RTaO4 family stabilize either in a M fergusonite or M’ fergusonite structure at room temperature depending on the processing parameters. The main difference between the M fergusonite and M’ fergusonite structures is the oxygen coordination around the Ta atom. In a M fergusonite structure, the Ta atom is surrounded by an oxygen tetrahedra, whereas an oxygen octahedra is formed in a M’ fergusonite structure. In the M fergusonite phase, doubling of the b axis has been observed compared to M′ structure, while the other two unit cell axes have almost the same value in both structures. In recent times, RTaO4 family compounds are being investigated extensively due to their potential applications in the field of scintillators and laser materials, owing to their high thermal stability and good chemical stability [13,14]. These compounds are also proposed as excellent alternatives to using Yttria-stabilized zirconia as ceramic thermal barrier coatings (TBCs) due to their lower thermal conductivity and better fracture toughness at high temperatures [15]. As is well known, most often it is the crystal structure that determines the properties of a material, and the structure may be altered by varying the thermodynamic parameters. Recently, by x-ray diffraction, the Raman spectroscopic technique and density functional theory (DFT)-based first principles calculations, we have shown the structural instability of EuTaO4 and GdTaO4 [16,17]. In continuation to this work, to understand the compression behavior of the RtaO4 family of compounds, we have performed density functional theory-based first-principle simulations on technologically important NdTaO4 and SmTaO4. Wenhui Xiao et al. [18] have reported that the M’ fergusonite structure is more stable compared to the M fergusonite structure. Therefore, in the present work, we report the results from DFT-based first principles calculations on the M’ fergusonite structure under compression. We have also calculated the equation of state for both the compounds using the third-order Birch–Murnaghan equation of state (BM-EOS). Compressibility analysis of the simulated volume of constituent polyhedral units RO8 and TaO6 indicates that the major contribution to the bulk modulus comes from RO8 polyhedra. This behavior validates Hazen and Finger’s proposed empirical model for predicting the bulk modulus, with contributions from the rare earth polyhedral unit as seen in RVO4, RWO4, RMoO4, RNbO4 compounds [6,7,19,20,21,22,23,24,25]. We have also calculated the pressure evolution of the Raman and IR modes, which is consistent with earlier reported results for EuTaO4 and GdTaO4 for the same structure [16,17]. Further, we have compared the results from this work with previously investigated EuTaO4 and GdTaO4 to establish the role of lanthanide contraction present in the RTaO4 family.
2. Computational Details
DFT-based first-principle simulations were carried out as implemented in Quantum Espresso [26] for determination of stable structures at ambient pressure as well as to investigate the influence of pressure on structural and vibrational properties. This is based on density functional theory, plane waves, and pseudopotentials. The projector-augmented wave (PAW) scheme [27], which describes electron–ion interactions, was employed as pseudo potential in self-consistency field calculation. Appropriate pseudopotentials are taken from the Pslibrary [28] considering 11 valence electrons for Nd (5s26s1.55p66p0.55d1) and Sm (5s26s1.55p66p0.55d1), 27 valence electrons for Ta (4f14 5d35p66s25s2) and 6 valence electrons for oxygen (2s2 2p4). A prescribed generalized gradient approximation (GGA) based on the parametrization proposed by Perdew, Burke, and Ernzherhof (PBE) [29] has been accounted for the for calculation of exchange and correlation energy for both the compounds with the lowest-energy M’ fergusonite structure. Wave functions in the Kohn–Sham equation are expanded in a plane wave basis set due to the major advantage of orthonormality and since it is easy to control the convergence with respect to the size of the basis with only one parameter Ecut. In our calculation, the plane wave basis was extended up to 70 Ry for both the compounds (NdTaO4 and SmTaO4) to achieve highly converged results in the PAW scheme after thoroughly going through the convergence test. A dense Monkhorst pack grid of 8 × 8 × 8 is used for Brillouin zone integrations. Geometric optimization of NdTaO4 and SmTaO4 structures has been achieved using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization algorithm [30], where the structures have been fully optimized to the equilibrium condition at ambient pressure, by minimizing the forces on the atoms and the stress tensor. Helmann–Feynman forces lower than 0.00003 eV/atom on each atom in the unit cell and maximum deviation among the diagonal components of the stress tensor on a unit cell lower than 0.1 GPa ensure a fully relaxed structure. The same steps were followed while calculating from ambient pressure to 50 GPa with an interval of 1 GPa. After obtaining the equilibrium structures at different pressures, phonon frequencies were calculated at the center of the Brillouin zone using density functional perturbation theory (DFPT) as implemented in the Quantum Espresso code [31]. Simulations were performed at zero temperature and under a hydrostatic environment. The stable structures and transition pressures were obtained by analyzing the enthalpy–pressure curve.
3. Results and Discussions
3.1. Ambient Structure
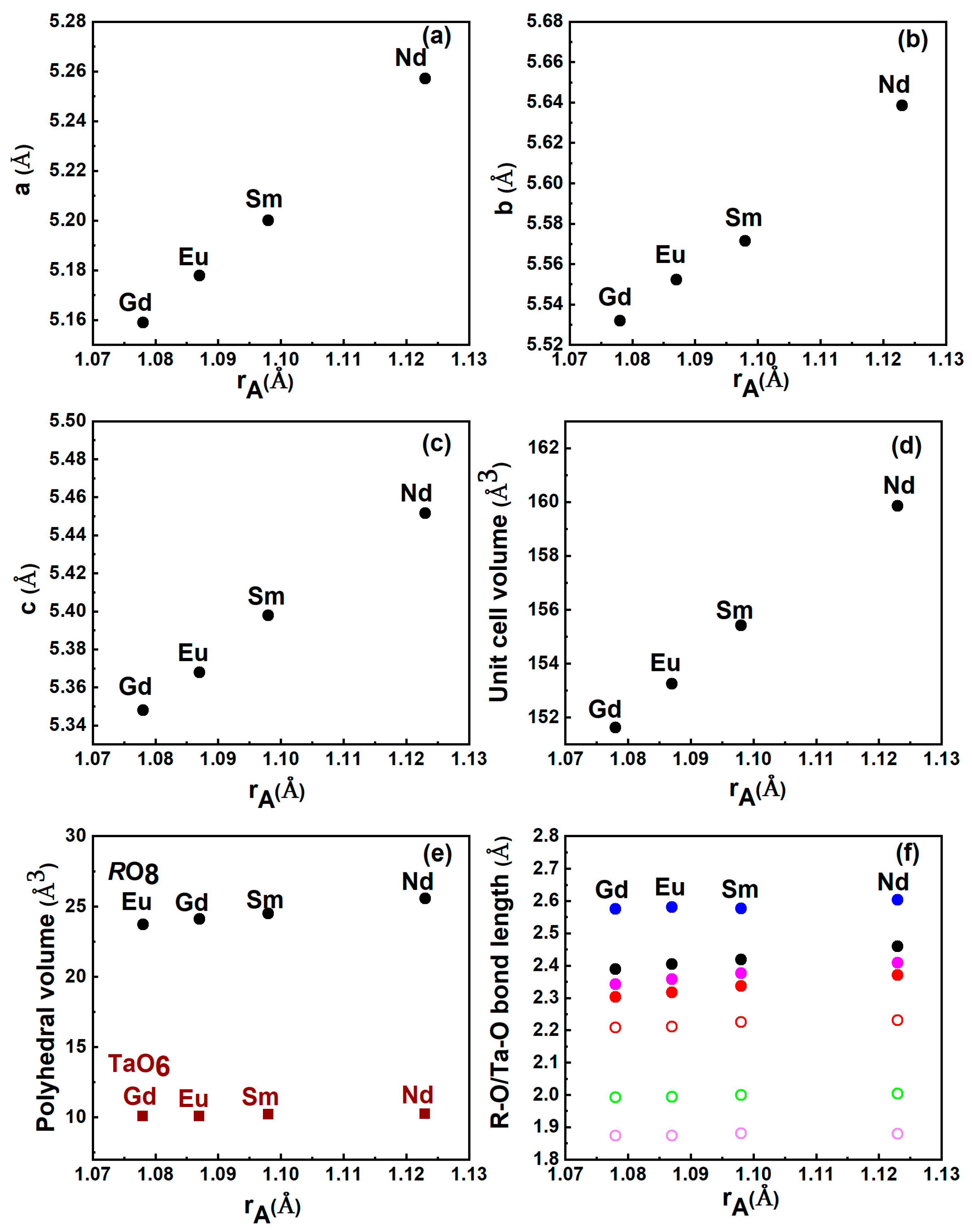
The optimized volume obtained at ambient pressure for both the compounds in the M’ fergusonite structure matches very well with previously reported experimental values [32]. Unit cell parameters obtained from the geometrically relaxed structure with constituent bond lengths along with the previously reported experimental data are given in Table 1. The equilibrium volume obtained for NdTaO4 is 159.1 Å3 and 155.4 Å3 for SmTaO4, which are within 1% and 0.3% of the experimentally observed volume. Figure 1 shows the polyhedral representation of NdTaO4 and SmTaO4 belonging to a P2/c (space group no.:13, Z = 2) structure with 2/m point group symmetry. The rare earth cation (Nd/Sm) is surrounded by eight oxygen, forming dodecahedra while oxygen coordination around the Ta cation is six. The formation of oxygen octahedra around tantalum is a distinctive signature of the M’ fergusonite structure that makes it different from the M fergusonite structure with space group I2/a (space group: 15). Each rare earth cation is bonded with eight oxygen with four different R-O bond distances while each Ta cation bonds to six oxygen with three different bond distances, making all the constituent units distorted. These distorted polyhedral units provide structural stability to these compounds against a large range of pressure/temperature, compared to zircon- or scheelite-structured compounds [6,7,19,20,21,22,23,24,25]. In the calculated M’ fergusonite structure, the Nd/Sm and Ta atoms occupy the 2e and 2f Wyckoff positions while oxygen atoms O1 and O2 occupy the 4g position. In Figure 2, we have plotted ambient pressure lattice parameters, the unit cell volume along with the constituent polyhedral volume (RO8, TaO6), and R-O and Ta-O bond lengths for both the compounds along with previously reported data for EuTaO4 and GdTaO4 for a better understanding of the role of the rare earth cation. An increasing linear trend has been observed in all the parameters with lanthanum cationic radii except the volume of TaO6 octahedra and its constituent bond lengths. This indicates the major influence of the R cation on structural parameters. In all four RTaO4 compounds, the polyhedral volume of all distorted TaO6 octahedra remains almost the same.
