Dihalogen and Pnictogen Bonding in Crystalline Icosahedral Phosphaboranes


1
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nam. 2, 16610 Prague 6, Czech Republic
2
Institute of Inorganic Chemistry of the Czech Academy of Sciences, 25068 Husinec-Řež, Czech Republic
*
Authors to whom correspondence should be addressed.
Crystals 2018, 8(10), 390; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst8100390
Submission received: 6 September 2018
/
Revised: 4 October 2018
/
Accepted: 9 October 2018
/
Published: 13 October 2018
(This article belongs to the Special Issue σ- and π-Hole Interactions in Crystals)
Abstract
:Noncovalent interactions in the single crystal of 3,6-Cl2-closo-1,2-P2B10H8 and in the crystal of closo-1,7-P2B10Cl10•toluene were analyzed by means of quantum chemical computations. The crystal packing in the second crystal was dominated by numerous B-Cl···Cl-B dihalogen and strong B-P···π pnictogen bonds, the latter of which were characterized by a small length of 3.08 Å and a large interaction energy value, exceeding −10 kcal mol−1.

1. Introduction
Phosphorus atoms can be relatively easily incorporated into the icosahedral closo-B12H122− skeleton, which is achieved experimentally by reacting a boron hydride with an open pentagonal belt, e.g. B10H14, with PCl3 [1,2]. Such a reaction yields closo-1,2-P2B10H10 and its mono- and di-chloro derivatives. Among them, 3,6-Cl2-closo-1,2-P2B10H8 (1) has been crystallized after separation from the reaction mixture. The parent phosphaborane can be thermally rearranged to obtain the isomeric closo-1,7-P2B10H10 [3]. However, when the pyrolysis reaction of B2Cl4 with PCl3 takes place, the terminal hydrogens of the latter cage are completely substituted by chlorines, resulting in closo-1,7-P2B10Cl10 (2). The molecular diagrams of 1 and 2 with numbering are shown in Figure 1. Compound 2 is prone to crystallization with toluene, yielding crystals of 2•C6H5CH3 in this process [4].
The phosphorus atoms of closo-1,2-P2B10H10 and closo-1,7-P2B10H10 are incorporated into the skeleton via multicenter 3-center-2-electron (3c2e) and 4-center-2-electron (4c2e) types of bonding [5]. The electron distribution is in disagreement with the classical electronegativity concept in multicenter bonding [5,6] and it results in areas of highly positive electrostatic potential, called σ-holes [7], on phosphorus atoms [8]. The concept of σ-hole, developed by Politzer et al., was originally used to describe halogen (X) bonds, where the σ-hole was centered on the carbon–halogen σ-bond axis. A partially positive σ-hole on a partially negative X atom could thus explain the interesting ability of an X atom to interact simultaneously with electrophiles and nucleophiles.
Closo-phosphaboranes are not the only heteroboranes with highly positive σ-holes. Indeed, various chalcogen and X-bonds have been reported for closo-thiaboranes and halogenated carboranes [9,10,11]. In addition, pnictogen (Pn) bonds are known for various nido-Pn2C2B7HnX9−n (Pn = P, As, Sb) molecules [12,13]. All these σ-hole interactions have been reported for single crystalline materials. In order to expand the body of Pn-bonding in closo structural motifs, we have selected a known crystal structure of 1 [1]. Additionally, we selected the crystal of 2 cocrystalized with toluene [4] for quantum chemical analysis, the first candidate for a non-single-crystalline material in boron cluster chemistry stabilized by σ-hole interactions.
2. Materials and Methods
The molecular electrostatic potential (ESP) surfaces of isolated molecules 1 and 2 were computed at the Hartree-Fock (HF) level with the correlation-consistent polarized valence double-zeta (cc-pVDZ) basis set using the Gaussian09 [14] and Molekel4.3 [15,16] programs. As a one-electron property, ESP is correctly described by the non-correlated HF method and the double-zeta basis set size is sufficient for its computation [17].
Around each unique molecule in the studied crystals, we created clusters by considering the surrounding molecules. The molecules that had any atom within 5 Å of the central molecule formed a cluster that was called the first layer. The second layer was obtained by considering molecules that had any atom within 5 Å of the first layer. Hydrogen atoms in the central molecule and in the first layer were optimized using the density functional theory (DFT) with the resolution of identity (RI) approximation, the empirical dispersion (D3), the Becke, Lee, Yang and Parr functional (BLYP), and the double-zeta valence polarized (DZVP) basis set [18]. Hydrogen atoms of the second layer were optimized by the hybrid DFT-D3/PM6-D3H4X approach. The DFT-D3/BLYP/DZVP method was used for the first layer and the corrected semiempirical quantum mechanical PM6-D3H4X method [19] for the second layer. All heavy atoms were frozen in crystallographic positions. The obtained clusters were used for energy calculations. Two-body interaction energy (ΔE2) was defined as the energy difference between the energy of the dimer and the sum of monomer energies.
