Theoretical Study of the Electronic, Magnetic, Mechanical and Thermodynamic Properties of the Spin Gapless Semiconductor CoFeMnSi


Abstract
:1. Introduction
2. Computational Methodology
3. Results and Discussions
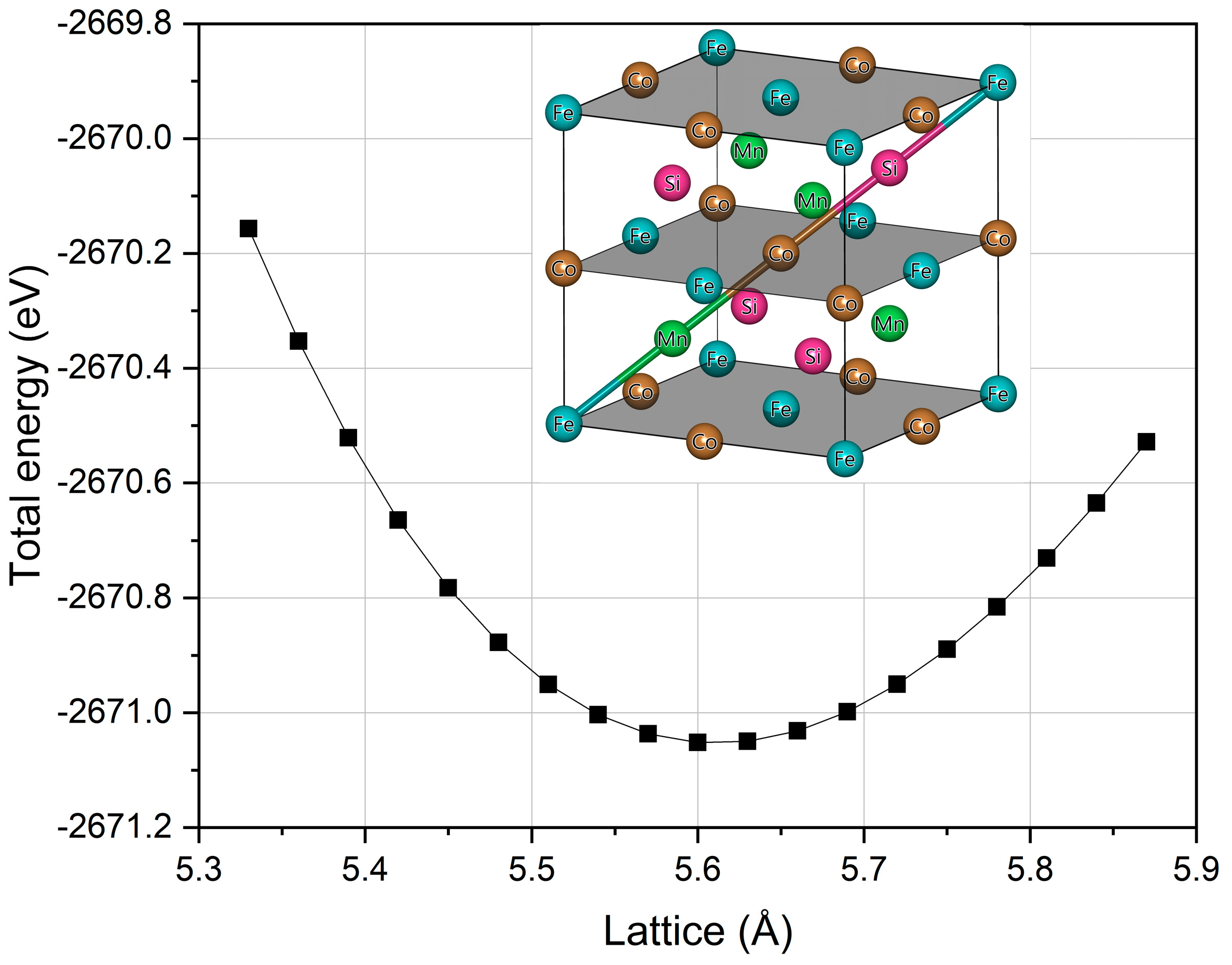
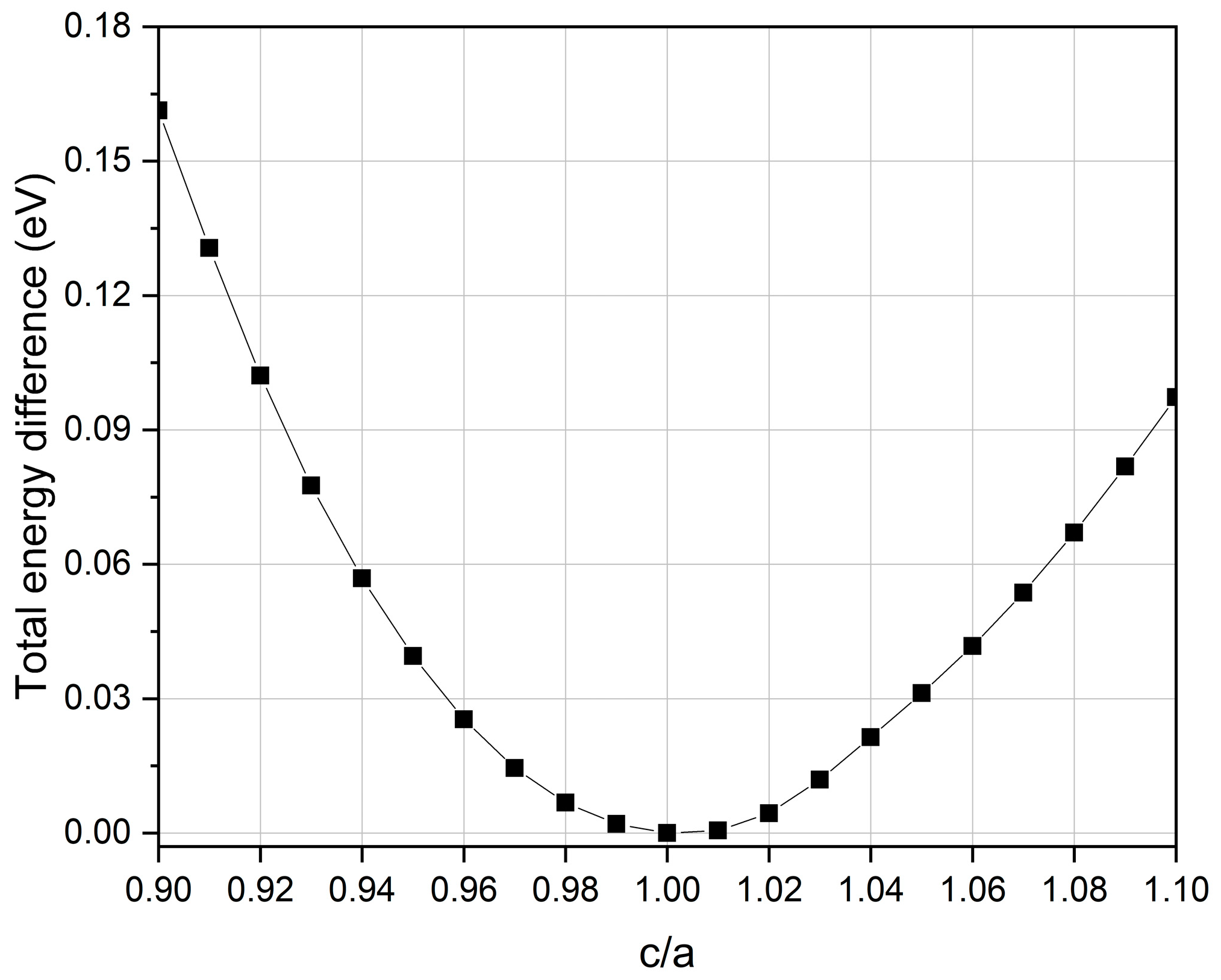
3.1. Crystal Structure and Equilibrium Lattice
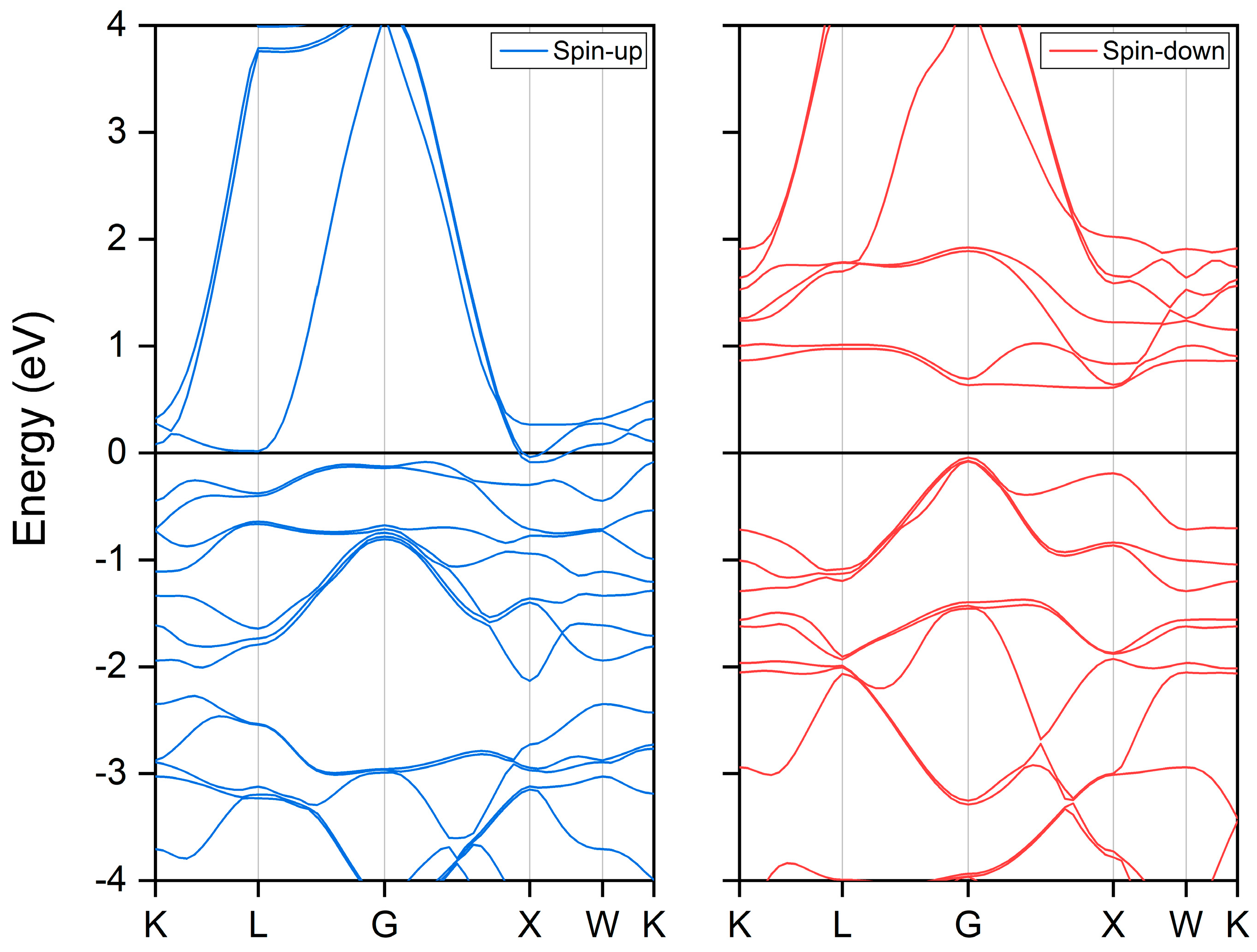
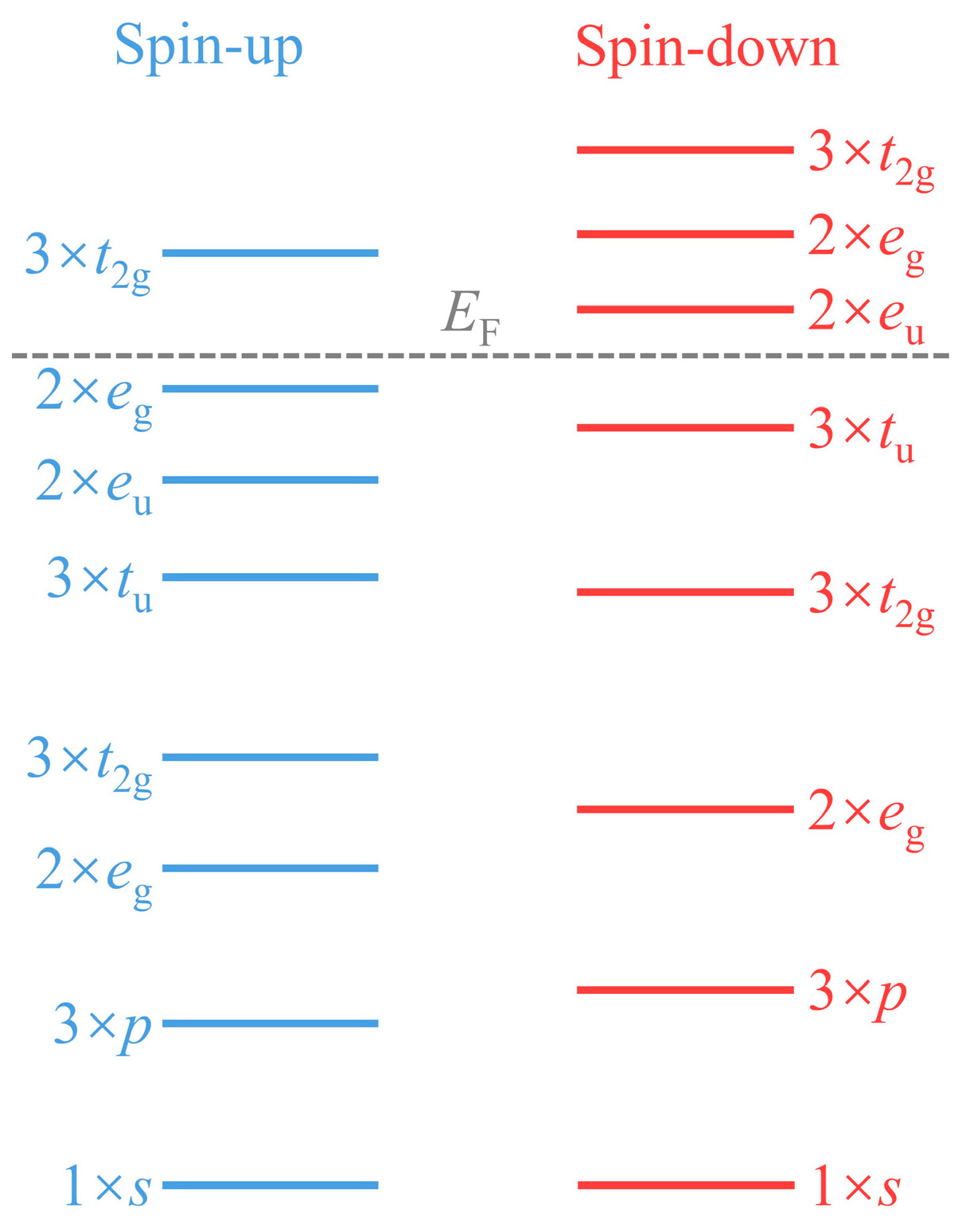
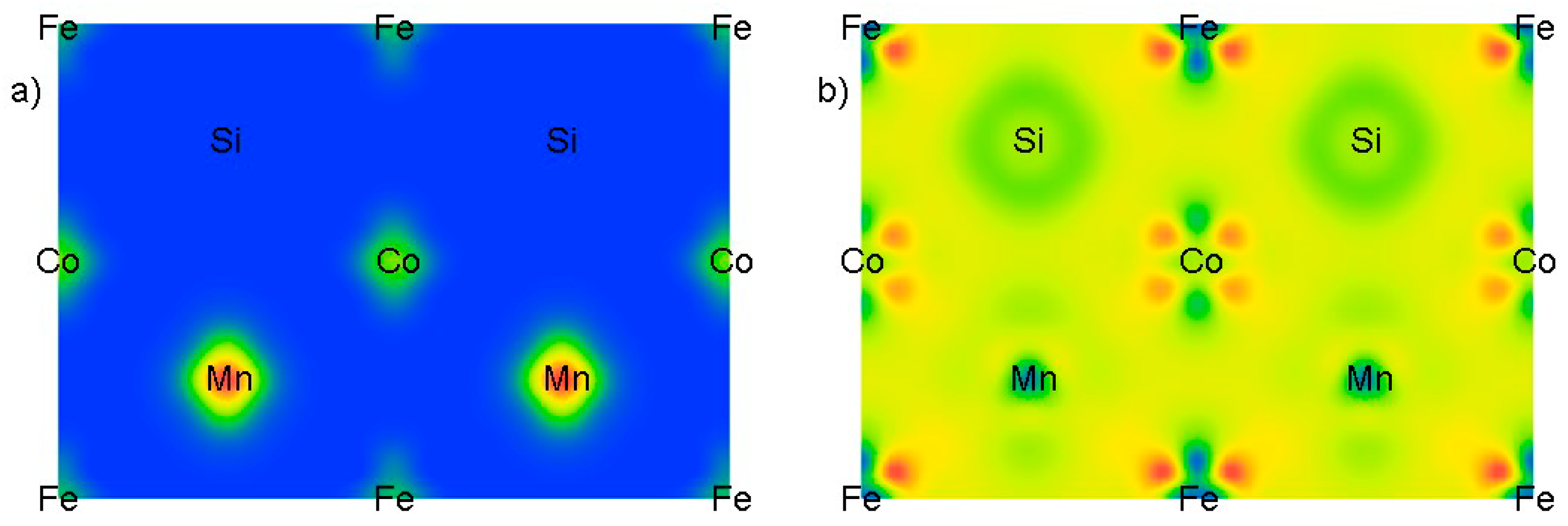
3.2. Electronic and Magnetic Properties
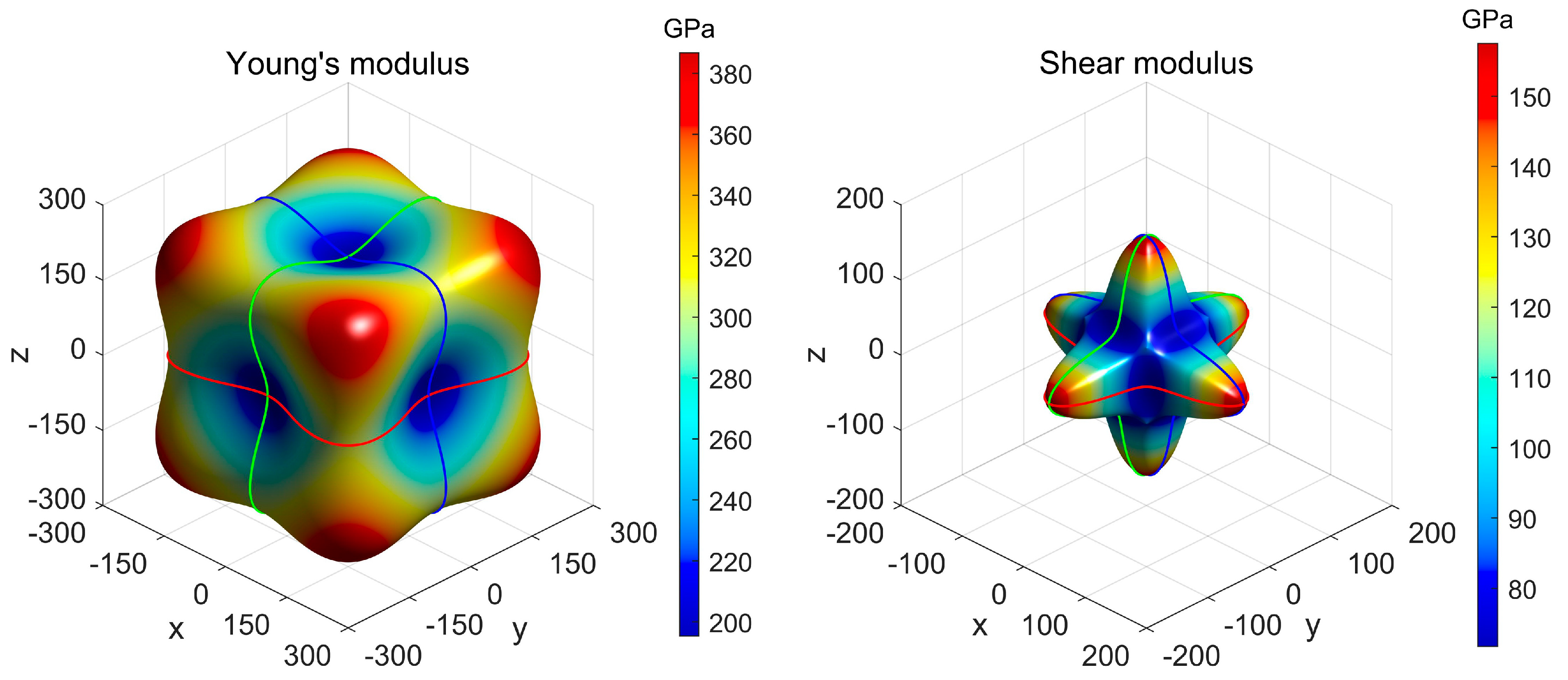
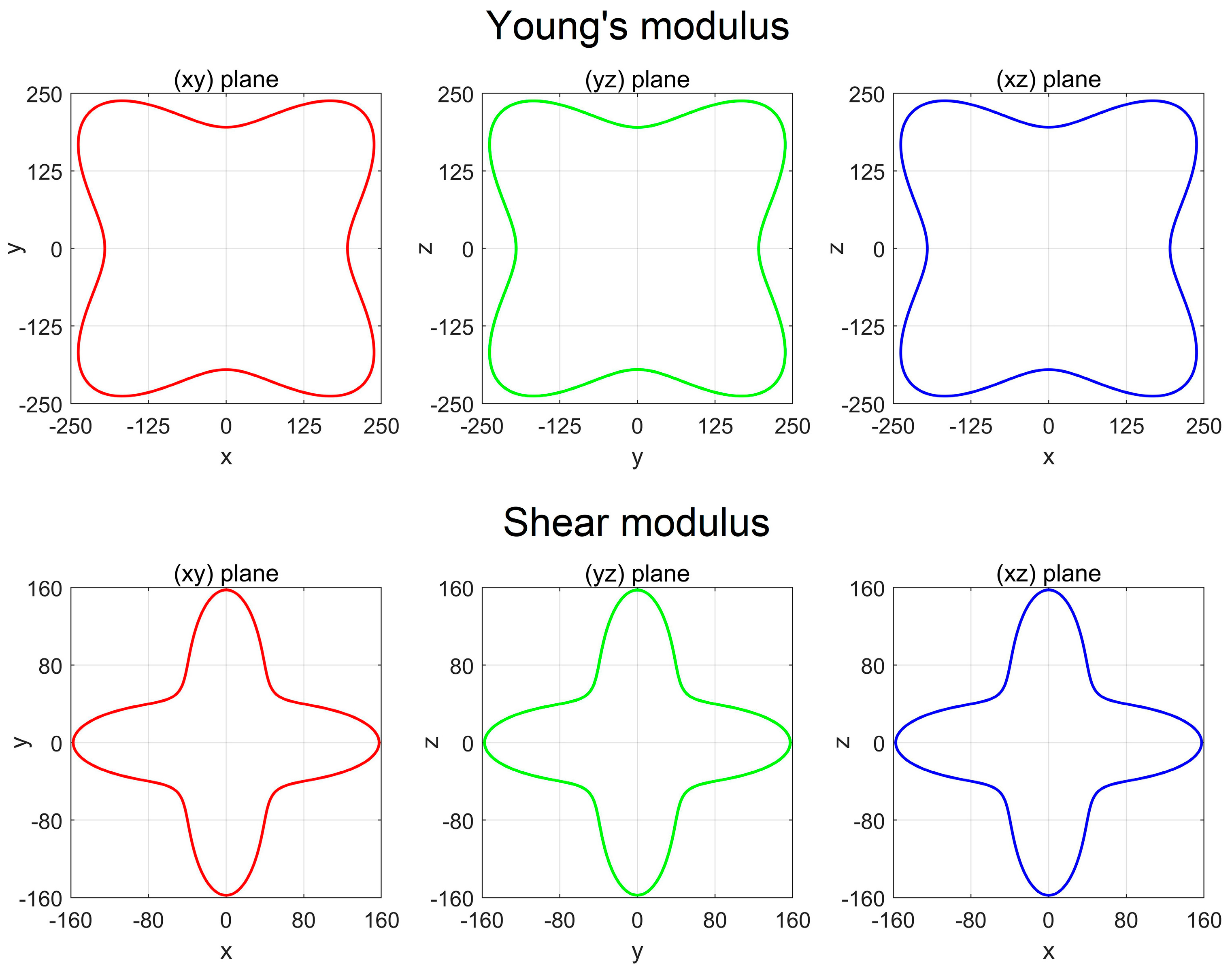
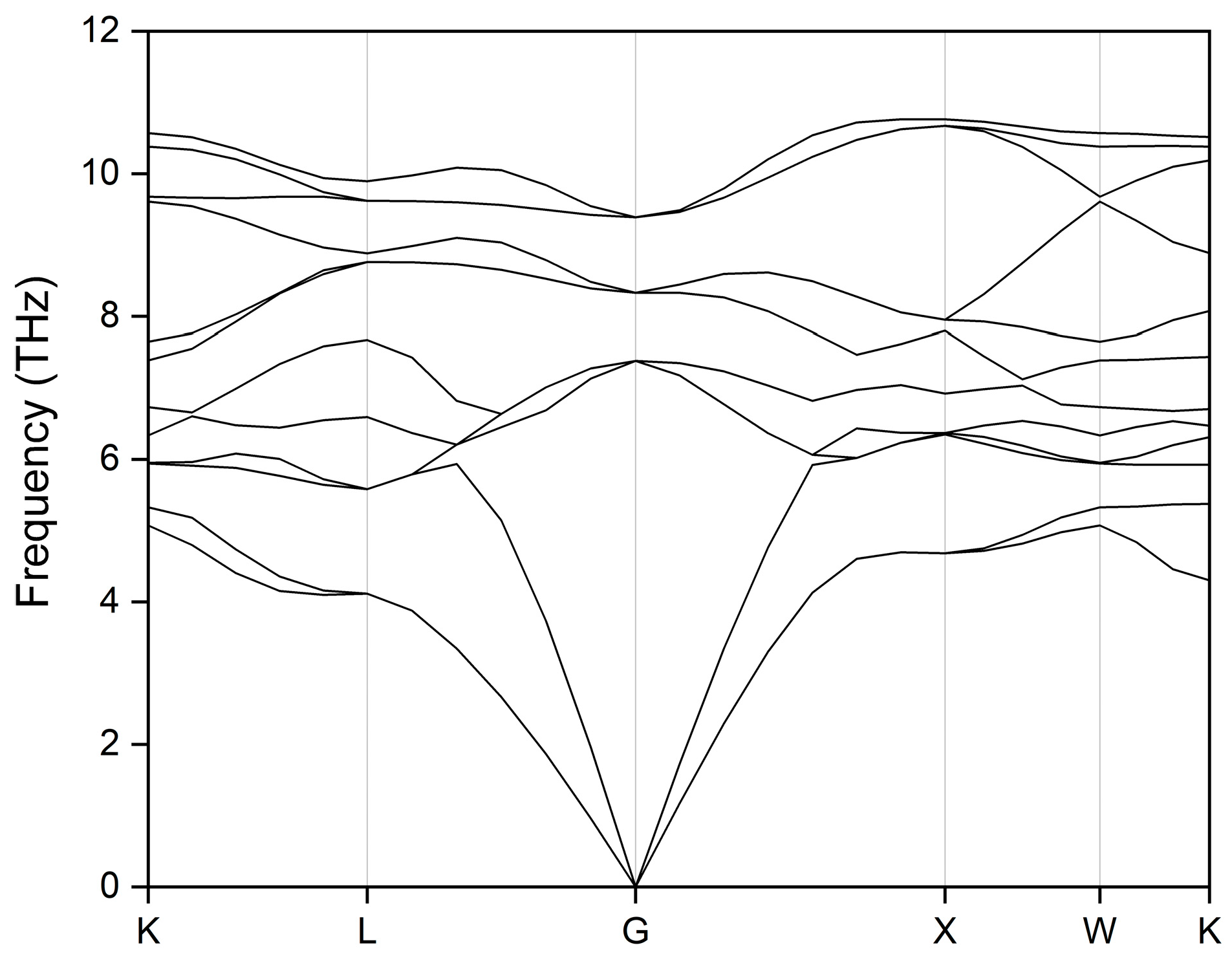
3.3. Mechanical Property and Dynamic Stability
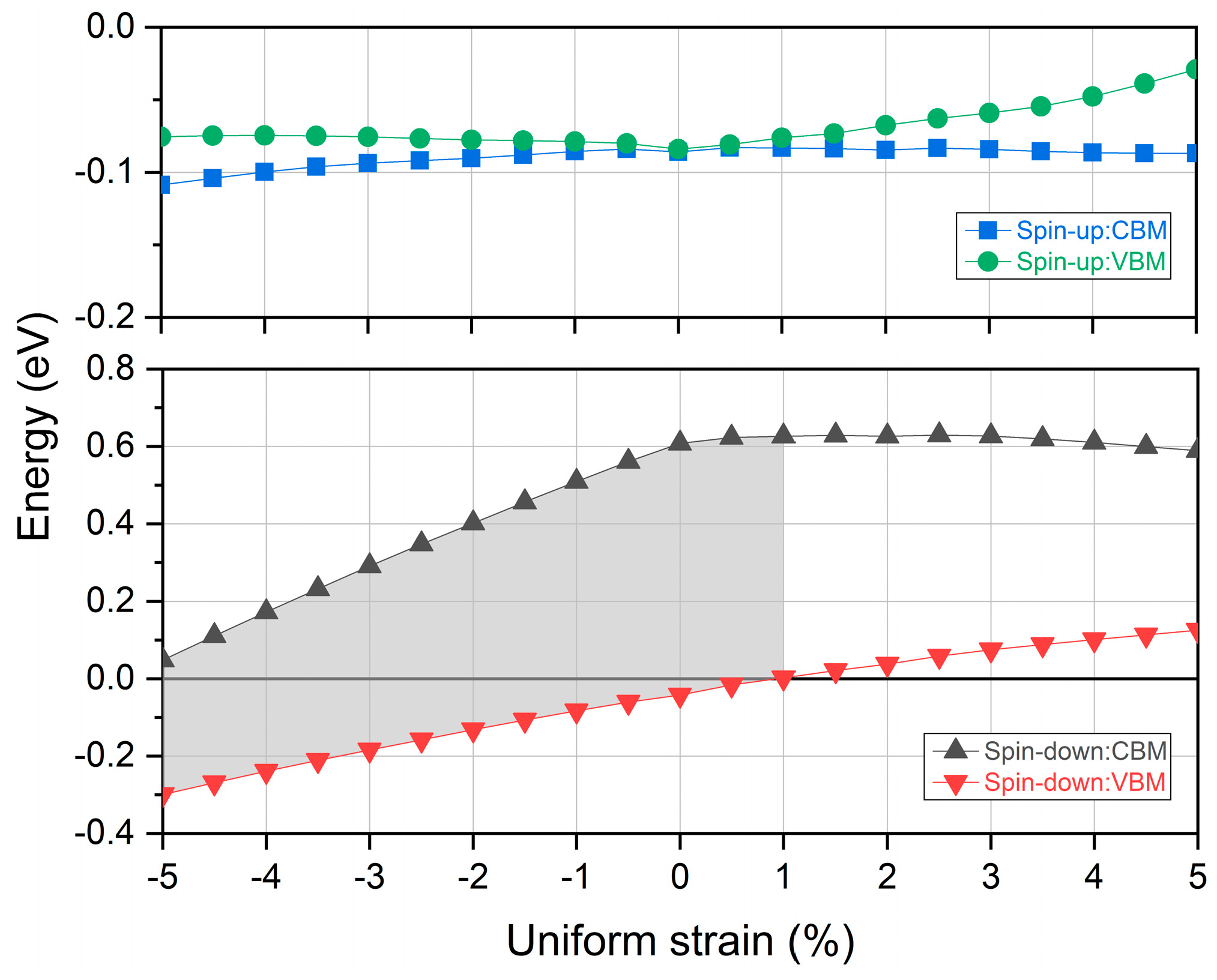
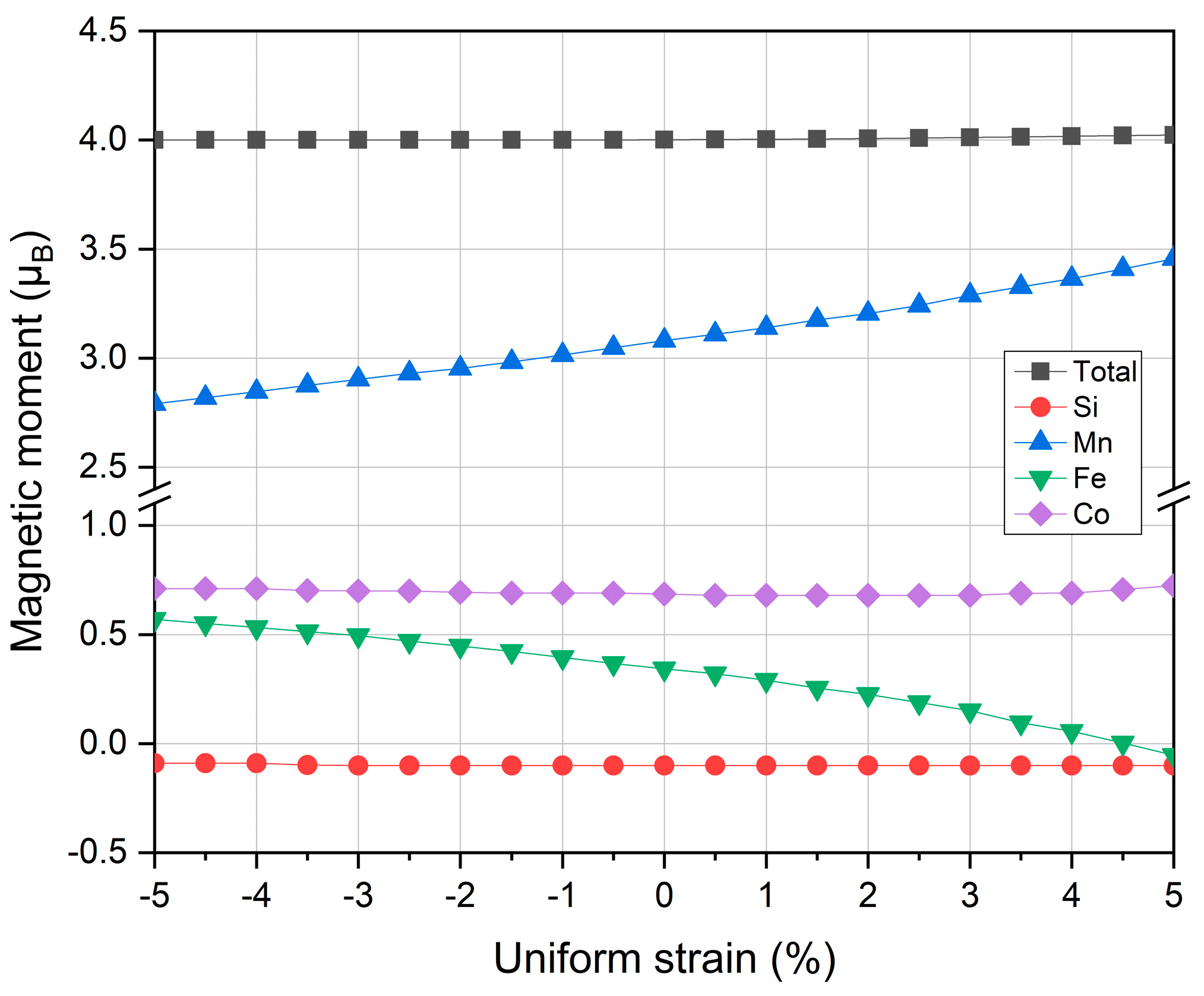
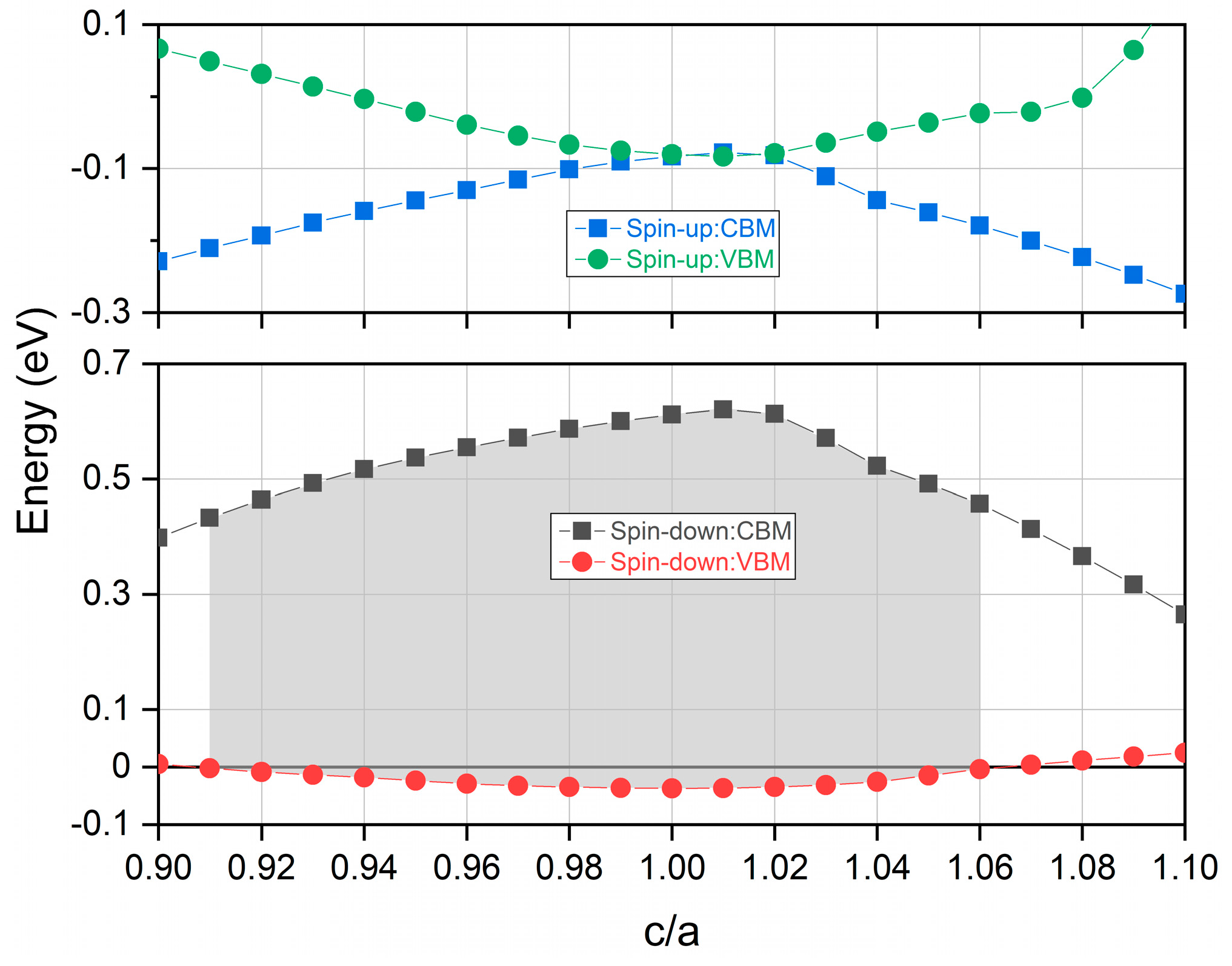
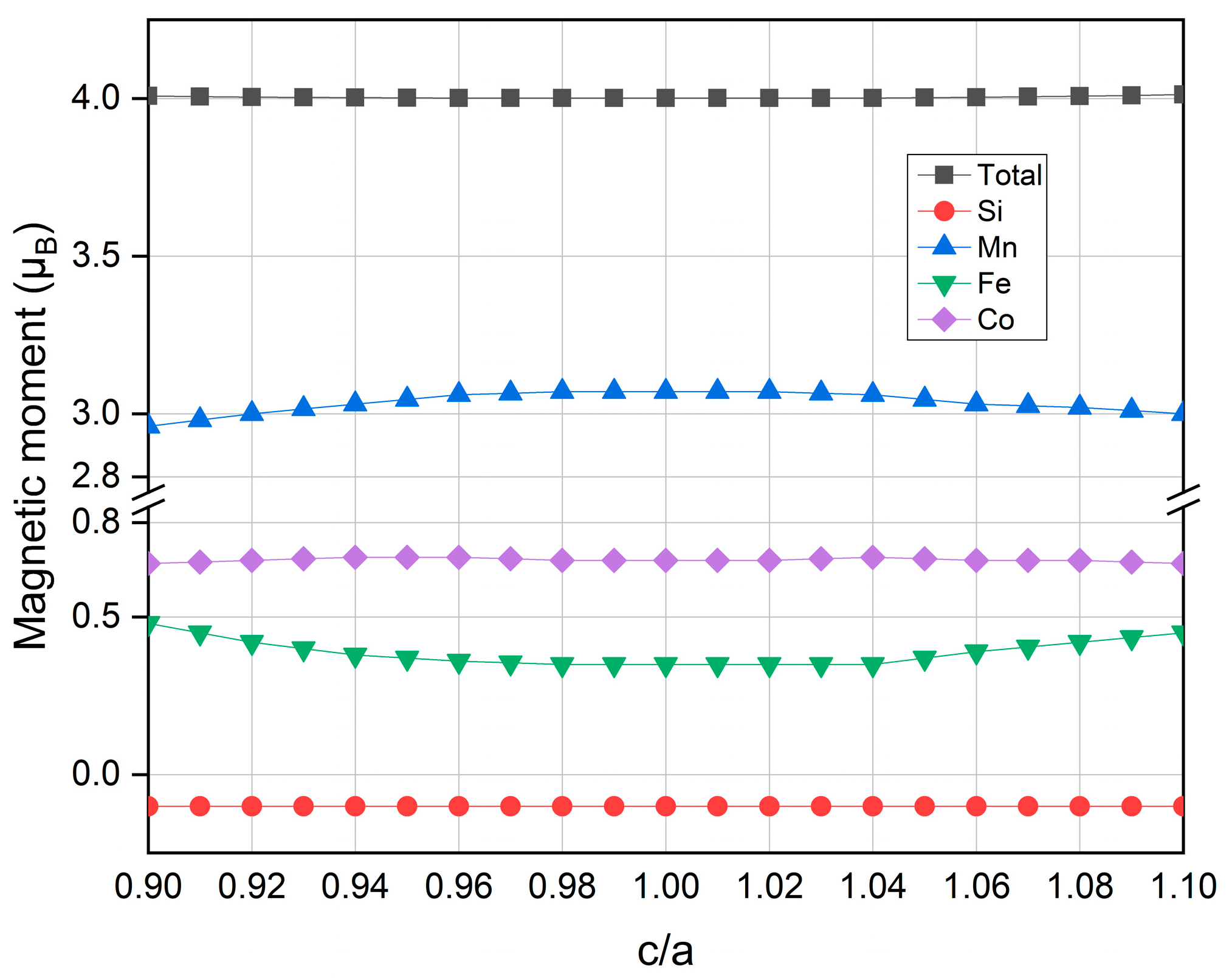
3.4. Strain Effects
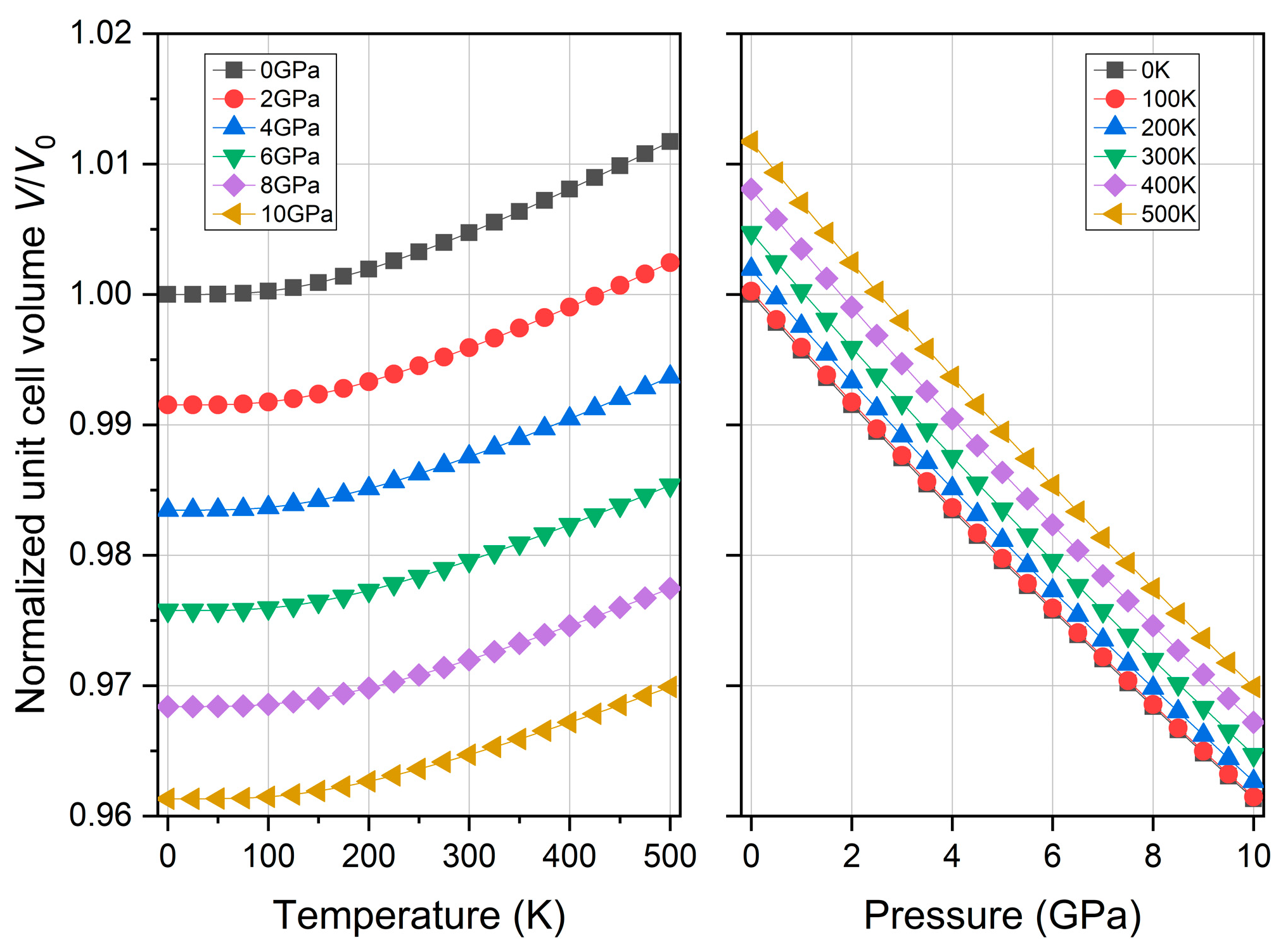
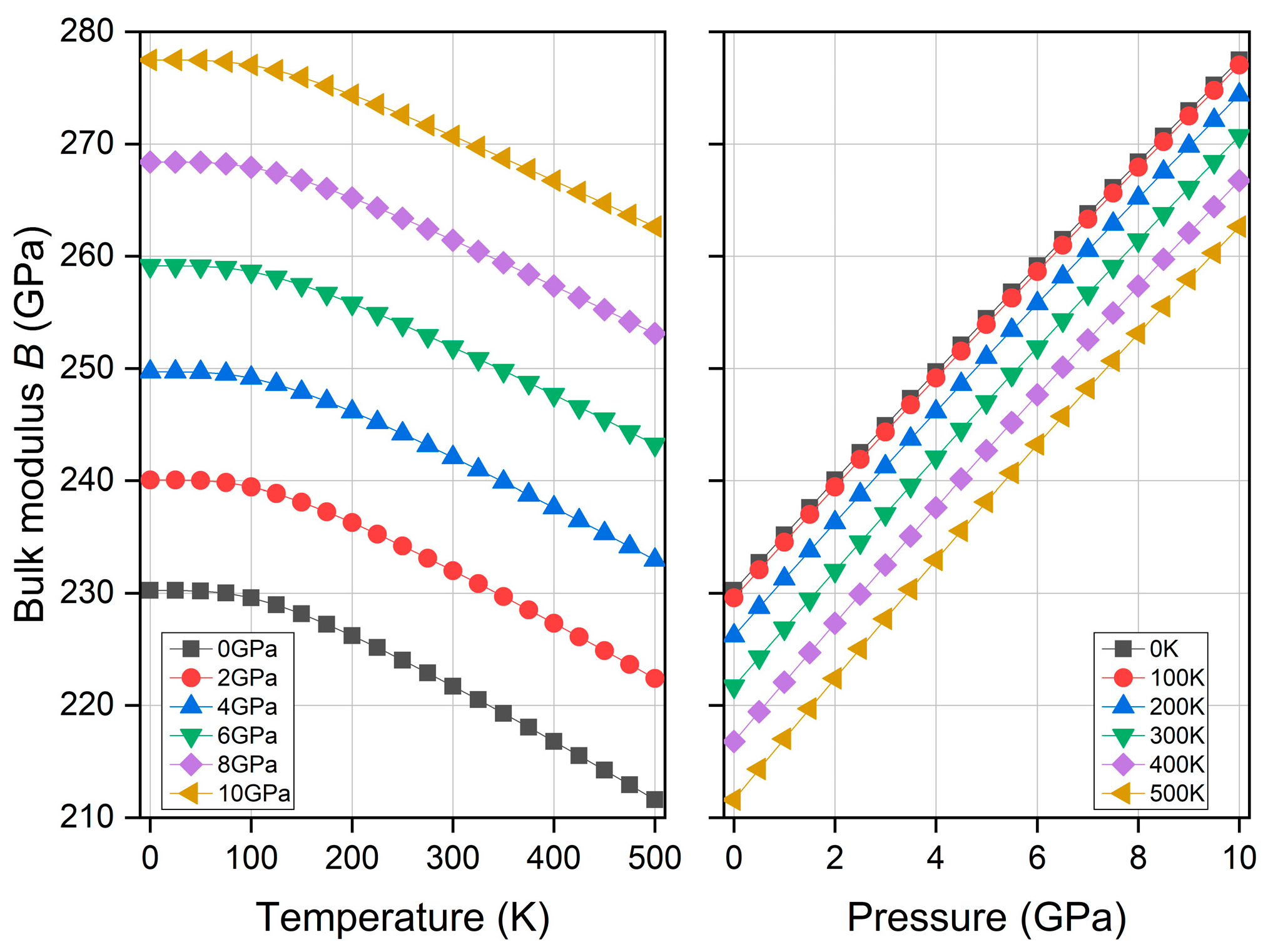
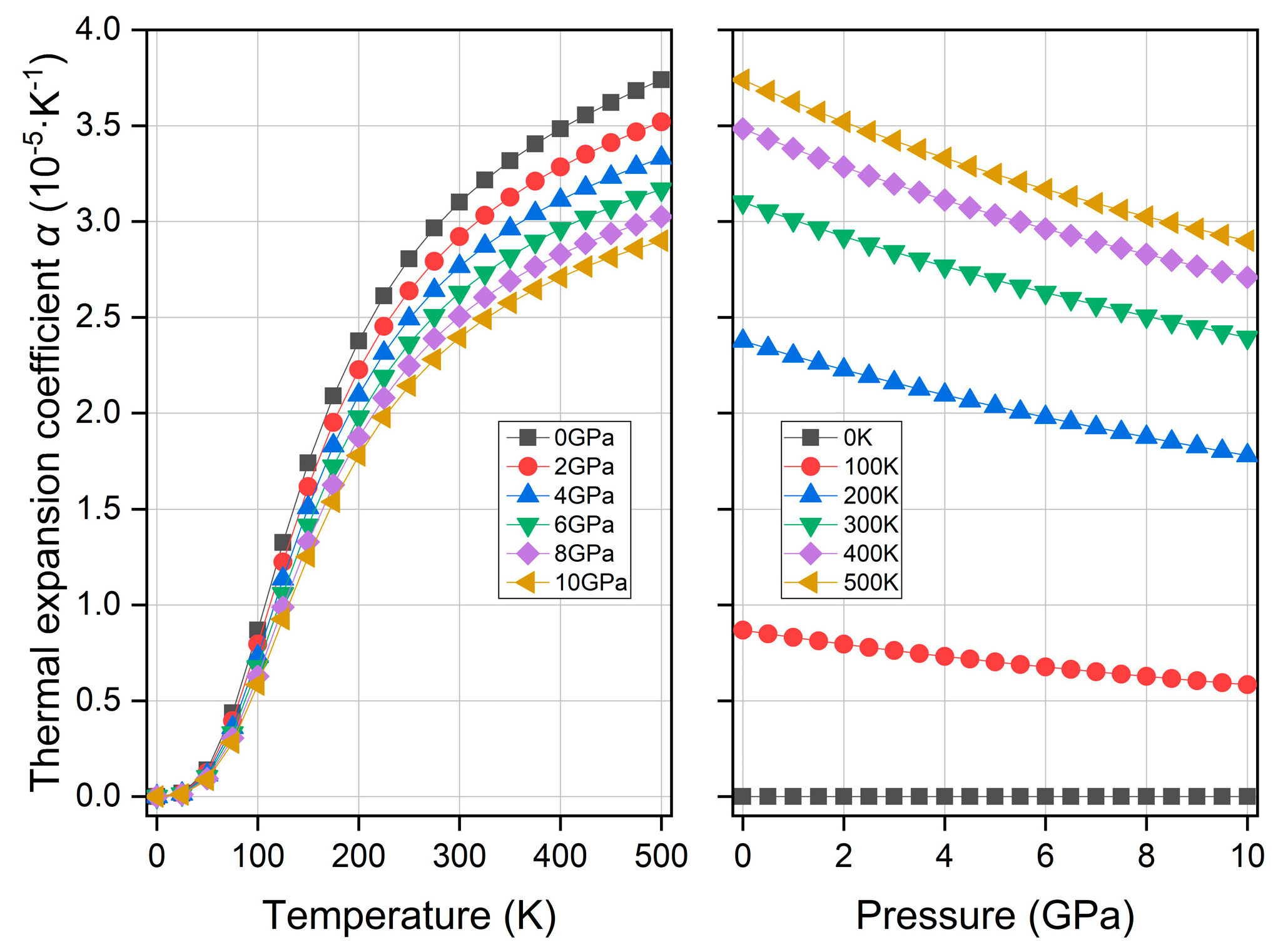
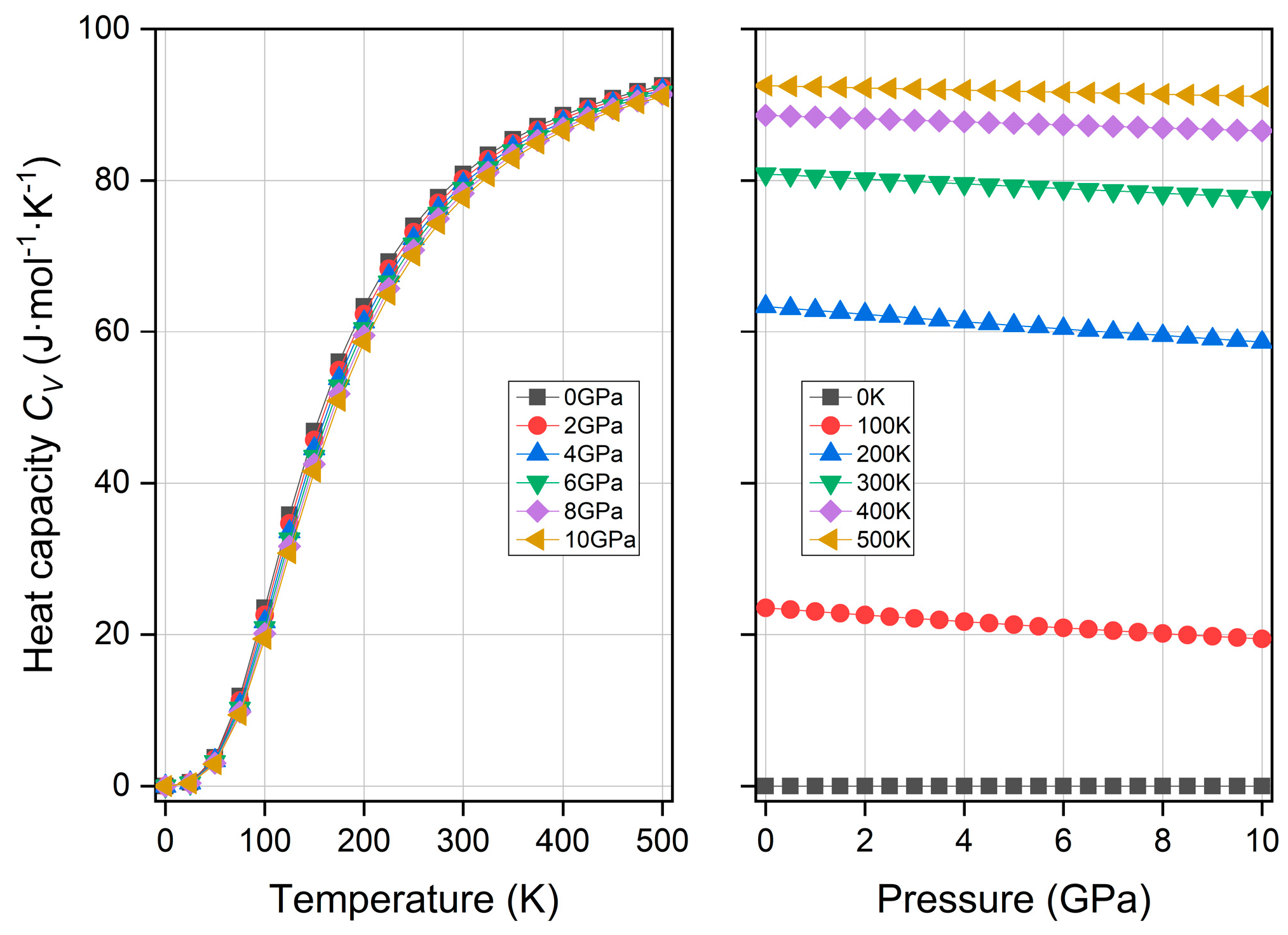
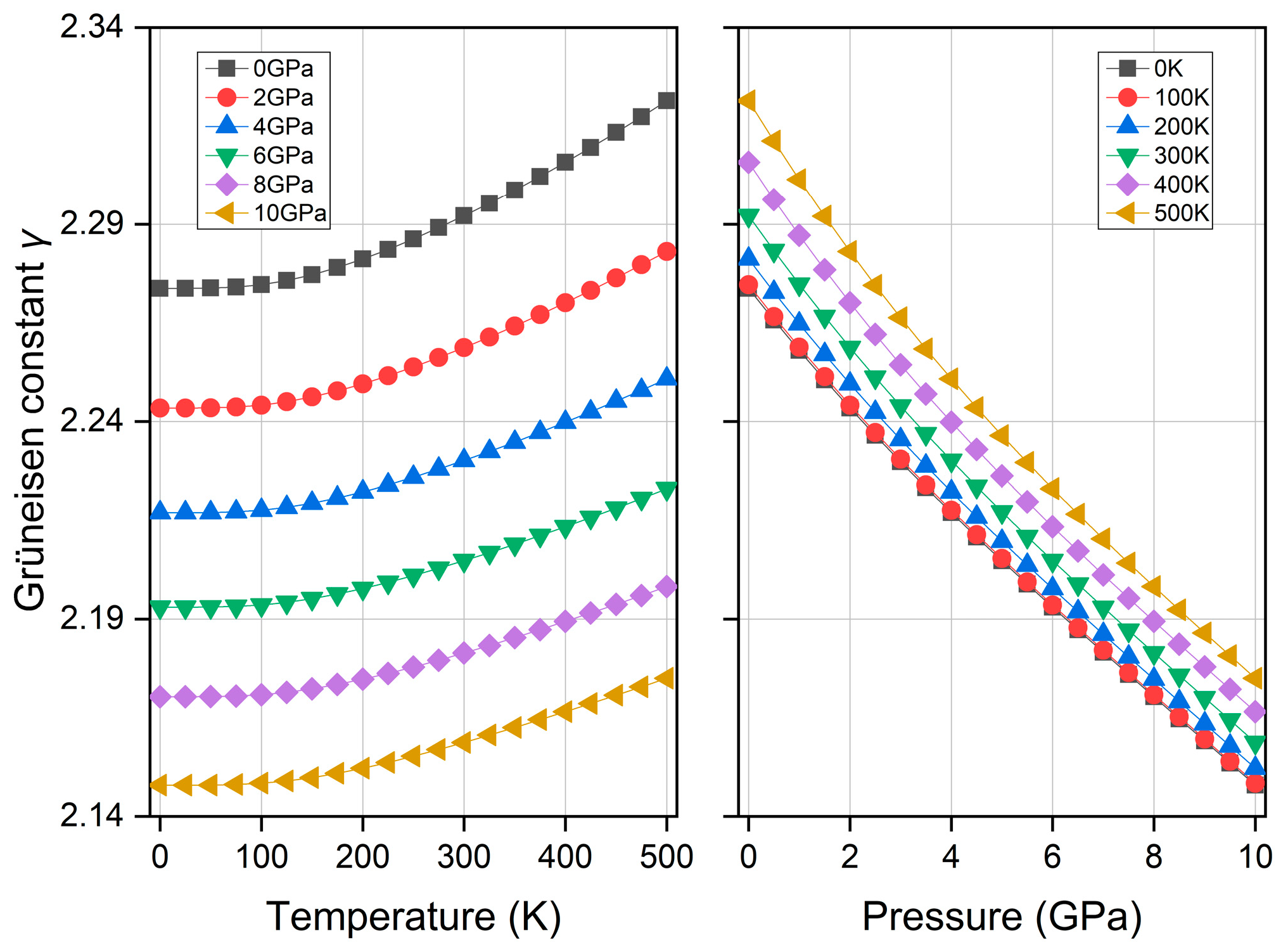
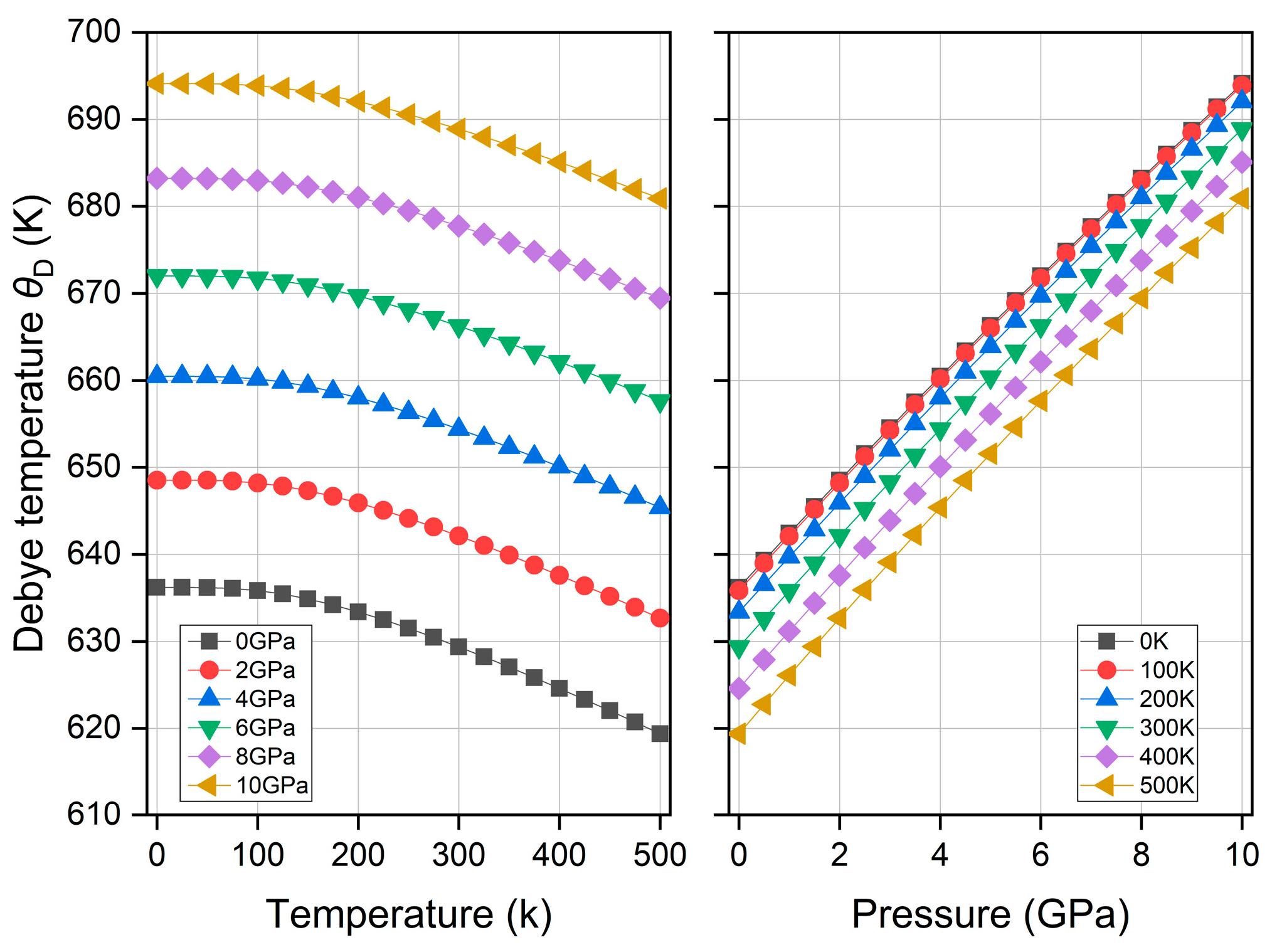
3.5. Thermodynamic Property
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.L. Proposal for a new class of materials: Spin gapless semiconductors. Phys. Rev. Lett. 2008, 100, 156404. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.W.; Fu, X.; Wan, C.H.; Yuan, Z.H.; Han, X.F.; Quang, N.V.; Cho, S. Spin gapless semiconductor like Ti2MnAl film as a new candidate for spintronics application. Physica. Status Solidi-Rapid Res. Lett. 2015, 9, 641–645. [Google Scholar] [CrossRef]
- Han, Y.L.; Khenata, R.; Li, T.Z.; Wang, L.Y.; Wang, X.T. Search for a new member of parabolic-like spin-gapless semiconductors: The case of diamond-like quaternary compound CuMn2InSe4. Results Phys. 2018, 10, 301–303. [Google Scholar] [CrossRef]
