
Investigating Genetic Diversity and Genomic Signatures of Hatchery-Induced Evolution in Gilthead Seabream (Sparus aurata) Populations

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and SNP Dataset
2.2. Genetic Diversity and Population Structure
2.3. Outlier Analyses
2.4. SNP Annotation
3. Results
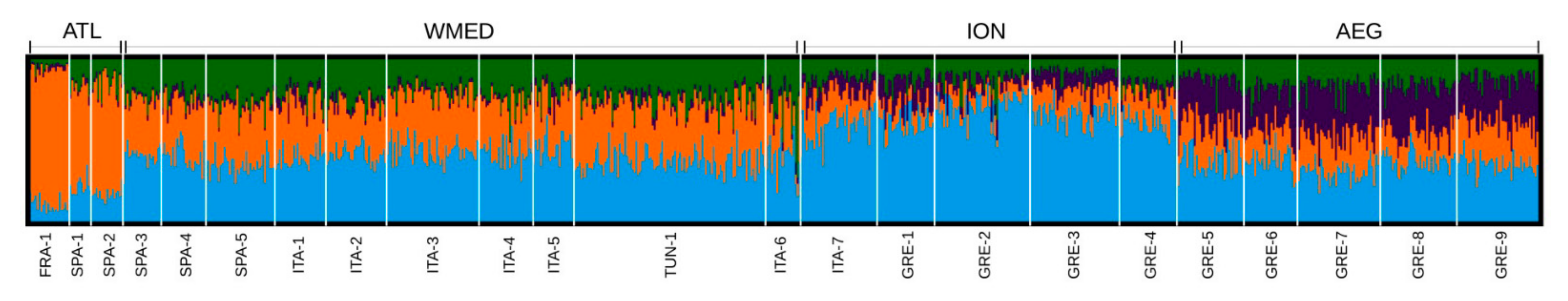
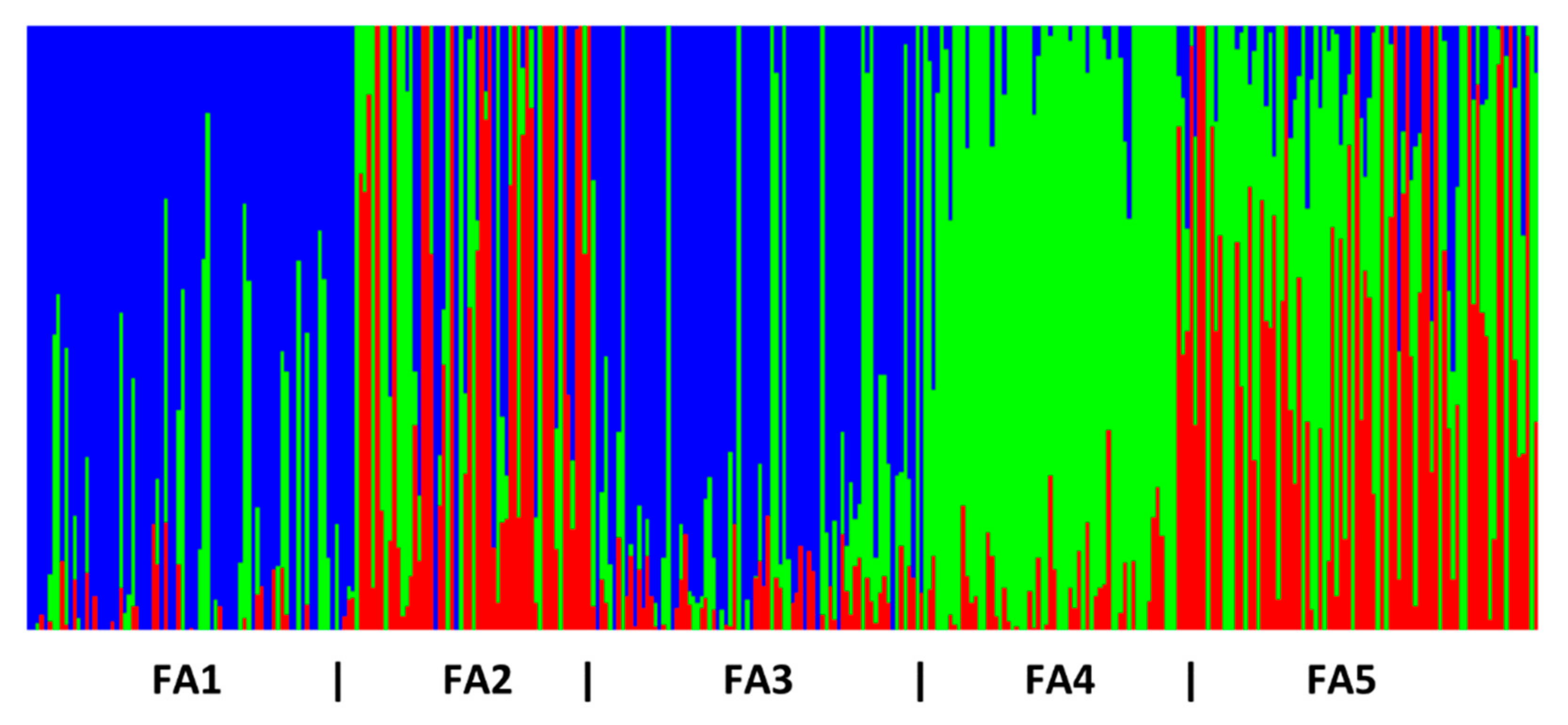
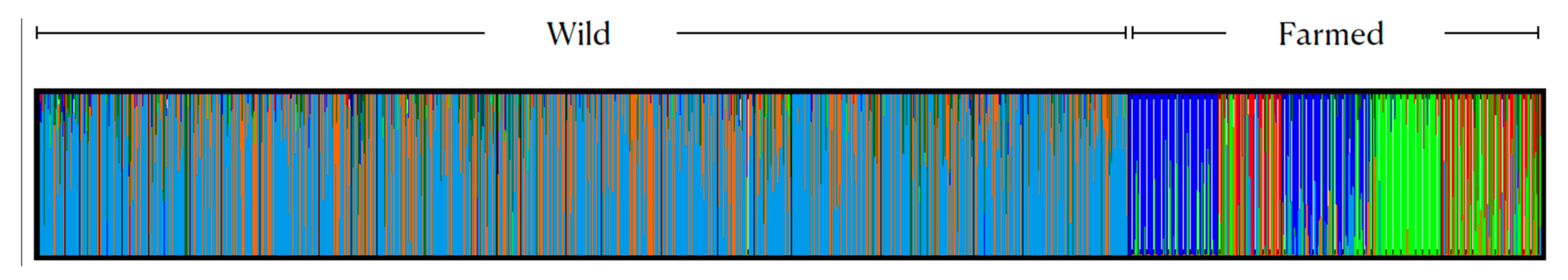
3.1. Genetic Diversity and Population Genetic Structure
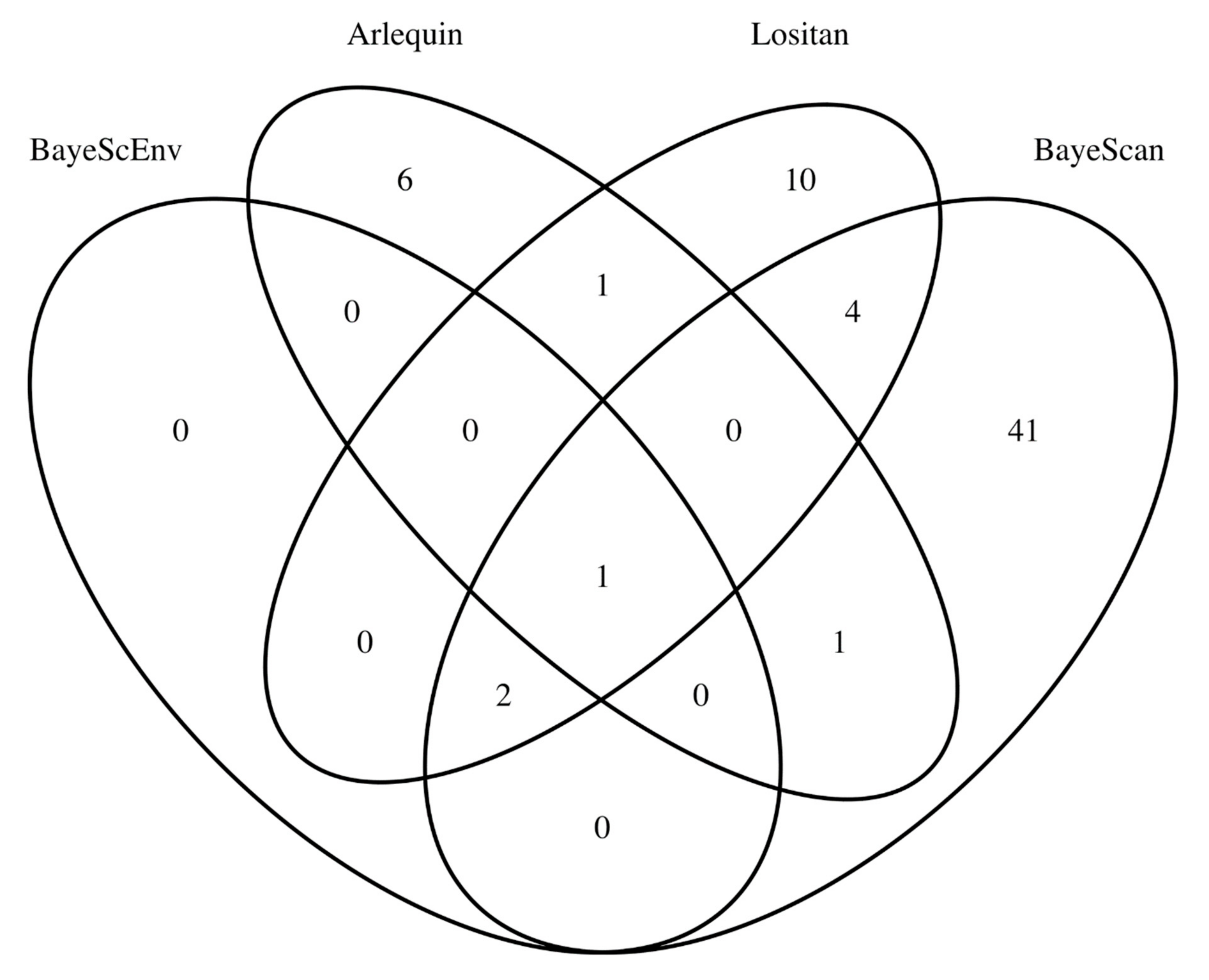