ΔE2(AB) = E(AB) − E(A) − E(B)
Three-body energy (ΔE3) was defined as the energy difference between the total energy of the trimer and the sum of both monomer energies and all ΔE2 values.
ΔE3(ABC) = E(ABC) − E(A) − E(B) − E(C) − ΔE2(AB) − ΔE2(BC) − ΔE2(AC)
Expanding the ΔE2 energies, the ΔE3(ABC) could thus be expressed as:
ΔE3(ABC) = E(ABC) − E(AB) − E(AC) − E(BC) + E(A) + E(B) + E(C)
The sum of the ΔE2 values (∑ΔE2(A)) of the central molecule A was computed for residues in the first and second layers. The whole layers were labeled as Q and were formed by residues marked as B to N).
Finally, the interaction of the central molecule A with the surrounding molecules was computed as:
ΔE(AQ) = E(AQ) − E(A) − E(Q)
The many-body energy (ΔEMB) was computed as the difference ΔE(AQ) and ∑ΔE2(A).
ΔEMB(AQ) = ΔE(AQ) − ∑ΔE2(A)
The energies were determined using the DFT-D3 method with the Tao, Perdew, Staroverov and Scuseria (TPSS) functional and the triple-zeta TZVPP basis set. Three-body dispersion was not used for the TPSS functional because it had been shown that the ΔE3 of TPSS was too repulsive and the three-body dispersion only made the overall errors worse [20]. The benchmark ΔE2 were determined by the MP2.5 method [21] with the complete basis set (CBS). MP2.5/CBS was calculated as the sum of the Møller–Plesset perturbation theory to the second order (MP2) with CBS and the scaled third-order energy contribution (the scaling factor of 0.5) using the augmented correlation-consistent polarized double-zeta (aug-cc-pVDZ) basis set. For MP2/CBS, an extrapolation from aug-cc-pVDZ to aug-cc-pVTZ was used [22]. Counterpoise corrections for the basis set superposition error (BSSE) and RI approximations were used for the MP2.5 calculations. ΔE2 values were decomposed by symmetry-adapted perturbation-theory (SAPT) methodology. The simplest truncation of SAPT (SAPT0) decomposition [23] was performed with the recommended jun-cc-pVDZ basis sets (i.e. cc-pVDZ on hydrogen and aug-cc-pVDZ on heavier atoms) [24]. Turbomole (7.0) [25], PSI4 [26], MOPAC2016 [27] and Cuby4 [28] were used.
3. Results and Discussion
3.1. The Properties of Isolated Molecules
To obtain a deeper insight into the noncovalent interactions of phosphaboranes, we computed the ESP surfaces of 1 and 2 and compared them with analogous compounds in the literature. The parent closo-1,2-P2B10H10 compound was reported to have highly positive σ-holes with a magnitude (VS,max) of 22.6 kcal mol−1 and a large dipole moment of 2.7 D [8]. The introduction of two electronegative Cl atoms in compound 1 resulted in even more positive σ-holes (VS,max of about 25.2 kcal mol−1, see Figure 2 and Table 1). The dipole moment of 1 was, however, smaller (1.2 D) because the vector addition of the two B–Cl bond dipole moments [29] was in the opposite direction with respect to the vector pointing out from the midpoint of the P(1)–P(2) vector towards the center of the cluster. The Cl atoms of 1 had slightly positive σ-holes (VS,max 2.3 kcal mol−1) and the minimum value of the ESP molecular surface (VS,min) of the Cl(3,6) atom was −9.5 kcal mol−1. The Cl atoms of 1 are thus expected to be better H-bond acceptors than X-bond donors.
A complete substitution of terminal hydrogens by chlorines considerably increased the dipole moment to 3.4 D (eight B–Cl bond dipole moments were added to the vector from the P(1)–P(7) midpoint towards the center of the cluster) and VS,max values up to 30.2 kcal mol−1 in the case of 2. It can thus be considered as a very good Pn-bond donor. Additionally, the Cl(2,3) atoms of 2 had positive σ-holes with a VS,max value of 13.1 kcal mol−1; the VS,min of the Cl(9,10) atoms was −13.2 kcal mol−1. Therefore, the Cl atoms of 2 can act as both X-bond donors and H-bond acceptors.