- Manna, K.; Sun, Y.; Muechler, L.; Kubler, J.; Felser, C. Heusler, Weyl and Berry. Nat. Rev. Mater. 2018, 3, 244–256. [Google Scholar] [CrossRef] [Green Version]
- Skaftouros, S.; Ozdogan, K.; Sasioglu, E.; Galanakis, I. Search for spin gapless semiconductors: The case of inverse Heusler compounds. Appl. Phys. Lett. 2013, 102, 022402. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jin, Y.J. A spin-gapless semiconductor of inverse Heusler Ti2CrSi alloy: First-principles prediction. J. Magn. Magn. Mater. 2015, 385, 55–59. [Google Scholar] [CrossRef]
- Wang, X.; Peleckis, G.; Zhang, C.; Kimura, H.; Dou, S.X. Colossal Electroresistance and Giant Magnetoresistance in Doped PbPdO2 Thin Films. Adv. Mater. 2009, 21, 2196. [Google Scholar] [CrossRef]
- Wang, X.T.; Cheng, Z.X.; Wang, J.L.; Wang, X.L.; Liu, G.D. Recent advances in the Heusler based spin-gapless semiconductors. J. Mater. Chem. C 2016, 4, 7176–7192. [Google Scholar] [CrossRef]
- Wang, X.T.; Li, T.Z.; Cheng, Z.X.; Wang, X.L.; Chen, H. Recent advances in Dirac spin-gapless semiconductors. Appl. Phys. Rev. 2018, 5, 041103. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.Z.; Liu, E.K.; Du, Y.; Li, G.J.; Liu, G.D.; Wang, W.H.; Wu, G.H. A new spin gapless semiconductors family: Quaternary Heusler compounds. Epl-Europhys Lett. 2013, 102, 17007. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Cao, J.; Khenata, R.; Cheng, Z.; Kuang, M.; Wang, X. Strain effect for the newly discovered spin-gapless diamond-like quaternary-type semiconductor CuMn2InSe4. J. Alloy. Compd. 2019, 793, 302–313. [Google Scholar] [CrossRef]
