Transcriptomic Landscape of Lower Grade Glioma Based on Age-Related Non-Silent Somatic Mutations

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of Age-Related Non-Silent Mutations in Lower Grade Glioma (LGG) and Glioblastoma (GBM)
2.2. Histological Tumour Grade Depends on Age at Diagnosis in the IDH1 Wild-Type Group but Not in the Mutant Group
2.3. Transcriptomic Landscape of Tumours of Different Histological Grades Harbouring IDH1 Mutations
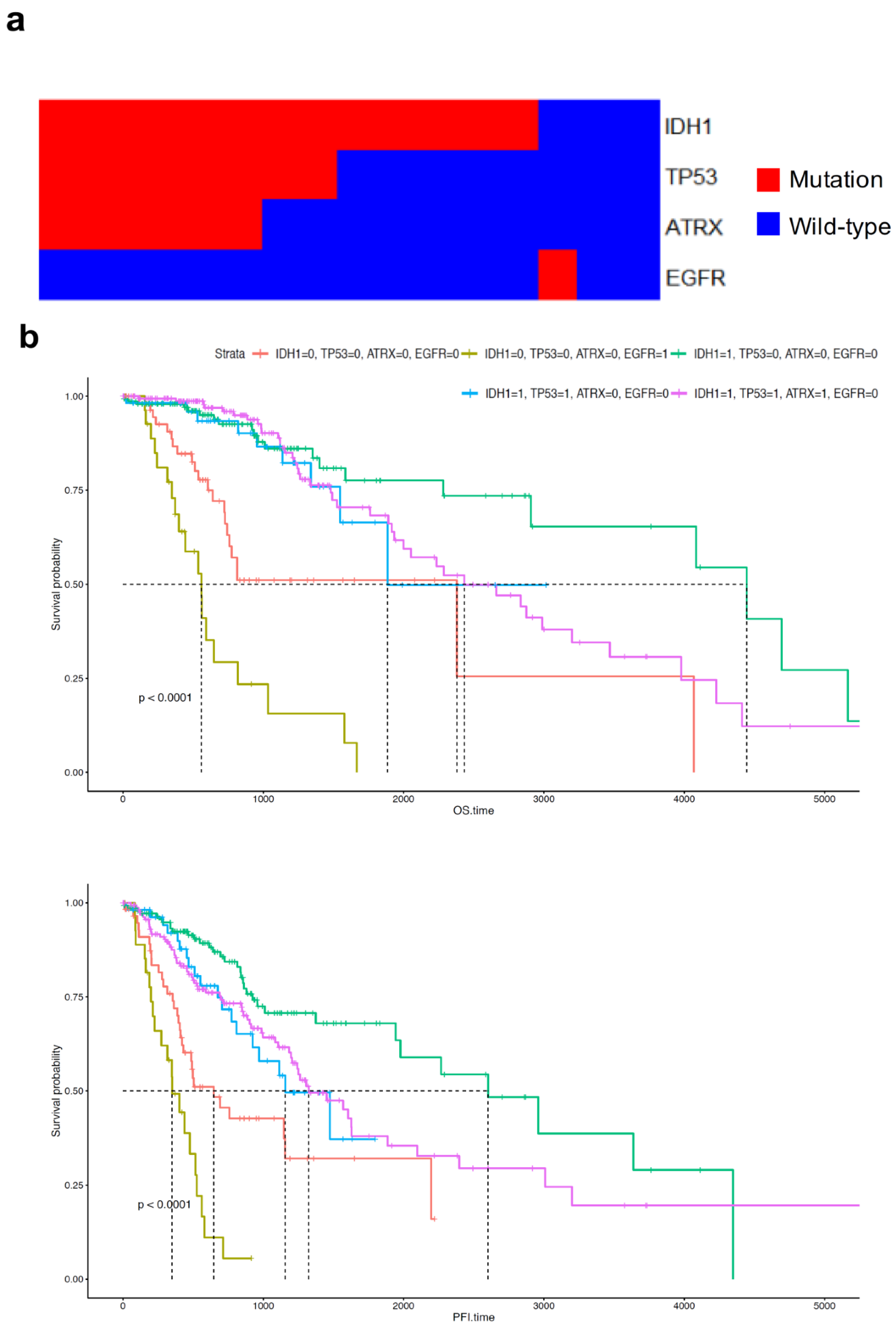
2.4. Distribution and Prognosis of Age-Dependent Non-Silent Somatic Mutations in LGG
2.5. ATRX and TP53 Mutation Occurred in Patients with Early Onset Age of G2 and G3 Glioma and IDH1 Mutation, and the Gene Expression Patterns of Mutant ATRX and TP53 in LGG Differed from Those of Their Wild-Type Counterpart
2.6. Telomerase Reverse Transcriptase (TERT) Expression Was Suppressed in LGG and GBM with ATRX Non-Silent Mutations
2.7. EGFR Mutation Occurred at Late Onset Age in Patients with G3 Tumour and Was Associated with Poor Prognosis of GBM Patients but Not Was Not Clustered in Transcriptomic Analysis
3. Discussion
4. Materials and Methods
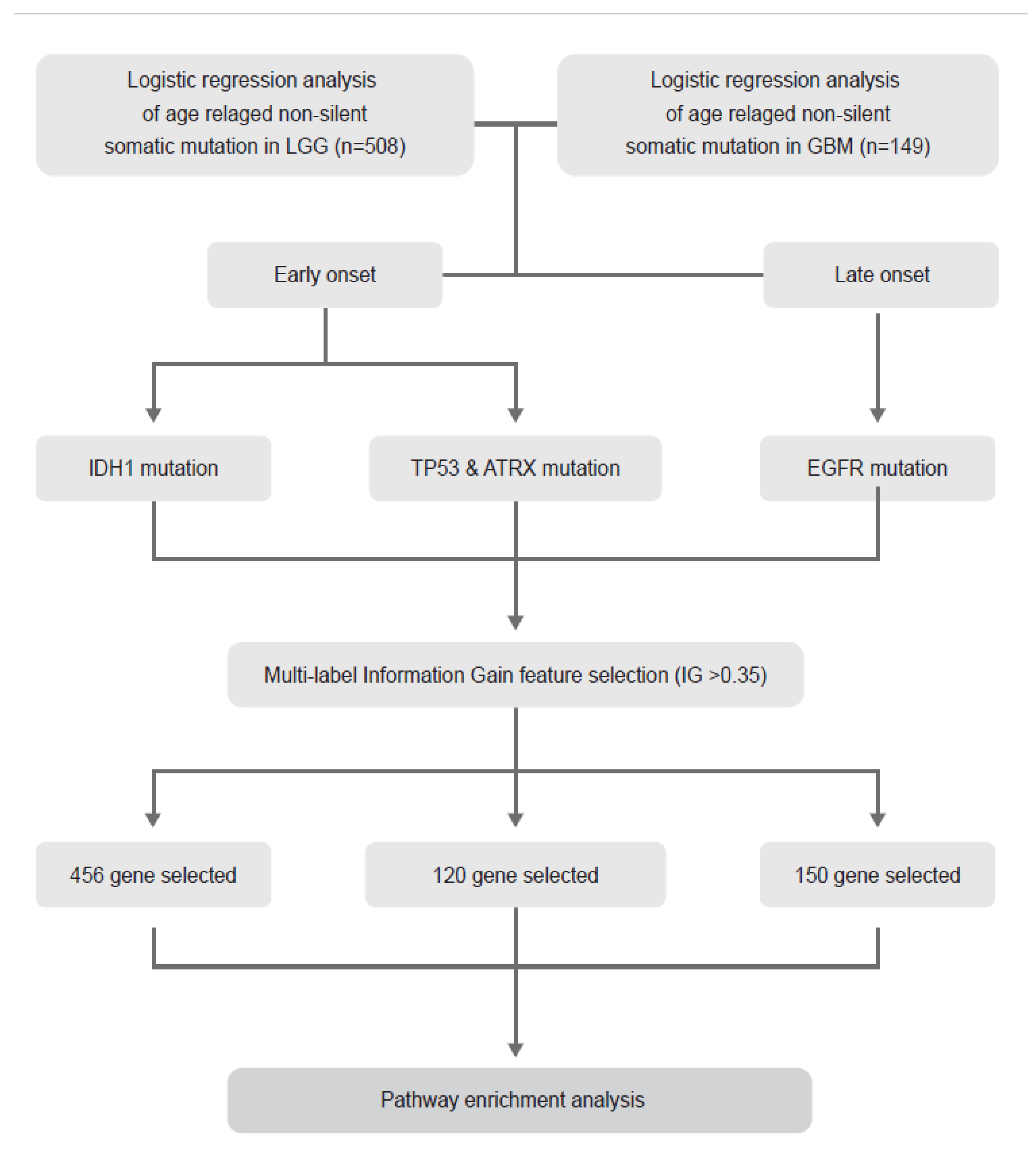
4.1. Description of TCGA Data and Entire Analysis Process
4.2. Extraction of Candidate Somatic Mutations Related to Age
4.3. Comparing Age with Mutation Status and Grade
4.4. Survival Analysis
4.5. Multi-Label IG Based Feature Selection
4.6. Hierarchical Clustering and Heatmap with Annotations of Information
4.7. Pathway Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.; Butowski, N.; Chang, S. Recent advances in therapy for glioblastoma. Arch. Neurol. 2010, 67, 279–283. [Google Scholar] [CrossRef] [Green Version]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [Green Version]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Jean-Quartier, C.; Jeanquartier, F.; Ridvan, A.; Kargl, M.; Mirza, T.; Stangl, T.; Markaĉ, R.; Jurada, M.; Holzinger, A. Mutation-based clustering and classification analysis reveals distinctive age groups and age-related biomarkers for glioma. BMC Med Inform. Decis. Mak. 2021, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grist, S.; McCarron, M.; Kutlaca, A.; Turner, D.; Morley, A. In vivo human somatic mutation: Frequency and spectrum with age. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 1992, 266, 189–196. [Google Scholar] [CrossRef]