3.2. Structural Behavior under Compression
3.2.1. The Low-Pressure Phase
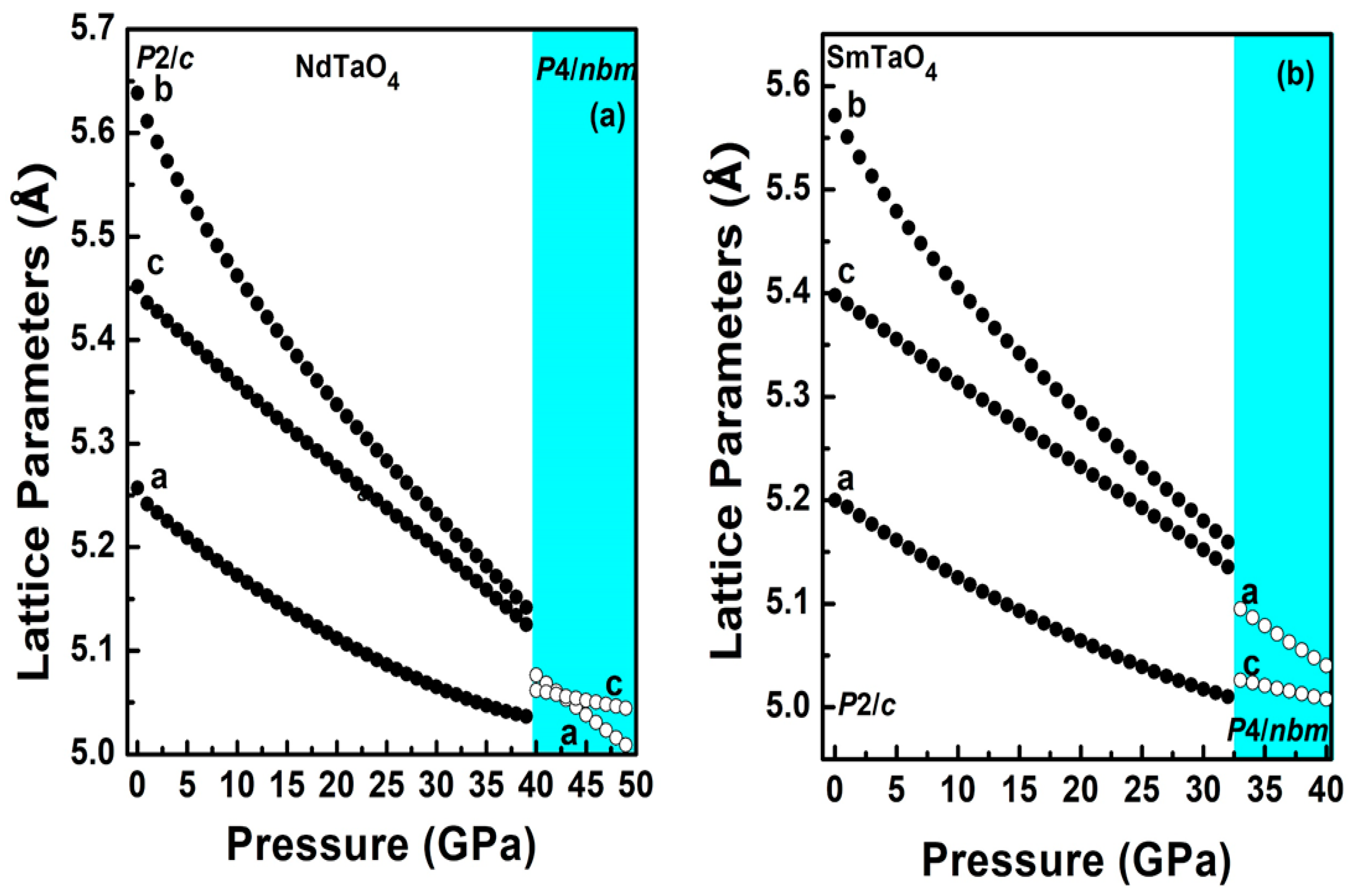
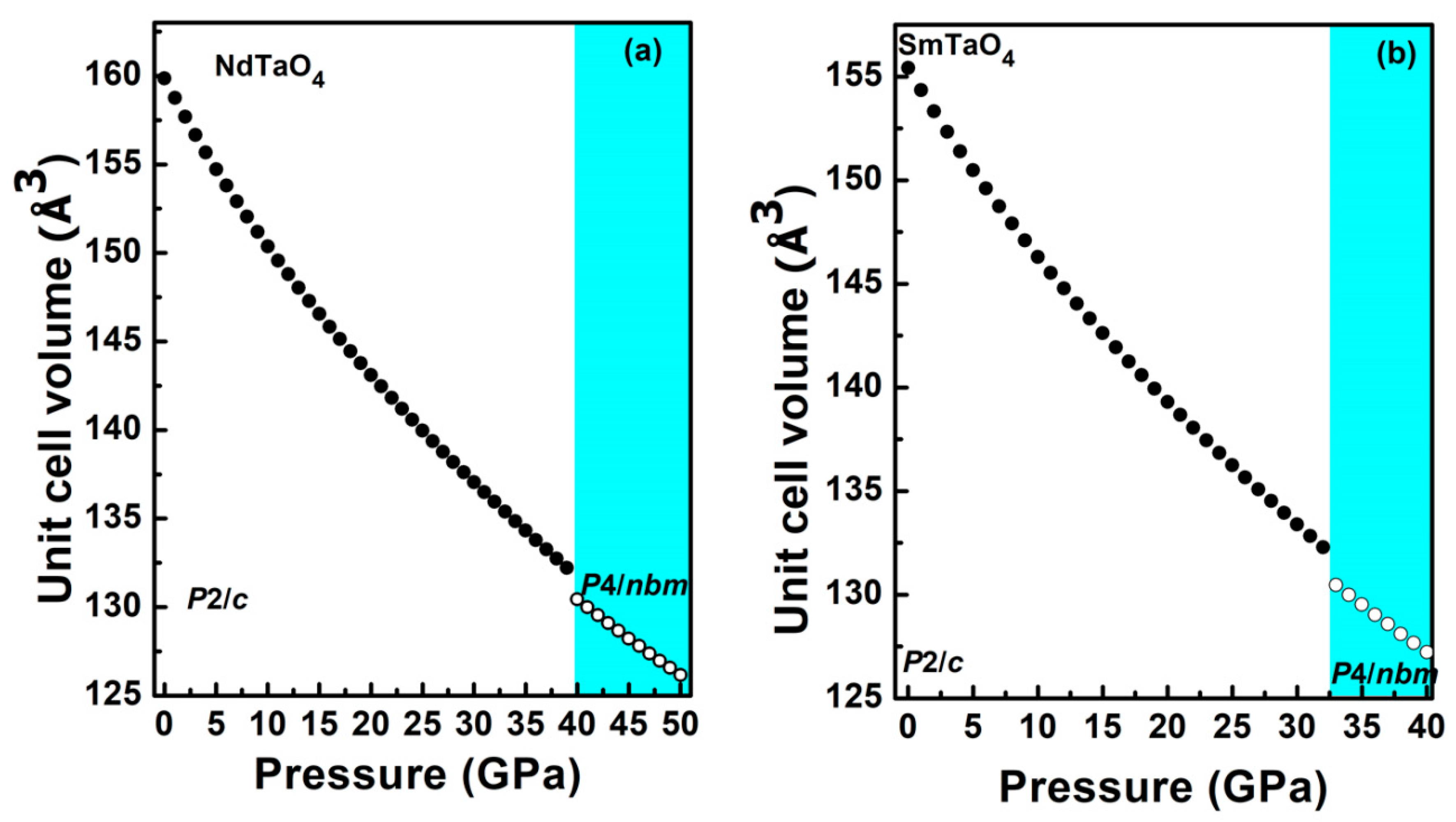
Simulated unit cell parameters for both the compounds, NdTaO4 and SmTaO4, have been plotted at different pressures in Figure 3a,b. The anisotropic compressibility of the b axis is clearly seen, which is 2 fold as compressible compared to the other two axes. This particular behavior has been observed in almost all RBO4 compounds irrespective of their ambient structure [5,6,7,9]. The axial compressibility obtained by fitting the calculated lattice parameters to third-order BM-EOS [33] for NdTaO4 is Ka = 1.89 × 10−3 GPa−1, Kb= 3.79 × 10−3 GPa−1 and Kc = 1.75 × 10−3 GPa−1; and SmTaO4 are Ka = 1.66 × 10−3 GPa−1, Kb = 3.64 × 10−3 GPa−1 and Kc = 1.60 × 10−3 GPa−1, respectively. The structural arrangement of relatively more compressible RO8 polyhedral units along the b axis could be the reason for anisotropic compression as has been reported in other rare earth metal oxides. Similar behavior has also been observed in EuTaO4 and GdTaO4 [16,17]. It is interesting to note that a decreasing trend is observed in the axial compressibility as we go from higher ionic radii to lower ionic radii of the lanthanide cation, indicating the more incompressible behavior of RTaO4 compounds with a lower ionic radii lanthanide cation. Figure 4 shows the pressure evolution of the unit cell volume for both the compounds. The bulk modulus obtained by fitting the simulated pressure–volume data to third-order BM-EOS is 145.1 and 147.2 GPa for NdTaO4 and SmTaO4, respectively, which is in good agreement with the bulk modulus reported for the RTaO4 family of compounds. The bulk modulus reported for NdVO4 [34] in the zircon and scheelite structures is 124.2 and 136 GPa, respectively, which is similar to the obtained bulk modulus of NdTaO4 in the present studies. A similar bulk modulus is observed for NdNbO4 (138.32 GPa) [18]. Similarly, for SmTaO4, the obtained bulk modulus in the present work is similar to that reported for other SmBO4 compounds [35]. This indicates that it is NdO8/SmO8 polyhedral units that mostly contribute to the bulk modulus. A similar bulk modulus for various RBO4 compounds has already been reported and reaffirms that it is indeed the lower valence polyhedral units (RO8) that mainly contribute to the bulk modulus [16,17]. This can be validated using Hazen and Finger’s proposed empirical model B0 = N × Z/(dR-O)3, where B0 is the bulk modulus, N is the dimensional-less proportional constant (610 for tantalates and niobates), Z is the formal charge of the R cation and dR-O is the average cation–anion distance, which only considers the rare earth polyhedral unit for predicting the bulk modulus [36]. For NdTaO4 with dNd-O= 2.460 Å and Z = 3, the calculated bulk modulus using Hazen and Finger’s equation is 122.9 GPa, which is similar to the simulated value obtained in the present work. For the SmTaO4 compound, the bulk modulus obtained using dSm-O = 2.460 Å is 127.4 GPa.