- Yang, T.; Hao, L.Y.; Khenata, R.; Wang, X.T. Strain Conditions for the Inverse Heusler Type Fully Compensated Spin-Gapless Semiconductor Ti2MnAl: A First-Principles Study. Materials 2018, 11, 2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; Liu, Z.H.; Liu, E.K.; Liu, G.D.; Ma, X.Q.; Wu, G.H. Towards fully compensated ferrimagnetic spin gapless semiconductors for spintronic applications. Epl-Europhys Lett. 2015, 111, 37009. [Google Scholar] [CrossRef]
- Ouardi, S.; Fecher, G.H.; Felser, C.; Kubler, J. Realization of spin gapless semiconductors: The Heusler compound Mn2CoAl. Phys. Rev. Lett. 2013, 110, 100401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainsla, L.; Mallick, A.I.; Raja, M.M.; Coelho, A.A.; Nigam, A.K.; Johnson, D.D.; Alam, A.; Suresh, K.G. Origin of spin gapless semiconductor behavior in CoFeCrGa: Theory and Experiment. Phys. Rev. B 2015, 92, 045201. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.Y.; Dai, X.F.; Wang, L.Y.; Liu, R.; Wang, X.T.; Li, P.P.; Cui, Y.T.; Liu, G.D. Ti2MnZ (Z=Al, Ga, In) compounds: Nearly spin gapless semiconductors. AIP Adv. 2014, 4, 047113. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, P.; Kharel, P.; Gilbert, S.; Staten, B.; Hurley, N.; Fuglsby, R.; Huh, Y.; Valloppilly, S.; Zhang, W.; Yang, K.; et al. Investigation of spin-gapless semiconductivity and half-metallicity in Ti2MnAl-based compounds. Appl. Phys. Lett. 2016, 108, 141901. [Google Scholar] [CrossRef] [Green Version]
- Birsan, A.; Kuncser, V. First principle investigations of the structural, electronic and magnetic properties of predicted new zirconium based full-Heusler compounds, Zr2MnZ (Z = Al, Ga and In). J. Magn. Magn. Mater. 2016, 406, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Yousuf, S.; Hien, N.D.; Batoo, K.M.; Raslan, E.H.; Gupta, D.C. Insight into structural, electronic and thermoelectric properties of Zr2MnX (X = Ga, In) Heuslers. Mater. Res. Express 2019, 6, 046530. [Google Scholar] [CrossRef]