3.2. Interactions in the Single Crystal of 1 and the Crystal of 2•Toluene
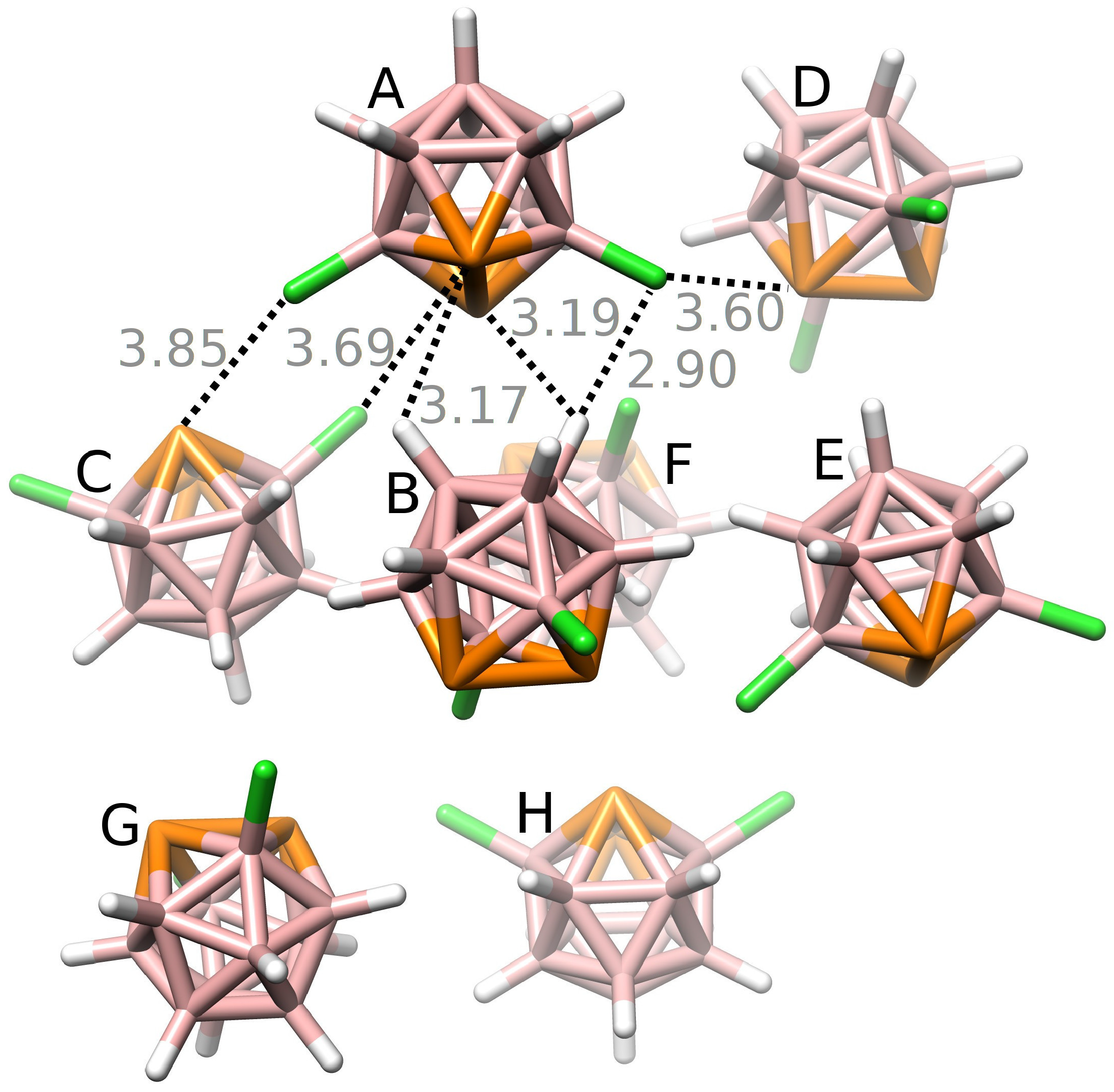
Initially, the crystal structure of 1 was analyzed. There are eight molecules in the unit cell (see Figure 3). First, all pairwise interactions were evaluated by computing interaction energy (ΔE2) values by the DFT-D3 method using various functionals and basis sets. For the most stable motifs, highly accurate MP2.5/CBS ΔE2 values were computed. Additionally, ΔE2 was decomposed into different terms by using the SATP technique. The obtained results are summarized in Table 2. The SAPT0/jun-cc-pVDZ results were in reasonable agreement with the MP2.5/CBS results (the root-mean-square error (RMSE) of 0.41 kcal mol−1). Among the DFT-D3 methods, the best agreement with MP2.5/CBS results, i.e. the RMSE of 0.47 kcal mol−1, was found for the TPSS/TZVPP level. The other tested functionals and basis sets had a bigger RMSE, i.e. 1.31, 1.29, 1.19 and 1.10 kcal mol−1 for BLYP/def2-QZVP, BLYP/DZVP, B3LYP/DZVP and TPSS/DZVP, respectively. The ΔE2 values ranged from −3.35 to −4.23 kcal mol−1 at the MP2.5/CBS level. These values are large considering that there were no intermolecular distances shorter than the sum of van der Waals radii (ΣrvdW) [30] in the crystal. The highly negative ΔE2 values were caused by the large dispersion contribution, which strongly dominated the interaction (65–71% of the total attractive energy). The electrostatic interaction was also important (21–27% of the total attractive energy). The smallest attractive term was induction (7–8% of the total attractive energy).