3.2.2. The High-Pressure Phase
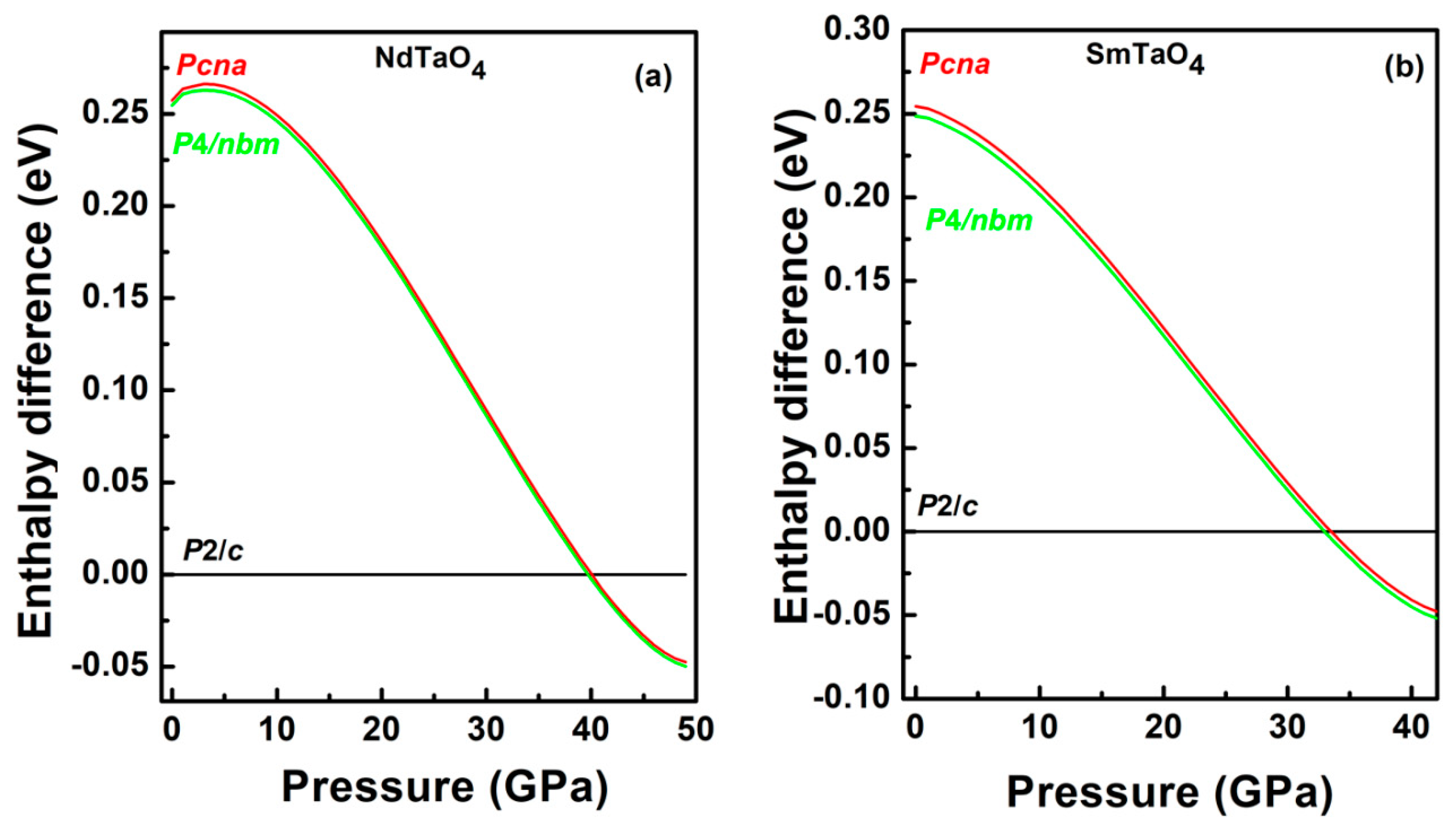
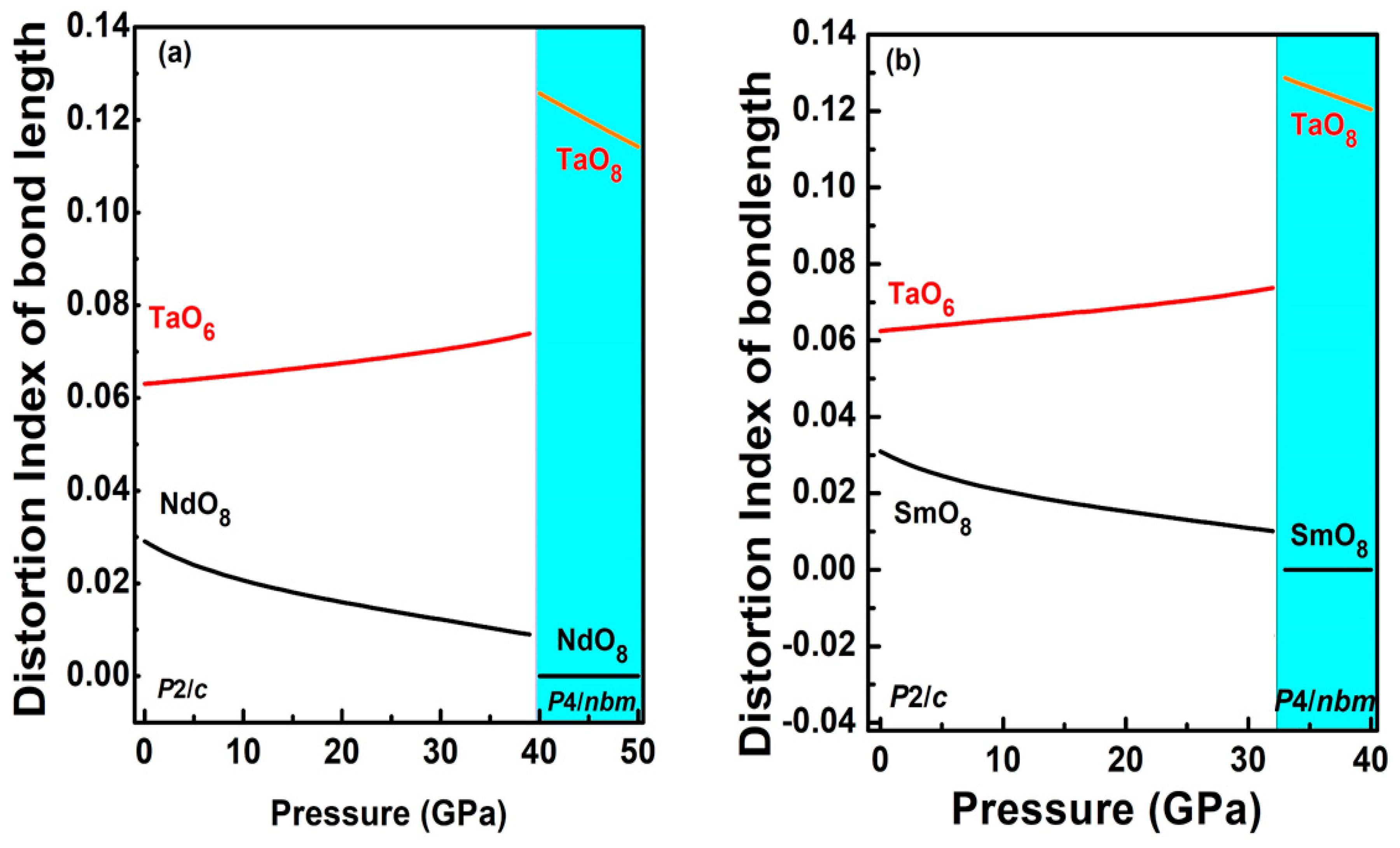
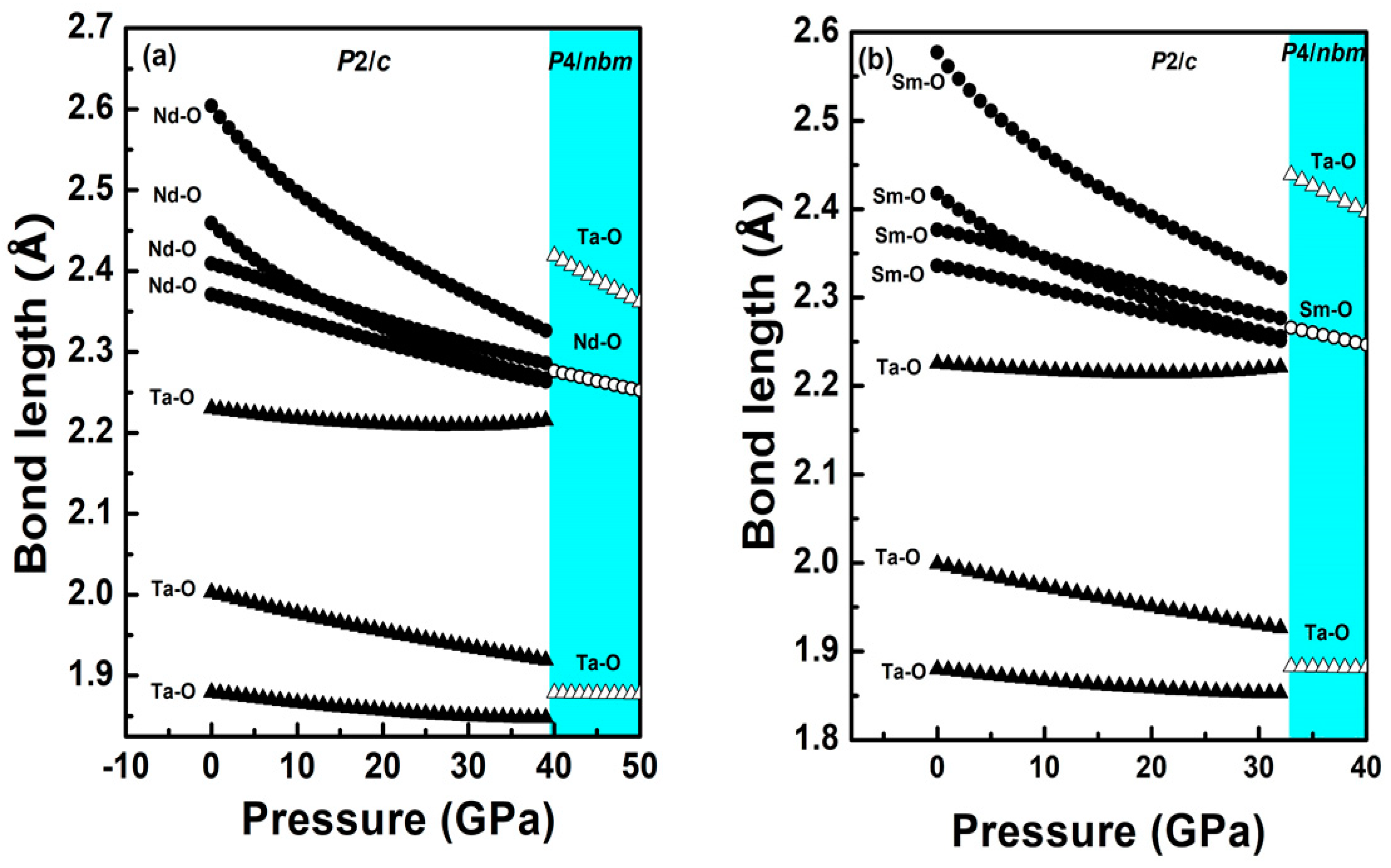
Earlier, based on our X-ray diffraction and Raman spectroscopic measurements along with DFT-based first principles calculations on EuTaO4 and GdTaO4, an isostructural transition was reported with a sudden drop in the monoclinic angle from 96° to 90° at the transition. This isostructural monoclinic phase could also be described as the pseudo orthorhombic structure due to all unit cell angles of 90°. Therefore, orthorhombic Pcna was tested as an alternative description against the isostructural monoclinic phase and our calculation reveals that, at high pressure, both the structures are energetically favorable. Since NdTaO4 and SmTaO4 are found to be synthesized in the same space group (P2/c) as EuTaO4 and GdTaO4, the same orthorhombic space group has been tested as the possible high-pressure phase for both the compounds in the present work. For NdTaO4, the orthorhombic structure with the Pcna space group becomes energetically favorable at approximately 40 GPa as seen in Figure 5a, which depicts enthalpy difference as a function of pressure (the monoclinic phase P2/c has been taken as a reference). Similar behavior has been observed at approximately 33.5 GPa for SmTaO4 (Figure 5b). This orthorhombic structure can also be alternatively described by a pseudo tetragonal structure due to almost the same value of lattice parameter a and c as seen in calculation. Therefore, the higher-symmetry tetragonal structure P4/nbm with 4/mmm point group symmetry was also tested against the Pcna structure and indeed tetragonal P4/nbm is a lower-energy structure at approximately 33 GPa for SmTaO4 and approximately 40 GPa for NdTaO4 as seen in Figure 5a,b. The phase transition in both the compounds is associated with a nearly 1.3% volume collapse at transition pressure along with change in oxygen coordination around the Ta cation from ambient six to eight at phase transition. In Figure 3, we have plotted lattice parameters of the high-pressure tetragonal phase (HP) along with unit cell parameters corresponding to the low-pressure monoclinic phase (LP). The axial compressibility obtained by fitting the calculated lattice parameters to third-order BM-EOS for NdTaO4 and SmTaO4 in the HP phases is Ka = 3.75 × 10−3 GPa−1, Kc = 1.02 × 10−3 GPa−1 and Ka = 3.4 × 10−3 GPa−1, Kc = 1.04 × 10−3 GPa−1, respectively. Unit cells for both the compounds also undergo anisotropic compression in the high-pressure tetragonal structure as observed in the low-pressure monoclinic structure. The simulated pressure volume data fitted with the third-order Birch–Murnaghan (BM) equation of state (EOS) yields a bulk modulus of 123.74 GPa for NdTaO4 and 130.60 GPa for SmTaO4. It is interesting to note that the high-pressure phase has a lower bulk modulus than the low-pressure phase, indicating a more compressible high-pressure phase in spite of volume collapse at phase transition. This can be understood by analyzing the compressional behavior of the constituent polyhedral unit in both the LP phase and the HP phase. In low-pressure phase, the compounds NdTaO4 and SmTaO4 are made up of highly compressible NdO8 and SmO8 polyhedra as evident