- Patel, P.D.; Shinde, S.; Gupta, S.D.; Dabhi, S.D.; Jha, P.K. The first principle calculation of structural, electronic, magnetic, elastic, thermal and lattice dynamical properties of fully compensated ferrimagnetic spin-gapless heusler alloy Zr2MnGa. Comput. Condens. Matter 2018, 15, 61–68. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Y.; Yuan, H.; Liu, J.; Chen, H. Theoretical investigation on thermoelectric properties of spin gapless semiconductor Cr2ZnSi. Appl. Phys. A 2018, 124, 841. [Google Scholar] [CrossRef]
- Jakobsson, A.; Mavropoulos, P.; Sasioglu, E.; Blugel, S.; Lezaic, M.; Sanyal, B.; Galanakis, I. First-principles calculations of exchange interactions, spin waves, and temperature dependence of magnetization in inverse-Heusler-based spin gapless semiconductors. Phys. Rev. B 2015, 91, 174439. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wu, Y.; Li, T.; Khenata, R.; Yang, T.; Wang, X. Electronic, Magnetic, Half-Metallic, and Mechanical Properties of a New Equiatomic Quaternary Heusler Compound YRhTiGe: A First-Principles Study. Materials 2018, 11, 797. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.L.; Bouhemadou, A.; Khenata, R.; Cheng, Z.X.; Yang, T.; Wang, X.T. Prediction of possible martensitic transformations in all-d-metal Zinc-based Heusler alloys from first-principles. J. Magn. Magn. Mater. 2019, 471, 49–55. [Google Scholar] [CrossRef]
- Yang, T.; Cao, J.T.; Wang, X.T. Structural, Electronic, Magnetic, Mechanic and Thermodynamic Properties of the Inverse Heusler Alloy Ti2NiIn Under Pressure. Crystals 2018, 8, 429. [Google Scholar] [CrossRef] [Green Version]