The sum of ΔE2 values (∑ΔE2(1)) was −48.45 kcal mol−1 for the first layer. The consideration of the second layer made the ∑ΔE2(1) more negative by 3.38 kcal mol−1. Three-body energy ΔE3(ABC), ΔE3(ACD) and ΔE3(BCD) values were relatively small (below 2% of ΔE2). They ranged from −0.01 to 0.22 kcal mol−1 at the DFT-D3/TPSS/TZVPP level. Similar ΔE3 results were obtained at the DFT-D3/B3LYP/DZVP level (ΔE3 ranged from −0.05 to 0.14 kcal mol−1).
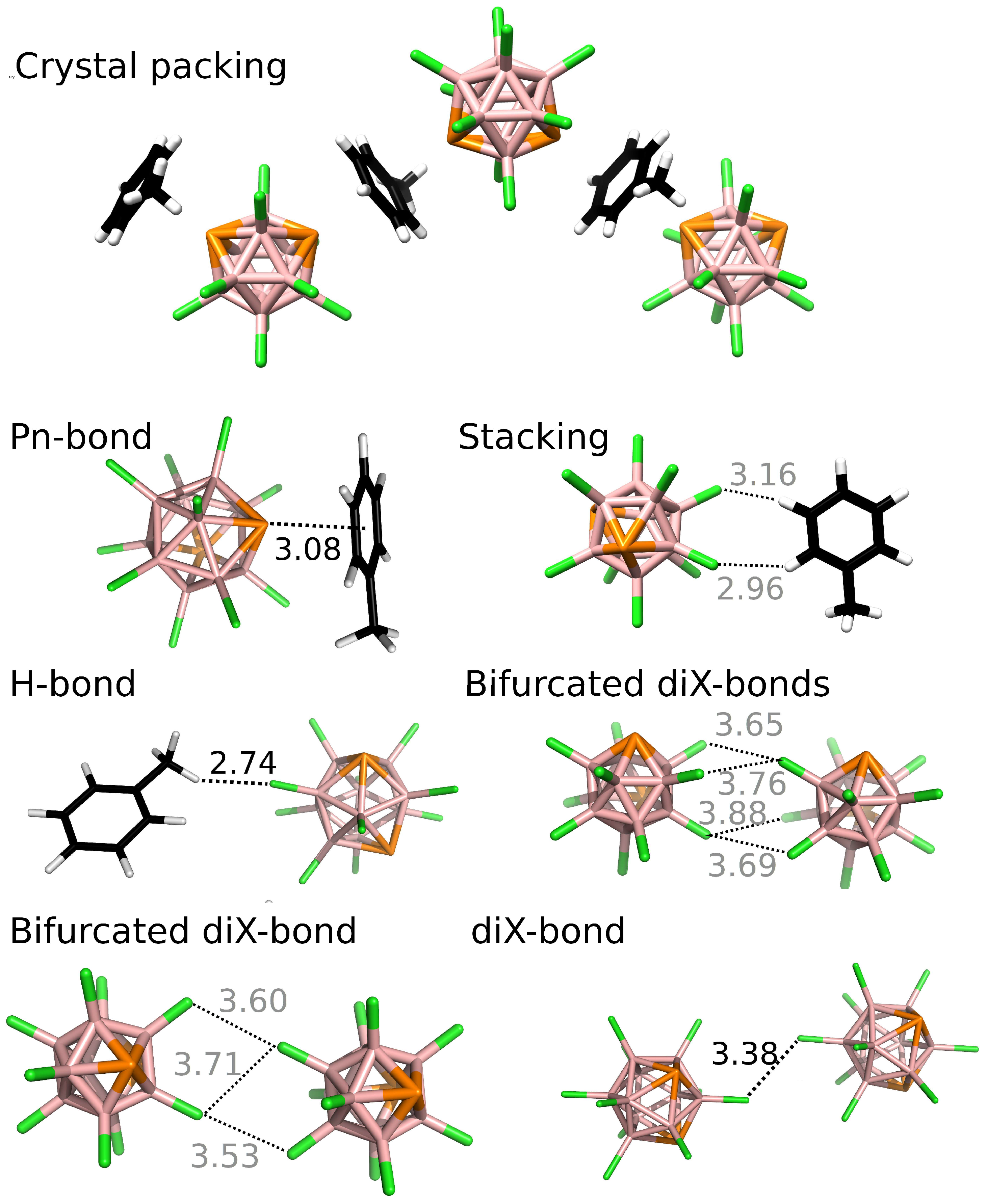
Secondly, we studied interactions in the crystal of 2•toluene. The results showed that the most negative ΔE2 value was exhibited by the B-P···π Pn-bond. The Pn-bond was characterized by a small length of 3.08 Å (88% of ΣrvdW) and its ΔE2 value exceeded −10 kcal mol−1 at the MP2.5/CBS level (see Table 3 and Figure 4). The SAPT decomposition revealed that electrostatic and induction contributions were more important in this Pn-bond. They represented 35% and 14% of the total attractive energy of this Pn-bond, respectively, but they did not exceed 27% and 10% of the total attractive energy in the other interactions reported in this study. Pn···π interactions in neutral complexes are well known, especially for heavier Pn atoms (As, Sb and Bi). For example, AsCl3 and SbCl3 formed Pn···π Pn-bonds with substituted benzenes that had a Pn···benzenecentroid separation of 3.14 and 3.24 Å (88 and 86% of ΣrvdW), respectively [31,32]. Phosphorus···π interactions are typically only observed in cationic phosphorus complexes, as exemplified by the interaction between the Mes*NP+ cation and benzene, which has a P···benzenecentroid separation of 3.00 Å (86% of ΣrvdW) [33]. The reported Pn-bond can be further related to analogous σ-hole interactions in heteroboranes. For example, the C-Br···π chalcogen bond reported for brominated carboranes had a length of 3.46 Å (98% of ΣrvdW) [34] and the ΔE value of the isolated X-bond was estimated to be about −4 kcal mol−1 [11]. The B-S···π chalcogen bond found in phenyl-substituted thiaboranes had a length of 3.24 Å (93% of ΣrvdW) and a ΔE of −8.6 kcal mol−1 [9]. The Sb2···H-B Pn-bond with a length of 2.78 Å (88% of ΣrvdW) and a ΔE2 of −6.46 kcal mol−1 was found in the single crystal of the stibacarbaborane molecule [12]. The ΔE2 values of hypothetical closo-1,2-P2B10H10···benzene, closo-1,2-As2B10H10···benzene, and AsCl3··benzene Pn-bonded complexes were computed and reported to be −4.8, −5.6 and −6.5 kcal mol−1, respectively [8,35].