from their value of bulk modulus being 117.3 and 115 GPa respectively, while TaO6 octahedra in both the compounds is highly incompressible with incompressibility modulus being 262.6 and 258.7 GPa for NdTaO4 and SmTaO4 respectively. The modulus of incompressibility has been obtained using calculated pressure and polyhedral volume fitted to third-order BM-EOS. A large difference in the bulk modulus at the LP phase affirms that indeed lower valence rare earth polyhedra significantly contribute to the compressibility of the compound in the low-pressure monoclinic phase, which validates Hazen and Finger’s proposed empirical model for predicting the bulk modulus, taking contribution from rare earth polyhedral unit as seen in RVO4, RWO4, RMoO4, RNbO4 compounds [6,7,19,20,21,22,23,24,25]. In Figure 6a,b, we have plotted the distortion index of bond lengths for the polyhedral units of both NdTaO4 and SmTaO4 compounds, which show that RO8 polyhedra become fully symmetric under compression, while that of TaO6 more distorted and distortion in the bond length increases sharply at phase transition with the formation of a TaO8 polyhedral unit. The distortion index of the bond length as defined by Baur [37] has been computed using VESTA software [38]. For the NdTaO4 compound, the bulk modulus obtained for constituent polyhedral NdO8 and TaO8 in the HP phase is 105.9 and 99.8 GPa, respectively, whereas the bulk modulus for the SmTaO4 compound obtained for constituent polyhedral SmO8 and TaO8 in the HP phase is 109.4 and 111.42 GPa, respectively. The similar bulk modulus indicates equal contribution to the compressibility of the high-pressure unit cell unlike the low-pressure phase. Incompressible TaO6 octahedra change to very compressible TaO8 at phase transition due to the increase in oxygen coordination under pressure and can be cited as among the reasons for the lower bulk modulus of the high-pressure phase. It should also be noted that the volume collapse at transition pressure is much smaller (~1.3%), which also supports the more compressible high-pressure unit cell. In Table 2, we have tabulated the atom positions along with their unit cell lattice parameters at ambient pressure and phase transition pressure for both the compounds. It can be clearly seen that all the constituent atoms show significant atomic rearrangement in position when compared to their ambient pressure. A more packed high-pressure unit cell is a consequence of the more effective packing of oxygen anions surrounding the Ta cation. To analyze pressure effects from this perspective, we calculated the pressure evolution of R-O and Ta-O bond distances for both the LP and HP phases. Results are summarized in Figure 7a,b, which shows calculated bond distances plotted against pressure. Figure 7a,b clearly shows that for both the compounds, at LP phase, the two largest bond distances between Nd-O decrease at a faster rate than the other six Nd-O bond lengths, whereas the Ta-O bond lengths show lower compressibility than the Nd-O bond lengths. In the high-pressure tetragonal phase, NdO8/SmO8 polyhedra become fully symmetric and the largest Ta-O bond shows most compressibility among all the constituent bonds, indicating the contribution of TaO8 towards the compressibility of the unit cell.