- Bainsla, L.; Suresh, K.G. Equiatomic quaternary Heusler alloys: A material perspective for spintronic applications. Appl. Phys. Rev. 2016, 3, 031101. [Google Scholar] [CrossRef]
- Bainsla, L.; Mallick, A.I.; Coelho, A.A.; Nigam, A.K.; Varaprasad, B.S.D.C.S.; Takahashi, Y.K.; Alam, A.; Suresh, K.G.; Hono, K. High spin polarization and spin splitting in equiatomic quaternary CoFeCrAl Heusler alloy. J. Magn. Magn. Mater. 2015, 394, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Bainsla, L.; Mallick, A.I.; Raja, M.M.; Nigam, A.K.; Varaprasad, B.S.D.C.S.; Takahashi, Y.K.; Alam, A.; Suresh, K.G.; Hono, K. Spin gapless semiconducting behavior in equiatomic quaternary CoFeMnSi Heusler alloy. Phys. Rev. B 2015, 91, 104408. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.X.; Chen, Z.B.; Gao, Y.C. Phase stability, magnetic, electronic, half-metallic and mechanical properties of a new equiatomic quaternary Heusler compound ZrRhTiIn: A first-principles investigation. J. Phys. Chem. Solids 2018, 116, 72–78. [Google Scholar] [CrossRef]
- Wang, X.; Khachai, H.; Khenata, R.; Yuan, H.; Wang, L.; Wang, W.; Bouhemadou, A.; Hao, L.; Dai, X.; Guo, R.; et al. Structural, electronic, magnetic, half-metallic, mechanical, and thermodynamic properties of the quaternary Heusler compound FeCrRuSi: A first-principles study. Sci Rep. 2017, 7, 16183. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.F.; Liu, G.D.; Fecher, G.H.; Felser, C.; Li, Y.X.; Liu, H.Y. New quarternary half metallic material CoFeMnSi. J. Appl. Phys. 2009, 105, 07E901. [Google Scholar] [CrossRef]
- Alijani, V.; Ouardi, S.; Fecher, G.H.; Winterlik, J.; Naghavi, S.S.; Kozina, X.; Stryganyuk, G.; Felser, C.; Ikenaga, E.; Yamashita, Y.; et al. Electronic, structural, and magnetic properties of the half-metallic ferromagnetic quaternary Heusler compounds CoFeMnZ (Z = Al, Ga, Si, Ge). Phys. Rev. B 2011, 84, 224416. [Google Scholar] [CrossRef]