The computed ∑ΔE2 values were used to evaluate the importance of the B-P···π Pn-bond for the crystal packing of 2•toluene. The ∑ΔE2(toluene) and ∑ΔE2(2) were −31.55 and −57.12 kcal mol−1 for the first layer, respectively. The B-P···π Pn-bonds thus formed 63 and 35 % of ∑ΔE2(toluene) and ∑ΔE2(2), respectively. The consideration of the second layer made ∑ΔE2(toluene) and ∑ΔE2(2) more negative by about 1.98 and 5.13 kcal mol−1, which lowered the relative contribution of Pn-bonds to 60% and 32%, respectively. Additionally, the computed ΔE3 values of the Pn-bonds were more repulsive than for those of the other interactions. The ΔE3(2toluene2) and ΔE3(toluene2toluene) sandwich-like motifs stabilized by the Pn-bond were 1.34 and 0.36 kcal mol−1, respectively, at the DFT-D3/TPSS/TZVPP level. The ΔE3 thus further reduced the strength of the Pn-bonding by about 7%. The other interactions were reduced by less than 2% by the ΔE3. The role of many-body energies was further examined by computing the ΔEMD for the toluene cluster (the first layer considered). The obtained value of 2.64 kcal mol−1 represented 8% of the ∑ΔE2(toluene) value. Summarizing the results shown above, the Pn-bond formed about 56 and 32% of the computed binding energy for the toluene and 2, respectively. These results indicate the large importance of Pn-bonding, especially for toluene. Interestingly, the diX-bonds represented about 42% of the computed binding of 2, which was even more than that of Pn-bonding in this case. Even though the diXbonds were considerably weaker than the Pn-bonds (see Table 3), they were much more numerous, which resulted in an overall large contribution to the computed binding energy.
The shortest intermolecular Cl···Cl separation in the crystal structure was 3.38 Å (96% of ΣrvdW). The diX-bond had ΔE2 of −2.65 kcal mol−1 at the MP2.5/CBS level. The geometrical arrangement of this interaction had very similar Θ1 and Θ2 angles (i.e. B-Cl···Cl angles of 125° and 127°), which is typical of diX-bonds [36]. Additionally, H-bonding can also be found in the crystal of 2•toluene. The H-bond had the least negative ΔE2 value among the interaction motifs in this study and the H···Cl distance was 2.74 Å (96% of ΣrvdW).
In summary, we analyzed noncovalent interactions in the single crystal of 1 and in the crystal of 2•toluene by quantum chemical protocols. The analysis revealed numerous diXbonds and unusually strong B-P···π pnictogen bonds, which dominated the crystal packing of the crystal of 2•toluene. The Pn-bond length was significantly below ΣrvdW and the ΔE value overcoming −10 kcal mol−1.
Author Contributions
Methodology, J.F. and D.H.; Writing Draft Preparation, J.F. and D.H.; Visualization, J.F.; Project Administration, J.F. and D.H.