3.3. Vibrational Properties under Compression
The primitive unit cell of the M’ fergusonite structure has two formula units, giving rise to a total of 36 phonon modes for both the compounds belonging to the P2/c space group with 2/m point group symmetry. Out of a total of 36 phonon modes, 18 modes are Raman active (8Ag + 10Bg), 15 modes are IR active (7Au + 8Bu) and 3 are low-frequency acoustic modes. The A and B modes are one-dimensional irreducible representations which are symmetric and antisymmetric with respect to the principle axis of symmetry. The assignment of Raman and IR modes is performed in accordance with DFPT as implemented in Quantum ESPRESSO. Ambient pressure Raman and IR modes have been tabulated in Table 3 and Table 4, respectively. We have also included previously published Raman mode frequencies from theoretical calculations for the EuTaO4 and GdTaO4 compounds [16,17]. The frequency distribution of the Raman modes is quite similar to the Raman mode distribution in the wolframite structure, which belongs to the same space group P2/c as M’ fergusonite [39,40]. A total of twelve low-frequency Raman modes are present in the frequency region 100–400 cm−1, two Raman modes are in the 400–600 cm−1 range and four are in the higher-frequency side, 600–800 cm−1. It is interesting to note that we have not observed any frequency gap as observed in the Raman spectrum of the zircon or scheelite structure [41]. This can be understood by group–subgroup relationships among zircon–scheelite–fergusonite structures by virtue of the reduction in point group symmetry from 4/mmm to 4/m to 2/m, which in turn increases the allowed numbers of Raman modes and hence fills the frequency gap. All the calculated Raman modes can be categorized as internal modes or external modes. Internal modes, lying in the higher frequency region correspond to TaO6 octahedra while, modes at lower frequency are external modes describing the movement of rigid TaO6 unit against the lanthanide cation [42]. Out of a total of six internal modes of TaO6 octahedra, the 2 Ag and 2 Bg modes lie in the higher–frequency region that is from 600 to 800 cm−1, and two with Ag symmetry appear in the 390–500 cm−1 range [42]. Identified internal modes are marked by an asterisk in Table 3. The highest-frequency Ag mode, which appears at approximately 762 cm−1 for NdTaO4 and 771 cm−1 for SmTaO4, describes the symmetric stretching mode of TaO6 octahedra. The frequency of the majority of the Raman active modes are observed to increase with a decrease in the lanthanum ionic radii except the two Ag modes (108 cm−1 for NdTaO4 and 107cm−1 for SmTaO4; 220 cm−1 for NdTaO4 and 219 cm−1 for SmTaO4) whereas the frequency of the two Bg modes (at 119 and 138 cm−1) remains unaltered by the change in lanthanide cationic radii. This observation is valid when we extend our comparison with the published Raman mode frequencies of EuTaO4 and GdTaO4. Table 3 lists the pressure evolution of all the Raman active modes in P2/c structure obtained by quadratic fitting of data points under pressure. No Raman mode softening has been seen in both the compounds due to the absence of negative pressure coefficients. Hardening of all the Raman active modes has been associated with the monoclinic fergusonite phase of the other compound such as rare earth niobates GdNbO4 and EuNbO4, as well as rare earth tantalates GdTaO4 and EuTaO4 [16,17,22,23]. For rare earth vanadate, mode softening has been observed in the zircon or scheelite phase, but no mode softening has been reported in the fergusonite structure consistent with our current observation in the present work [41,43]. There is crossover between the Bg and Ag modes located at 380–400 cm−1 due to a nearly 3-fold higher pressure coefficient of the Bg mode than the Ag mode. No other mode crossover has been observed in spite of the large difference in the pressure coefficient of the Raman modes. In the HP phase, stabilized in the tetragonal structure, calculation predicts the presence of the 11 Raman active modes (5 Eg + 2A1g + 1B1g + 3B2g). The pressure evolution of the Raman active modes are shown in Table 5. In the HP phase, all modes show positive pressure coefficients except three modes, which show nonlinear behavior under compression.
As seen in Table 4 and Table 5 out of 15 IR modes show a decreasing trend when we go from the NdTaO4 to SmTaO4 compound, whereas one Bu mode (257 cm−1) remains unaltered. The remaining nine modes show an increase in frequency with lower lanthanide radii. From Table 4, it can be clearly seen that one IR active mode (Au) shows a nonlinear response in frequency under compression. The presence of the negative pressure coefficient of the IR active mode (Bu) indicates phonon softening in the compound with pressure. Phonon softening was cited as among the crucial trigger points for pressure-induced instability in metal oxides [39]. The other 13 IR modes have a positive pressure coefficient. According to our calculation, the high-pressure phase for both the compounds have seven IR active modes (4Eu + 3A2u). IR active modes at transition pressure, along with pressure coefficients, are summarized in Table 6. It can be clearly seen in Table 6 that all the modes show a positive pressure coefficient except the lowest-frequency A2u mode, which shows a nonlinear response under pressure.
4. Conclusions
To conclude, the compressional behavior of the M’ fergusonite-structured NdTaO4 and SmTaO4, investigated through DFT-based first-principle simulations indicate pressure-induced first-order phase transition from monoclinic to tetragonal structure. The transition is accompanied by an increase in oxygen coordination around the Ta cation from six to eight and a nearly 1.3% volume reduction at transition pressure (40 GPa for NdTaO4 and 33 GPa for SmTaO4). In the low-pressure monoclinic phase, the compressibility of the unit cell has a major contribution from rare earth polyhedra, whereas both the rare earth polyhedra and tantalum polyhedra exhibit a similar contribution towards the compressibility of the unit cell for both the compounds at the HP tetragonal phase, which in turn explains the lower bulk modulus obtained in the HP phase. The pressure evolution of phonon modes has been evaluated in both the LP phase and the HP phase. No Raman mode softening has been seen in both the compounds, although one particular IR mode has been observed to show red shift under pressure, possibly leading to instability in the compounds. Earlier reported experimental high-pressure studies on EuTaO4 and GdTaO4 show isostructural first-order reversible phase transition at approximately 20 GPa, although theoretical calculations predict an equally probable orthorhombic Pcna as an alternative description of the HP phase near 43 and 40 GPa, respectively. The experimental and theoretical phase transition pressures differ due to the crucial dependence of a non-hydrostatic stress environment on phase transition. Therefore, a high-pressure study on NdTaO4 and SmTaO4 by experimental techniques is desirable for a better understanding of structural and vibrational changes under compression, which is out of scope in the present work.
Author Contributions
Conceptualization, methodology, validation, writing—review and editing, visualization, supervision: A.B.G. (corresponding Author); Formal analysis, investigation, writing—original draft preparation, visualization: S.B. (First author); Data curation, visualization: A.T. (second author). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available from the corresponding author on reasonable request.