- Klaer, P.; Balke, B.; Alijani, V.; Winterlik, J.; Fecher, G.H.; Felser, C.; Elmers, H.J. Element-specific magnetic moments and spin-resolved density of states in CoFeMnZ (Z = Al, Ga; Si, Ge). Phys. Rev. B 2011, 84, 144413. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, H.; Yuan, H.; Zhou, Y.; Chen, X. The effect of disorder on electronic and magnetic properties of quaternary Heusler alloy CoFeMnSi with LiMgPbSb-type structure. J. Magn. Magn. Mater. 2015, 378, 7–15. [Google Scholar] [CrossRef]
- Fu, H.R.; Li, Y.L.; Ma, L.; You, C.Y.; Zhang, Q.; Tian, N. Structures, magnetism and transport properties of the potential spin-gapless semiconductor CoFeMnSi alloy. J. Magn. Magn. Mater. 2019, 473, 16–20. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys.-Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Blanco, M.A.; Francisco, E.; Luana, V. GIBBS: Isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model. Comput. Phys. Commun. 2004, 158, 57–72. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Abbasi-Perez, D.; Luana, V. GIBBS2: A new version of the quasiharmonic model code. II. Models for solid-state thermodynamics, features and implementation. Comput. Phys. Commun. 2011, 182, 2232–2248. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Luana, V. GIBBs2: A new version of the quasi-harmonic model code. I. Robust treatment of the static data. Comput. Phys. Commun. 2011, 182, 1708–1720. [Google Scholar] [CrossRef]
- Faleev, S.V.; Ferrante, Y.; Jeong, J.; Samant, M.G.; Jones, B.; Parkin, S.S.P. Unified explanation of chemical ordering, the Slater-Pauling rule, and half-metallicity in full Heusler compounds. Phys. Rev. B 2017, 95, 045140. [Google Scholar] [CrossRef] [Green Version]