Funding
This work was supported by the research project RVO 61388963 of the Czech Academy of Sciences. This research was funded by the Czech Science Foundation grant number 17-08045S.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Grüner, B.; Hnyk, D.; Císařová, I.; Plzák, Z.; Štíbr, B. Phosphaborane Chemistry. Syntheses and Calculated Molecular Structures of Mono- and Di-chloro Derivatives of 1,2-Diphospha-closo-dodecaborane(10). J. Chem. Soc. Dalton Trans. 2002, 15, 2954–2959. [Google Scholar] [CrossRef]
- Štíbr, B.; Holub, J.; Bakardjiev, M.; Hnyk, D.; Tok, O.L.; Milius, W.; Wrackmeyer, B. Phosphacarborane Chemistry: The Synthesis of the Parent Phosphadicarbaboranes nido-7,8,9-PC2B8H11 and [nido-7,8,9-PC2B8H10]−, and Their 10-Cl Derivatives—Analogs of the Cyclopentadiene Anion. Eur. J. Inorg. Chem. 2002, 9, 2320–2326. [Google Scholar] [CrossRef]
- Little, J.L.; Whitesell, M.A.; Chapman, R.W.; Kester, J.G.; Huffman, J.C.; Todd, L.J. Synthesis of Some 10-, 11-, and 12-Atom Phosphaboranes. Crystal Structure of 2-(Trimethylamine)-1-PB11H10. Inorg. Chem. 1993, 32, 3369–3372. [Google Scholar] [CrossRef]
- Keller, W.; Sawitzki, G.; Haubold, W. Synthesis of Halogenated Polyhedral Phosphaboranes. Crystal Structures of closo-1,7-P2B10Cl10. Inorg. Chem. 2000, 39, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Melichar, P.; Hnyk, D.; Fanfrlík, J. Systematic Examination of Classical and Multi-Center Bonding in Heteroborane Clusters. Phys. Chem. Chem. Phys. 2018, 20, 4666–4675. [Google Scholar] [CrossRef] [PubMed]
- Hnyk, D.; Všetečka, V.; Drož, L.; Exner, O. Charge Distribution within 1,2-Dicarba-closo-dodecaborane: Dipole Moments of its Phenyl Derivatives. Collect. Czech. Chem. Commun. 2001, 66, 1375–1379. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Pecina, A.; Lepšík, M.; Hnyk, D.; Hobza, P.; Fanfrlík, J. Chalcogen and Pnicogen Bonds in Complexes of Neutral Icosahedral and Biccaped Square-Antiprismatic Heteroboranes. J. Phys. Chem. A 2015, 119, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Přáda, A.; Padělková, Z.; Pecina, A.; Macháček, J.; Lepšík, M.; Holub, J.; Růžička, A.; Hnyk, D.; Hobza, P. The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Hnyk, D. Chalcogens Act as Inner and Outer Heteroatoms in Borane Cages with Possible Consequences for σ-Hole Interactions. CrystEngComm 2016, 47, 8973–9162. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Holub, J.; Růžičková, Z.; Řezáč, J.; Lane, P.D.; Wann, D.A.; Hnyk, D.; Růžička, A.; Hobza, P. Competition between Halogen, Hydrogen and Dihydrogen Bonding in Brominated Carboranes. ChemPhysChem 2016, 17, 3373–3376. [Google Scholar] [Green Version]
- Holub, J.; Melichar, P.; Růžičková, Z.; Vrána, J.; Wann, D.A.; Fanfrlík, J.; Hnyk, D.; Růžička, A. A Novel Stibacarbaborane Cluster with Adjacent Antimony Atoms Exhibiting Unique Pnictogen Bond Formation that Dominates Its Crystal Packing. Dalton Trans. 2017, 46, 13714–13719. [Google Scholar] [CrossRef] [PubMed]
- Holub, J.; Růžičková, Z.; Hobza, P.; Fanfrlík, J.; Hnyk, D.; Růžička, A. Various Types of Non-covalent Interactions Contributing Towards Crystal Packing of Halogenated Diphospha-dicarborane with an Open Pentagonal Belt. New J. Chem. 2018, 42, 10481–10483. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Flűkiger, P.; Lűthi, H.P.; Portmann, S.; Weber, J. MOLEKEL 4.3; Swiss Center for Scientific Computing: Manno, Switzerland, 2000. [Google Scholar]
- Portmann, S.; Luthi, H.P. MOLEKEL: An Interactive Molecular Graphic Tool. CHIMIA Int. J. Chem. 2000, 54, 766–770. [Google Scholar]
- Riley, K.E.; Tran, K.A.; Lane, P.; Murray, J.S.; Politzer, P. Comparative Analysis of Electrostatic Potential Maxima and Minima on Molecular Surfaces, as Determined by Three Methods and a Variety of Basis Sets. J. Comput. Sci. 2016, 17, 273–284. [Google Scholar] [CrossRef]