Acknowledgments
S.B. and A.T. acknowledge T. Sakuntala for providing motivation and encouragement.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haugsrud, R.; Norby, T. Proton conduction in rare-earth ortho-niobates and ortho-tantalates. Nat. Mater. 2006, 5, 193–196. [Google Scholar] [CrossRef]
- Yang, H.; Teng, Y.; Ren, X.; Wu, L.; Liu, H.; Wang, S.; Xu, L. Synthesis and crystalline phase of monazite-type Ce1−xGdxPO4 solid solutions for immobilization of minor actinide curium. J. Nucl. Mater. 2014, 444, 39–42. [Google Scholar] [CrossRef]
- Heuser, J.; Bukaemskiy, A.; Neumeier, S.; Neumann, A.; Bosbach, D. Raman and infrared spectroscopy of monazite-type ceramics used for nuclear waste conditioning. Prog. Nucl. Energy 2014, 72, 149–155. [Google Scholar] [CrossRef]
- Errandonea, D.; Manjon, F.J. Pressure effects on the structural and electronic properties of ABX4 scintillating crystals. Prog. Mater. Sci. 2008, 53, 711–773. [Google Scholar] [CrossRef]
- Errandonea, D.; Garg, A.B. Recent progress on the characterization of the high-pressure behaviour of AVO4 orthovanadates. Prog. Mater. Sci. 2018, 97, 123–169. [Google Scholar] [CrossRef]
- López-Moreno, S.; Rodríguez-Hernández, P.; Muñoz, A.; Romero, A.H.; Errandonea, D. First-principles calculations of elec-tronic, vibrational, and structural properties of scheelite EuWO4 under pressure. Phys. Rev. B 2011, 84, 064108. [Google Scholar] [CrossRef]
- Errandonea, D.; Santamaria-Perez, D.; Achary, S.N.; Tyagi, A.K.; Gall, P.; Gougeon, P. High-pressure x-ray diffraction study of CdMoO4 and EuMoO4. J. Appl. Phys. 2011, 109, 043510. [Google Scholar] [CrossRef]
- Siqueira, K.P.F.; Moreira, R.L.; Dias, A. Synthesis and Crystal Structure of Lanthanide Orthoniobates Studied by Vibrational Spectroscopy. Chem. Mater. 2010, 22, 2668–2674. [Google Scholar] [CrossRef]
- Lacomba-Perales, R.; Errandonea, D.; Meng, Y.; Bettinelli, M. High-pressure stability and compressibility of APO4 (A = La, Nd, Eu, Gd, Er, and Y) orthophosphates: An x-ray diffraction study using synchrotron radiation. Phys. Rev. B 2010, 81, 064113. [Google Scholar] [CrossRef]
- Errandonea, D.; Pellicer-Porres, J.; Manjon, F.J.; Segura, A.; Ferrer Roca, C.; Kumar, R.; Tschauner, O.; Lopez-Solano, J.; Ro-driguez, P.; Radescu, S.; et al. Determination of the High-Pressure Crystal Structure of BaWO4 and PbWO4. Phys. Rev. B 2006, 73, 224103. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, K.P.F.; Dias, A. Effect of the processing parameters on the crystalline structure of lanthanide orthotantalates. Mater. Res. 2013, 17, 167–173. [Google Scholar] [CrossRef]
- Siqueira, K.P.F.; Carvalho, G.B.; Dias, A. Influence of the processing conditions and chemical environment on the crystal structures and phonon modes of lanthanide orthotantalates. Dalton Trans. 2011, 40, 9454–9460. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Lee, W.; Melis, A.; Elmughrabi, A.; Lee, K.; Park, C.; Yeom, J.-Y. A Review of Inorganic Scintillation Crystals for Extreme Environments. Crystals 2021, 11, 669. [Google Scholar] [CrossRef]
- Yang, H.; Peng, F.; Zhang, Q.; Guo, C.; Shi, C.; Liu, W.; Sun, G.; Zhao, Y.; Zhang, D.; Sun, D.; et al. A promising high-density scintillator of GdTaO4 single crystal. Crystengcomm 2014, 16, 2480–2485. [Google Scholar] [CrossRef]
- Wang, J.; Chong, X.; Zhou, R.; Feng, J. Microstructure and thermal properties of RETaO4 (RE = Nd, Eu, Gd, Dy, Er, Yb, Lu) as promising thermal barrier coating materials. Scr. Mater. 2017, 126, 24–28. [Google Scholar] [CrossRef]
- Banerjee, S.; Garg, A.B.; Poswal, H.K. Pressure driven structural phase transition in EuTaO4: Experimental and first principles investigations. J. Phys. Condens. Matter 2022, 34, 135401. [Google Scholar] [CrossRef]
- Banerjee, S.; Garg, A.B.; Poswal, H.K. Structural and vibrational properties of GdTaO 4 under compression: An insight from experiment and first principles simulations. J. Appl. Phys. 2023, 133, 025902. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, Y.; Pi, Z.; Zhang, F. Phase Stability and Mechanical Properties of the Monoclinic, Monoclinic-Prime and Tetragonal REMO4 (M = Ta, Nb) from First-Principles Calculations. Coatings 2022, 12, 73. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D.; Rodríguez-Hernández, P.; López-Moreno, S.; Muñoz, A.; Popescu, C. High-pressure structural behaviour of HoVO4: Combined XRD experiments and ab initio calculations. J. Phys. Condens. Matter 2014, 26, 265402. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D. High-pressure powder x-ray diffraction study of EuVO4. J. Solid State Chem. 2015, 226, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, B.; Tan, D.; Xiao, W.; Song, M. Phase transformations of zircon-type DyVO4 at high pressures up to 36.4 GPa: X-ray diffraction measurements. J. Alloy. Compd. 2021, 875, 159926. [Google Scholar] [CrossRef]
- Garg, A.B.; Rao, M.R.; Errandonea, D.; Pellicer-Porres, J.; Martinez-Garcia, D.; Popescu, C. Pressure-induced instability of the fergusonite phase of EuNbO4 studied by in situ Raman spectroscopy, x-ray diffraction, and photoluminescence spectroscopy. J. Appl. Phys. 2020, 127, 175905. [Google Scholar] [CrossRef]
- Pellicer-Porresa, J.; Garg, A.B.; Vázquez-Socorroa, D.; Martínez-García, D.; Popescu, C.; Errandonea, D. Stability of the fer-gusonite phase in GdNbO4 by high pressure XRD and Raman experiments. J. Solid State Chem. 2017, 251, 14–18. [Google Scholar] [CrossRef]
- Garg, A.B.; Liang, A.; Errandonea, D.; Rodríguez-Hernández, P.; Muñoz, A. Monoclinic–triclinic phase transition induced by pressure in fergusonite-type YbNbO4. J. Phys. Condens. Matter 2022, 34, 174007. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D.; Rodríguez-Hernández, P.; Muñoz, A. High-pressure monoclinic–monoclinic transition in fergu-sonite-type HoNbO4. J. Phys. Condens. Matter 2021, 33, 195401. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; de Gironcoli, S.; Delugas, P.; Ruffino, F.F.; et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 2020, 152, 154105. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Corso, A.D. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337–350. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional per-turbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Hartenbach, I.; Lissner, F.; Nikelski, T.; Meier, S.F.; Müller-Bunz, H.; Schleid, T. About Lanthanide Oxotantalates with the Formula MTaO4 (M = La–Nd, Sm–Lu). Z. Anorg. Und Allg. Chem. 2005 631, 2377–2382.
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Panchal, V.; Errandonea, D.; Manjón, F.; Muñoz, A.; Rodríguez-Hernández, P.; Achary, S.; Tyagi, A. High-pressure lattice-dynamics of NdVO. J. Phys. Chem. Solids 2017, 100, 126–133. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D.; Rodríguez-Hernández, P.; Muñoz, A. ScVO4 under non-hydrostatic compression: A new meta-stable polymorph. J. Phys. Condens. Matter 2017, 29, 055401. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M.; Finger, L.W. Bulk modulus-volume relationship for cation-anion polyhedra. J. Geophys. Res. Solid Earth 1979, 84, 6723–6728. [Google Scholar] [CrossRef]