- Modrek, A.S.; Golub, D.; Khan, T.; Bready, D.; Prado, J.; Bowman, C.; Deng, J.; Zhang, G.; Rocha, P.P.; Raviram, R. Low-grade astrocytoma mutations in IDH1, P53, and ATRX cooperate to block differentiation of human neural stem cells via repression of SOX2. Cell Rep. 2017, 21, 1267–1280. [Google Scholar] [CrossRef]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas-Boluda, A.; Vaquero, J.; Barrin, S.; Kantari-Mimoun, C.; Ponzo, M.; Renault, G.; Deptula, P.; Pogoda, K.; Bucki, R.; Cascone, I.; et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. BioRxiv 2020. [Google Scholar] [CrossRef]
- Kwaan, H.C.; Lindholm, P.F. Fibrin and fibrinolysis in cancer. Semin. Thromb. Hemost. 2019, 45, 413–422. [Google Scholar] [CrossRef]
- Melrose, J. Keratan sulfate (KS)-proteoglycans and neuronal regulation in health and disease: The importance of KS-glycodynamics and interactive capability with neuroregulatory ligands. J. Neurochem. 2019, 149, 170–194. [Google Scholar] [CrossRef] [Green Version]
- O’regan, L.; Blot, J.; Fry, A.M. Mitotic regulation by NIMA-related kinases. Cell Div. 2007, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Osmani, S.A.; Pu, R.T.; Morris, N.R. Mitotic induction and maintenance by overexpression of a G2-specific gene that encodes a potential protein kinase. Cell 1988, 53, 237–244. [Google Scholar] [CrossRef]
- Machida, Y.J.; Dutta, A. The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev. 2007, 21, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Qiao, R.; Weissmann, F.; Yamaguchi, M.; Brown, N.G.; VanderLinden, R.; Imre, R.; Jarvis, M.A.; Brunner, M.R.; Davidson, I.F.; Litos, G. Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2016, 113, E2570–E2578. [Google Scholar] [CrossRef] [Green Version]