- Hostaš, J.; Řezáč, J. Accurate DFT-D3 Calculations in a Small Basis Set. J. Chem. Theory Comput. 2017, 13, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; Hobza, P. Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; Huang, Y.; Hobza, P.; Beran, J.O.G. Benchmark Calculations of Three-Body Intermolecular Interactions and the Performance of Low-Cost Electronic Structure Methods. J. Chem. Theory Comput. 2015, 11, 3065–3079. [Google Scholar] [CrossRef] [PubMed]
- Pitonak, M.; Neogady, P.; Cerny, J.; Grimme, S.; Hobza, P. Scaled MP3 Non-Covalent Interaction Energies Agree Closely with Accurate CCSD(T) Benchmark Data. ChemPhysChem 2009, 10, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-Set Convergence in Correlated Calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of Symmetry Adapted Perturbation Theory (SAPT). I. Efficiency and Performance for Interaction Energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An Open-Source Ab Initio Electronic Structure Program. WIREs Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Stewart, J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J. Cuby: An Integrative Framework for Computational Chemistry. J. Comput. Chem. 2016, 37, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Macháček, J.; Plešek, J.; Holub, J.; Hnyk, D.; Všetečka, V.; Císařová, I.; Kaupp, M.; Štíbr, B. New Route to 1-Thia-closo-dodecaborane(11), closo-1-SB11H11, and Its Halogenation Reactions. The Effect of the Halogen on the Dipole Moments and the NMR Spectra and the Importance of Spin-Orbit Coupling for the 11B Chemical Shifts. Dalton Trans. 2006, 1024–1029. [Google Scholar]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidbaur, H.; Nowak, R.; Steigelmann, O.; Muller, G. π-Complexes of p-Block Elements: Synthesis and Structures of Adducts of Arsenic and Antimony Halides with Alkylated Benzenes. Chemische Berichte 1990, 123, 1221–1226. [Google Scholar] [CrossRef]
- Burford, N.; Clyburne, J.A.C.; Wiles, J.A.; Cameron, T.S.; Robertson, K.N. Tethered Diarenes as Four-Site Donors to SbCl3. Organometallics 1996, 15, 361–364. [Google Scholar] [CrossRef]
- Burford, N.; Clyburne, J.A.C.; Bakshi, P.K.; Cameron, T.S. η6-Arene Complexation to a Phosphenium Cation. J. Am. Chem. Soc. 1993, 115, 8829–8830. [Google Scholar] [CrossRef]
- McGrath, T.D.; Welch, A.J. Steric Effects in Heteroboranes. IV. 1-Ph-2-Br-1,2-closo-C2B10H10. Acta Cryst. 1995, C51, 649–651. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen Bonding versus Chalcogen and Pnicogen Bonding: A Combined Cambridge Structural Database and Theoretical Study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willet, R.D.; Peterson, K.A.; Twamley, B. The Nature of Halogen···Halogen Synthons: Crystalographic and Theoretical Studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The molecular diagrams and numbering of 3,6-Cl2-closo-1,2-P2B10H8 (1) and closo-1,7-P2B10Cl10 (2).
Figure 1.
The molecular diagrams and numbering of 3,6-Cl2-closo-1,2-P2B10H8 (1) and closo-1,7-P2B10Cl10 (2).

Figure 2.
The computed molecular electrostatic potential (ESP) surface and molecular diagrams for 3,6-Cl2-closo-1,2-P2B10H8 and closo-1,7-P2B10Cl10; the color range of the ESP in kcal mol−1.
Figure 2.
The computed molecular electrostatic potential (ESP) surface and molecular diagrams for 3,6-Cl2-closo-1,2-P2B10H8 and closo-1,7-P2B10Cl10; the color range of the ESP in kcal mol−1.

Figure 3.
The crystal packing of 3,6-Cl2-closo-1,2-P2B10H8 (1). Distances in Å. The color gray is used for distances larger than ΣrvdW. The positions of H atoms were optimized at the DFT-D3/BLYP/DZVP level.
Figure 3.
The crystal packing of 3,6-Cl2-closo-1,2-P2B10H8 (1). Distances in Å. The color gray is used for distances larger than ΣrvdW. The positions of H atoms were optimized at the DFT-D3/BLYP/DZVP level.

Figure 4.
The crystal packing and binding motifs of closo-1,7-P2B10Cl10•toluene. Distances in Å. The color gray is used for distances larger than ΣrvdW. The positions of H atoms were optimized at the DFT-D3/BLYP/DZVP level.
Figure 4.