- Baur, W.H. The geometry of polyhedral distortions. Predictive relationships for the phosphate group. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 1195–1215. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Errandonea, D.; Popescu, C.; Garg, A.B.; Botella, P.; Martinez-García, D.; Pellicer-Porres, J.; Rodríguez-Hernández, P.; Muñoz, A.; Cuenca-Gotor, V.; Sans, J.A. Pressure-induced phase transition and band-gap collapse in the wide-band-gap semiconductor InTaO4. Phys. Rev. B 2016, 93, 035204. [Google Scholar] [CrossRef]
- Errandonea, D.; Ruiz-Fuertes, J. A Brief Review of the Effects of Pressure on Wolframite-Type Oxides. Crystals 2018, 8, 71. [Google Scholar] [CrossRef]
- Marqueño, T.; Errandonea, D.; Pellicer-Porres, J.; Martinez-Garcia, D.; Santamaria-Pérez, D.; Muñoz, A.; Rodríguez-Hernández, P.; Mujica, A.; Radescu, S.; Achary, S.N.; et al. High-pressure polymorphs of gadolinium orthovanadate: X-ray diffraction, Raman spectroscopy, and ab initio calculations. Phys. Rev. B 2019, 100, 064106. [Google Scholar] [CrossRef]
- Ruiz-Fuertes, J.; Errandonea, D.; López-Moreno, S.; González, J.; Gomis, O.; Vilaplana, R.; Manjón, F.J.; Muñoz, A.; Rodríguez-Hernández, P.; Friedrich, A.; et al. High-pressure Raman spectroscopy and lat-tice-dynamics calculations on scintillating MgWO4: A comparison with isomorphic compounds. Phys. Rev. B 2011, 83, 214112. [Google Scholar] [CrossRef]
- Errandonea, D.; Achary, S.N.; Pellicer-Porres, J.; Tyagi, A.K. Pressure-Induced Transformations in PrVO4 and SmVO4 and Isolation of High-Pressure Metastable Phases. Inorg. Chem. 2013, 52, 5464–5469. [Google Scholar] [CrossRef] [PubMed]
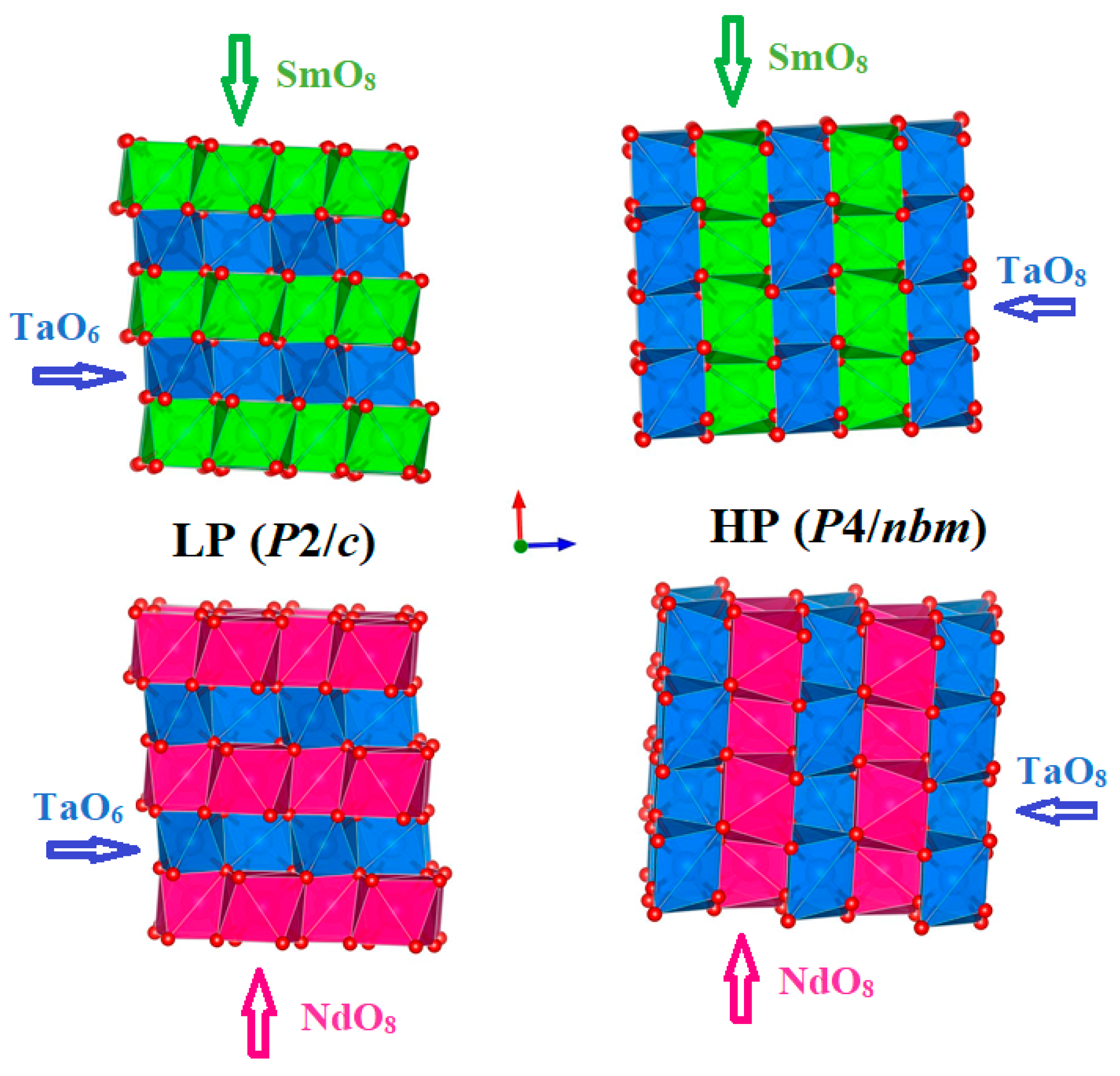
Figure 1.
Polyhedral representation of SmTaO4 (upper) and NdTaO4 (lower) in both LP (P2/c) and HP (P4/nbm) phases. Sm, Nd, Ta, and O atoms are green, pink, blue and red, respectively.
Figure 1.
Polyhedral representation of SmTaO4 (upper) and NdTaO4 (lower) in both LP (P2/c) and HP (P4/nbm) phases. Sm, Nd, Ta, and O atoms are green, pink, blue and red, respectively.

Figure 2.
Dependence of lattice parameters: (a) a, (b) b, (c) c, (d) unit cell volume V, (e) constituent polyhedral volume and (f) interatomic distances with the ionic radii of the lanthanide cation (Gd, Eu, Sm, and Nd).
Figure 2.
Dependence of lattice parameters: (a) a, (b) b, (c) c, (d) unit cell volume V, (e) constituent polyhedral volume and (f) interatomic distances with the ionic radii of the lanthanide cation (Gd, Eu, Sm, and Nd).

Figure 3.
Pressure dependence of unit cell lattice parameters for the compounds (a) NdTaO4 and (b) SmTaO4 in the low-pressure phase (solid circle) and the high-pressure phase (empty circle). The colored region describes the high-pressure tetragonal phase.
Figure 3.
Pressure dependence of unit cell lattice parameters for the compounds (a) NdTaO4 and (b) SmTaO4 in the low-pressure phase (solid circle) and the high-pressure phase (empty circle). The colored region describes the high-pressure tetragonal phase.

Figure 4.
Pressure dependence of the unit cell lattice volume for the compounds (a) NdTaO4 and (b) SmTaO4, of the low-pressure phase (solid circle) and the high-pressure phase (empty circle). The colored region describes the high-pressure tetragonal phase.
Figure 4.
Pressure dependence of the unit cell lattice volume for the compounds (a) NdTaO4 and (b) SmTaO4, of the low-pressure phase (solid circle) and the high-pressure phase (empty circle). The colored region describes the high-pressure tetragonal phase.

Figure 5.
Enthalpy difference (eV) versus pressure for (a) NdTaO4 and (b) SmTaO4. The ambient pressure monoclinic P2/c phase (black) has been taken as a reference for the both compounds. Red and green lines correspond to the Pcna (orthorhombic) and P4/nbm (tetragonal) structures, respectively.
Figure 5.
Enthalpy difference (eV) versus pressure for (a) NdTaO4 and (b) SmTaO4. The ambient pressure monoclinic P2/c phase (black) has been taken as a reference for the both compounds. Red and green lines correspond to the Pcna (orthorhombic) and P4/nbm (tetragonal) structures, respectively.

Figure 6.
Pressure dependence of the distortion index of the bond length of NdO8 and SmO8 (black), TaO6 (red) and TaO8 (orange) for the compounds (a) NdTaO4 and (b) SmTaO4, of the low-pressure phase (white region) and the high-pressure phase (colored region).
Figure 6.
Pressure dependence of the distortion index of the bond length of NdO8 and SmO8 (black), TaO6 (red) and TaO8 (orange) for the compounds (a) NdTaO4 and (b) SmTaO4, of the low-pressure phase (white region) and the high-pressure phase (colored region).

Figure 7.
(a) Nd-O bond lengths versus pressure in the LP phase (solid circle) and the HP phase (empty circle), Ta-O bond lengths versus pressure in the LP phase (solid triangle) and the HP phase (empty triangle); (b) Sm-O bond lengths versus pressure in LP phase (solid circle) and HP phase (empty circle), Ta-O bond lengths versus pressure in the LP phase (solid triangle) and the HP phase (empty triangle). The white region describes the LP phase while the colored region is for the HP phase.
Figure 7.
(a) Nd-O bond lengths versus pressure in the LP phase (solid circle) and the HP phase (empty circle), Ta-O bond lengths versus pressure in the LP phase (solid triangle) and the HP phase (empty triangle); (b) Sm-O bond lengths versus pressure in LP phase (solid circle) and HP phase (empty circle), Ta-O bond lengths versus pressure in the LP phase (solid triangle) and the HP phase (empty triangle). The white region describes the LP phase while the colored region is for the HP phase.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of simulated ambient pressure lattice parameters and constituent bond lengths of NdTaO4 and SmTaO4 with previously reported experimental values.
Table 1.
Comparison of simulated ambient pressure lattice parameters and constituent bond lengths of NdTaO4 and SmTaO4 with previously reported experimental values.
| Lattice Parameters | NdTaO4 | SmTaO4 | ||
|---|---|---|---|---|
| Experiment [32] | Calculated | Experiment [32] | Calculated | |
| a (Å) | 5.2437(4) | 5.257 | 5.2065(4) | 5.206 |
| b (Å) | 5.5969(4) | 5.638 | 5.5542(4) | 5.571 |
| c (Å) | 5.4275(4) | 5.451 | 5.3947(4) | 5.397 |
| β (degree) | 96.767(9) | 96.79 | 97.721(9) | 96.74 |
| Bond length | 2× | 2× | 2× | 2× |
| R-O2 (Å) | 2.454 | 2.459 | 2.414 | 2.4179 |
| R-O1(Å) | 2.371 | 2.371 | 2.346 | 2.33622 |
| R-O1(Å) | 2.608 | 2.603 | 2.586 | 2.57709 |
| R-O2(Å) | 2.408 | 2.409 | 2.377 | 2.37666 |
| Ta-O1(Å) | 1.864 | 1.879 | 1.874 | 1.88026 |
| Ta-O2(Å) | 2.201 | 2.230 | 2.211 | 2.22584 |
| Ta-O2(Å) | 1.996 | 2.003 | 1.990 | 1.99934 |
Table 2.