- Boekhout, M.; Wolthuis, R. Nek2A destruction marks APC/C activation at the prophase-to-prometaphase transition by spindle-checkpoint-restricted Cdc20. J. Cell Sci. 2015, 128, 1639–1653. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-M.; Lee, J. MET: Roles in epithelial-mesenchymal transition and cancer stemness. Ann. Transl. Med. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Lal, B.; Kwon, S.; Fan, X.; Saldanha, U.; Reznik, T.E.; Kuchner, E.B.; Eberhart, C.; Laterra, J.; Abounader, R. The scatter factor/hepatocyte growth factor: C-met pathway in human embryonal central nervous system tumor malignancy. Cancer Res. 2005, 65, 9355–9362. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Kiliti, A.J.; Zhang, K.; Vasani, N.; Mao, N.; Jordan, E.; Wise, H.C.; Bhattarai, T.S.; Hu, W.; Dorso, M. AKT1 E17K inhibits cancer cell migration by abrogating β-catenin signaling. Mol. Cancer Res. 2021, 19, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Ginty, D.D.; Segal, R.A. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr. Opin. Neurobiol. 2002, 12, 268–274. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.-J.; Roux, P.P.; Blenis, J.; Yoon, S.-O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellai, M.; Caldera, V.; Annovazzi, L.; Schiffer, D. The distribution and significance of IDH mutations in gliomas. In Evolution of the Molecular Biology of Brain Tumors and Therapeutic Implications; InTech: London, UK, 2013; pp. 299–342. [Google Scholar]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovčevska, I. Sequencing the next generation of glioblastomas. Crit. Rev. Clin. Lab. Sci. 2018, 55, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Oppel, F.; Tao, T.; Shi, H.; Ross, K.N.; Zimmerman, M.W.; He, S.; Tong, G.; Aster, J.C.; Look, A.T. Loss of atrx cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 2019, 15, e1008039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idilli, A.I.; Cusanelli, E.; Pagani, F.; Berardinelli, F.; Bernabé, M.; Cayuela, M.L.; Poliani, P.L.; Mione, M.C. Expression of tert prevents ALT in zebrafish brain tumors. Front. Cell Dev. Biol. 2020, 8, 65. [Google Scholar] [CrossRef]