The crystal packing and binding motifs of closo-1,7-P2B10Cl10•toluene. Distances in Å. The color gray is used for distances larger than ΣrvdW. The positions of H atoms were optimized at the DFT-D3/BLYP/DZVP level.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The magnitudes of the σ-holes (VS,max) of the P and Cl atoms.
| Compound | Atom | VS,max [kcal mol−1] |
|---|---|---|
| 3,6-Cl2-closo-1,2-P2B10H8 (1) | P(1,2) | 2 × 25.2; 25.1 |
| Cl(3,6) 1 | 2.3 | |
| closo-1,7-P2B10Cl10 (2) | P(1,7) | 30.2; 2 × 28.9; 2 × 27.8 |
| Cl(2,3) | 13.1 | |
| Cl(9,10) 2 | 1.5 |
1 The minimum value of the electrostatic potential (ESP) molecular surface of the Cl(3,6) atom is −9.5 kcal mol−1. 2 The minimum value of the ESP molecular surface of the Cl(9,10) atom is −13.2 kcal mol−1.
Table 2.
Computed DFT-D3/TPSS/TZVPP and MP2.5/CBS two-body interaction energies for the crystal of 3,6-Cl2-closo-1,2-P2B10H8 (1). Interaction energies were decomposed into electrostatic (Eelec), induction (Eind), dispersion (Edisp), and exchange (Eexch) contributions by SAPT0. Energies in kcal mol−1. The relative values in parentheses express the contribution to the sum of all attractive energy terms of SAPT0.
Table 2.
Computed DFT-D3/TPSS/TZVPP and MP2.5/CBS two-body interaction energies for the crystal of 3,6-Cl2-closo-1,2-P2B10H8 (1). Interaction energies were decomposed into electrostatic (Eelec), induction (Eind), dispersion (Edisp), and exchange (Eexch) contributions by SAPT0. Energies in kcal mol−1. The relative values in parentheses express the contribution to the sum of all attractive energy terms of SAPT0.
| Interaction | DFT-D3/MP2.5 | SAPT0/jun-cc-pVDZ | ||||
|---|---|---|---|---|---|---|
| Total | Eelec | Eind | Edisp | Eexch | ||
| A···B | −4.85/−4.23 | −4.83 | −2.28 (21%) | −0.81 (8%) | −7.67 (71%) | 5.94 |
| A···C | −4.09/−3.90 | −4.01 | −2.26 (27%) | −0.61 (7%) | −5.42 (65%) | 4.28 |
| A···D | −3.85/−3.35 | −3.70 | −2.11 (24%) | −0.65 (8%) | −5.87 (68%) | 4.92 |
Table 3.
Computed DFT-D3/TPSS/TZVPP and MP2.5/CBS two-body interaction energies for the crystal of closo-1,7-P2B10Cl10(2)•toluene. Interaction energies were decomposed into electrostatic (Eelec), induction (Eind), dispersion (Edisp), and exchange (Eexch) contributions by SAPT0. Energies in kcal mol−1. The relative values in parentheses express the contribution to the sum of all attractive energy terms of SAPT0.
Table 3.
Computed DFT-D3/TPSS/TZVPP and MP2.5/CBS two-body interaction energies for the crystal of closo-1,7-P2B10Cl10(2)•toluene. Interaction energies were decomposed into electrostatic (Eelec), induction (Eind), dispersion (Edisp), and exchange (Eexch) contributions by SAPT0. Energies in kcal mol−1. The relative values in parentheses express the contribution to the sum of all attractive energy terms of SAPT0.
| Interaction | DFT-D3/MP2.5 | SAPT0/jun-cc-pVDZ | ||||
|---|---|---|---|---|---|---|
| Total | Eelec | Eind | Edisp | Eexch | ||
| 2···toluene Pn-bond | −9.94/−10.55 | −11.56 | −9.80 (35%) | −3.96 (14%) | −14.61 (52%) | 16.81 |
| 2···toluene stacking | −2.51/−2.25 | −1.96 | −0.84 (22%) | −0.21 (6%) | −2.75 (72%) | 1.84 |
| 2···toluene H-bond | −0.87/−0.80 | −0.51 | −0.35 (16 %) | −0.21 (10%) | −1.58 (74%) | 1.63 |
| 2···2 bifurcated diX-bonds | −3.67 | −3.27 | −1.17 (16%) | −0.32 (4%) | −5.73 (79%) | 3.95 |
| 2···2 bifurcated diX-bond | −2.94 | −3.01 | −1.78 (26%) | −0.40 (6%) | −4.67 (68%) | 3.84 |
| 2···2 diX-bond | −2.32/−2.65 | −2.13 | −1.51 (25%) | −0.35 (6%) | −4.13 (69%) | 3.86 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fanfrlík, J.; Hnyk, D. Dihalogen and Pnictogen Bonding in Crystalline Icosahedral Phosphaboranes. Crystals 2018, 8, 390. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst8100390
AMA Style
Fanfrlík J, Hnyk D. Dihalogen and Pnictogen Bonding in Crystalline Icosahedral Phosphaboranes. Crystals. 2018; 8(10):390. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst8100390
Chicago/Turabian StyleFanfrlík, Jindřich, and Drahomír Hnyk. 2018. "Dihalogen and Pnictogen Bonding in Crystalline Icosahedral Phosphaboranes" Crystals 8, no. 10: 390. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst8100390
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.