- Galanakis, I.; Dederichs, P.H.; Papanikolaou, N. Slater-Pauling behavior and origin of the half-metallicity of the full-Heusler alloys. Phys. Rev. B 2002, 66, 174429. [Google Scholar] [CrossRef] [Green Version]
- Akriche, A.; Bouafia, H.; Hiadsi, S.; Abidri, B.; Sahli, B.; Elchikh, M.; Timaoui, M.A.; Djebour, B. First-principles study of mechanical, exchange interactions and the robustness in Co2MnSi full Heusler compounds. J. Magn. Magn. Mater. 2017, 422, 13–19. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, Z.; Liu, G.; Dai, X.; Khenata, R.; Wang, L.; Bouhemadou, A. Rare earth-based quaternary Heusler compounds MCoVZ (M = Lu, Y; Z = Si, Ge) with tunable band characteristics for potential spintronic applications. IUCrJ 2017, 4, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhao, W.; Cheng, Z.; Dai, X.; Khenata, R. Electronic, magnetic, half-metallic and mechanical properties of a new quaternary Heusler compound ZrRhTiTl: Insights from first-principles studies. Solid State Commun. 2018, 269, 125–130. [Google Scholar] [CrossRef]
- Yip, S.; Li, J.; Tang, M.J.; Wang, J.G. Mechanistic aspects and atomic-level consequences of elastic instabilities in homogeneous crystals. Mater. Sci. Eng. A 2001, 317, 236–240. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Yip, S.; Phillpot, S.; Wolf, D. Mechanical instabilities of homogeneous crystals. Phys. Rev. B Condens. Matter 1995, 52, 12627–12635. [Google Scholar] [CrossRef]
- Sin’ko, G.V.; Smirnov, N.A. Ab initio calculations of elastic constants and thermodynamic properties of bcc, fcc, and hcp Al crystals under pressure. J. Phys.-Condens. Matter 2002, 14, 6989–7005. [Google Scholar]