- Lafferty-Whyte, K.; Cairney, C.J.; Will, M.B.; Serakinci, N.; Daidone, M.-G.; Zaffaroni, N.; Bilsland, A.; Keith, W.N. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 2009, 28, 3765–3774. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.-J.; Wu, P.-H.; Ho, C.-T.; Way, T.-D.; Pan, M.-H.; Chen, H.-M.; Ho, Y.-S.; Wang, Y.-J. P53-dependent downregulation of hTERT protein expression and telomerase activity induces senescence in lung cancer cells as a result of pterostilbene treatment. Cell Death Dis. 2017, 8, e2985. [Google Scholar] [CrossRef] [Green Version]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Kwon, C.H.; Nakano, I. Detoxification of oxidative stress in glioma stem cells: Mechanism, clinical relevance, and therapeutic development. J. Neurosci. Res. 2014, 92, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Puca, E.; Silginer, M.; Hemmerle, T.; Pazahr, S.; Bink, A.; Weller, M.; Neri, D.; Roth, P. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, Y.; Bryant, S.H. FSelector: A Ruby gem for feature selection. Bioinformatics 2012, 28, 2851–2852. [Google Scholar] [CrossRef] [Green Version]
- Romanski, P.; Kotthoff, L.; Kotthoff, M.L. Package ‘FSelector’. 2013. Available online: http://cran/r-project.org/web/packages/FSelector/index.html (accessed on 12 September 2020).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Park, J.; Ahn, J.W.; Sim, J.M.; Kang, S.J.; Kim, S.; Hwang, S.J.; Han, S.-H.; Sung, K.S.; Lim, J. Transcriptomic Landscape of Lower Grade Glioma Based on Age-Related Non-Silent Somatic Mutations. Curr. Oncol. 2021, 28, 2281-2295. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol28030210
Park Y, Park J, Ahn JW, Sim JM, Kang SJ, Kim S, Hwang SJ, Han S-H, Sung KS, Lim J. Transcriptomic Landscape of Lower Grade Glioma Based on Age-Related Non-Silent Somatic Mutations. Current Oncology. 2021; 28(3):2281-2295. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol28030210
Chicago/Turabian StylePark, YoungJoon, JeongMan Park, Ju Won Ahn, Jeong Min Sim, Su Jung Kang, Suwan Kim, So Jung Hwang, Song-Hee Han, Kyoung Su Sung, and Jaejoon Lim. 2021. "Transcriptomic Landscape of Lower Grade Glioma Based on Age-Related Non-Silent Somatic Mutations" Current Oncology 28, no. 3: 2281-2295. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol28030210