Calculated structural parameters along with the atomic positions of NdTaO4 and SmTaO4 at ambient pressure and transition pressure.
Table 2.
Calculated structural parameters along with the atomic positions of NdTaO4 and SmTaO4 at ambient pressure and transition pressure.
| NdTaO4: LP Monoclinic Phase (P2/c) @ Ambient Pressure a = 5.2504 Å; b = 5.6312 Å; c = 5.4446 Å; β = 96.79° | |||
| Nd (2e) | 0.0000 | 0.23488 | 0.2500 |
| Ta (2f) | 0.5000 | 0.31247 | 0.7500 |
| O1 (4g) | 0.74508 | 0.90645 | 0.39125 |
| O2 (4g) | 0.27392 | 0.56319 | 0.49246 |
| NdTaO4: HP Tetragonal Phase (P4/nbm) @ 40 GPa a = 5.0765 Å; c = 5.0616 Å | |||
| Nd (2b) | 0.75000 | 0.75000 | 0.500 |
| Ta (2c) | 0.75000 | 0.25 | 0.0 |
| O (8m) | 0.45494 | 0.54506 | 0.23076 |
| SmTaO4: LP Monoclinic Phase (P2/c) @ Ambient Pressure a = 5.2025 Å; b = 5.5722 Å; c = 5.3985 Å; β = 96.74° | |||
| Sm (2e) | 0.0000 | 0.23438 | 0.2500 |
| Ta (2f) | 0.5000 | 0.30982 | 0.7500 |
| O1 (4g) | 0.74726 | 0.91026 | 0.39507 |
| O2 (4g) | 0.27135 | 0.56359 | 0.49281 |
| SmTaO4: HP Tetragonal Phase (P4/nbm) @ 33 GPa a = 5.0949 Å; c = 5.0262 Å | |||
| Sm (2b) | 0.75000 | 0.75000 | 0.500 |
| Ta (2c) | 0.75000 | 0.25 | 0.0 |
| O (8m) | 0.45372 | 0.54628 | 0.23465 |
Table 3.
Calculated Raman modes for NdTaO4 and SmTaO4 at ambient pressure along with their pressure coefficients (present work). Reported data of earlier studies on EuTaO4 and GdTaO4 have been included for comparison. Frequencies marked with an asterisk (*) correspond to internal modes of TaO6 octahedra.
Table 3.
Calculated Raman modes for NdTaO4 and SmTaO4 at ambient pressure along with their pressure coefficients (present work). Reported data of earlier studies on EuTaO4 and GdTaO4 have been included for comparison. Frequencies marked with an asterisk (*) correspond to internal modes of TaO6 octahedra.
| Raman Frequency | NdTaO4 | SmTaO4 | EuTaO4 [16] | GdTaO4 [17] | ||||
|---|---|---|---|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | ω | dω/dP | ω | dω/dP | |
| Bg | 99.3 | 1.42 | 101.6 | 1.83 | 102.3 | 1.25 | 100.6 | 1.64 |
| Ag | 108.7 | 1.40 | 107.5 | 2.45 | 107.1 | 1.14 | 103.5 | 1.84 |
| Bg | 119.6 | 1.28 | 119.7 | 1.53 | 119.8 | 1.10 | 119.1 | 1.27 |
| Bg | 138.7 | 2.22 | 138.4 | 2.95 | 138.5 | 1.67 | 138.5 | 2.49 |
| Bg | 164.3 | 0.88 | 168.7 | 1.43 | 172.0 | 0.94 | 171 | 0.92 |
| Ag | 176.7 | 0.76 | 177.2 | 1.05 | 177.6 | 0.54 | 177.9 | 0.41 |
| Ag | 220.6 | 4.07 | 219 | 4.7 | 216.9 | 4.17 | 211.1 | 4.74 |
| Bg | 257.1 | 2.41 | 261.9 | 2.99 | 263.6 | 2.65 | 261.6 | 2.66 |
| Ag | 257.6 | 2.85 | 262.5 | 3.04 | 264.3 | 2.70 | 263.4 | 2.18 |
| Bg | 314.1 | 4.22 | 321.9 | 5.19 | 325.2 | 4.62 | 322 | 4.78 |
| Bg | 383.5 | 4.20 | 392.5 | 5.3 | 396.0 | 3.96 | 395.3 | 4.80 |
| Ag* | 392.6 | 1.36 | 397.1 | 1.8 | 400.3 | 1.68 | 402.6 | 1.76 |
| Bg | 466.9 | 1.7 | 475.1 | 2.3 | 480.8 | 1.88 | 486.7 | 1.53 |
| Ag* | 472.9 | 1.42 | 478.7 | 1.97 | 482.2 | 1.84 | 487.6 | 1.61 |
| Bg* | 600.3 | 3.65 | 613 | 4.11 | 620.5 | 3.93 | 626.3 | 4.29 |
| Ag* | 609.3 | 3.62 | 620.8 | 4.05 | 627.6 | 3.77 | 633.4 | 4.15 |
| Bg* | 627.4 | 4.78 | 643.4 | 4.93 | 651.1 | 4.17 | 661.7 | 4.76 |
| Ag* | 762.2 | 3.55 | 771.5 | 3.58 | 777.1 | 3.11 | 785.9 | 3.69 |
Table 4.
Calculated IR modes for NdTaO4 and SmTaO4 at ambient pressure along with their pressure coefficients (present work).
Table 4.
Calculated IR modes for NdTaO4 and SmTaO4 at ambient pressure along with their pressure coefficients (present work).
| IR Frequency | NdTaO4 @ Ambient Pressure | SmTaO4 @ Ambient Pressure | ||
|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | |
| Au | 135.5 | 2.13 | 134.3 | 2.88 |
| Bu | 138.6 | 1.58 | 137 | 2.92 |
| Bu | 156.4 | 3.85 | 155.1 | 5.03 |
| Bu | 207.5 | −1.44 | 204.2 | −1.54 |
| Au | 249.1 | Nonlinear | 253.7 | Nonlinear |
| Bu | 257.2 | 2.24 | 257.2 | 2.97 |
| Bu | 275.8 | 2.24 | 281.9 | 2.39 |
| Au | 311.1 | 0.87 | 310.8 | 1.53 |
| Au | 354.7 | 3.33 | 359.4 | 3.81 |
| Bu | 382 | 3.26 | 387.3 | 3.59 |
| Bu | 480.7 | 4.12 | 491.5 | 4.65 |
| Au | 503.6 | 3.74 | 512.4 | 4.23 |
| Au | 561.3 | 4.33 | 572.8 | 4.68 |
| Bu | 607.5 | 4.23 | 616.7 | 4.66 |
| Au | 743.2 | 3.77 | 752.2 | 3.96 |
Table 5.
Calculated Raman modes for NdTaO4 and SmTaO4 at transition pressure along with their pressure coefficients (present work).
Table 5.
Calculated Raman modes for NdTaO4 and SmTaO4 at transition pressure along with their pressure coefficients (present work).
| Raman Frequency | NdTaO4(HP) @ 40 GPa | SmTaO4(HP) @ 33 GPa | ||
|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | |
| Eg | 103.1 | Nonlinear | 99.6 | Nonlinear |
| B2g | 163.6 | Nonlinear | 159.4 | Nonlinear |
| Eg | 208.7 | 1.1 | 199.9 | 1.2 |
| B1g | 357.5 | 0.92 | 343.9 | 1.26 |
| Eg | 412.3 | 1.93 | 397.2 | 2.16 |
| A1g | 412.9 | Nonlinear | 408.7 | Nonlinear |
| B2g | 513 | 2.77 | 501 | 2.73 |
| Eg | 546.6 | 2.9 | 533.6 | 3.09 |
| Eg | 741.8 | 1.55 | 738.4 | 1.85 |
| B2g | 750 | 1.85 | 743.8 | 2.09 |
| A1g | 823.7 | 1.12 | 815.7 | 1.25 |
Table 6.
Calculated IR modes for NdTaO4 and SmTaO4 at 40 and 33 GPa along with their pressure coefficients (present work).
Table 6.
Calculated IR modes for NdTaO4 and SmTaO4 at 40 and 33 GPa along with their pressure coefficients (present work).
| IR Frequency | NdTaO4(HP)40 GPa | SmTaO4(HP)33 GPa | ||
|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | |
| A2u | 109 | Nonlinear | 110.6 | Nonlinear |
| Eu | 136.2 | 1.92 | 126.2 | 2.24 |
| Eu | 183.2 | 1.36 | 183.8 | 1.33 |
| A2u | 281.2 | 1.80 | 264.1 | 1.89 |
| Eu | 509.3 | 2.26 | 494.6 | 2.52 |
| A2u | 669.1 | 0.62 | 659.6 | 0.85 |
| Eu | 699.9 | 1.53 | 687.7 | 1.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Banerjee, S.; Tyagi, A.; Garg, A.B. Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies. Crystals 2023, 13, 254. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst13020254
AMA Style
Banerjee S, Tyagi A, Garg AB. Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies. Crystals. 2023; 13(2):254. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst13020254
Chicago/Turabian StyleBanerjee, Saheli, Amit Tyagi, and Alka B. Garg. 2023. "Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies" Crystals 13, no. 2: 254. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst13020254
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.