- Wu, M.; Han, Y.; Bouhemadou, A.; Cheng, Z.; Khenata, R.; Kuang, M.; Wang, X.; Yang, T.; Yuan, H.; Wang, X. Site preference and tetragonal distortion in palladium-rich Heusler alloys. IUCrJ 2019, 6, 218–225. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lattice [Å] | Magnetic Moment [μB] | |||||
|---|---|---|---|---|---|---|---|
| Total | Fe | Mn | Co | Si | |||
| CoFeMnSi | Current | 5.611 | 4.00 | 0.34 | 3.08 | 0.69 | −0.11 |
| Reference [31] | 5.653 * | 3.99* | 0.576 | 2.649 | 0.878 | −0.07 | |
| Reference [28] | 5.658 * | 4.01 | 0.53 | 2.72 | 0.82 | ||
| Reference [45] | 5.67 * | 3.49 * | |||||
| Reference [32] | 5.611 | 4.00 | 0.52 | 2.70 | 0.89 | −0.11 | |
| Compound | C11 [GPa] | C12 [GPa] | C44 [GPa] | B [GPa] | G [GPa] | E [GPa] | υ | B/G | η | |
|---|---|---|---|---|---|---|---|---|---|---|
| CoFeMnSi | Current | 332.2 | 188.9 | 157.5 | 236.7 | 114.8 | 296.5 | 0.29 | 2.06 | 2.19 |
| Reference [32] | 317.0 | 189.0 | 167.0 | 231.0 | 114.0 | 293.0 | 0.29 | 2.04 | 2.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, X.; You, J.; Liu, P.-F.; Wang, Y. Theoretical Study of the Electronic, Magnetic, Mechanical and Thermodynamic Properties of the Spin Gapless Semiconductor CoFeMnSi. Crystals 2019, 9, 678. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9120678
Tan X, You J, Liu P-F, Wang Y. Theoretical Study of the Electronic, Magnetic, Mechanical and Thermodynamic Properties of the Spin Gapless Semiconductor CoFeMnSi. Crystals. 2019; 9(12):678. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9120678
Chicago/Turabian StyleTan, Xingwen, Jiaxue You, Peng-Fei Liu, and Yanfeng Wang. 2019. "Theoretical Study of the Electronic, Magnetic, Mechanical and Thermodynamic Properties of the Spin Gapless Semiconductor CoFeMnSi" Crystals 9, no. 12: 678. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9120678