3.2. SNP Outliers of Hatchery-Induced Evolution in Gilthead Seabream
4. Discussion
4.1. Assessing the Significance of Candidate SNPs for Domestication in Gilthead Seabream
4.2. Evidence on the Genetic Basis of Domestication in Gilthead Seabream
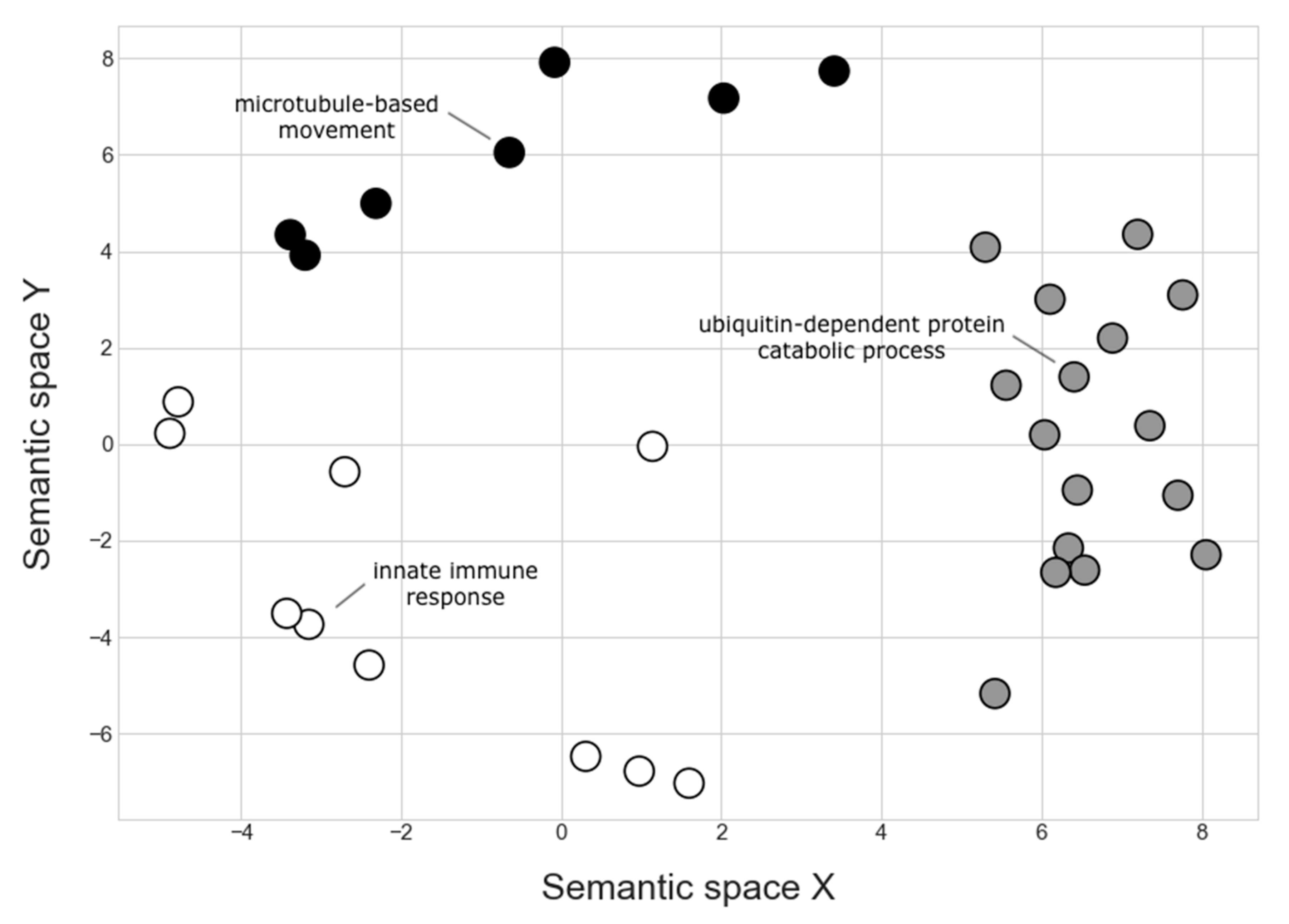
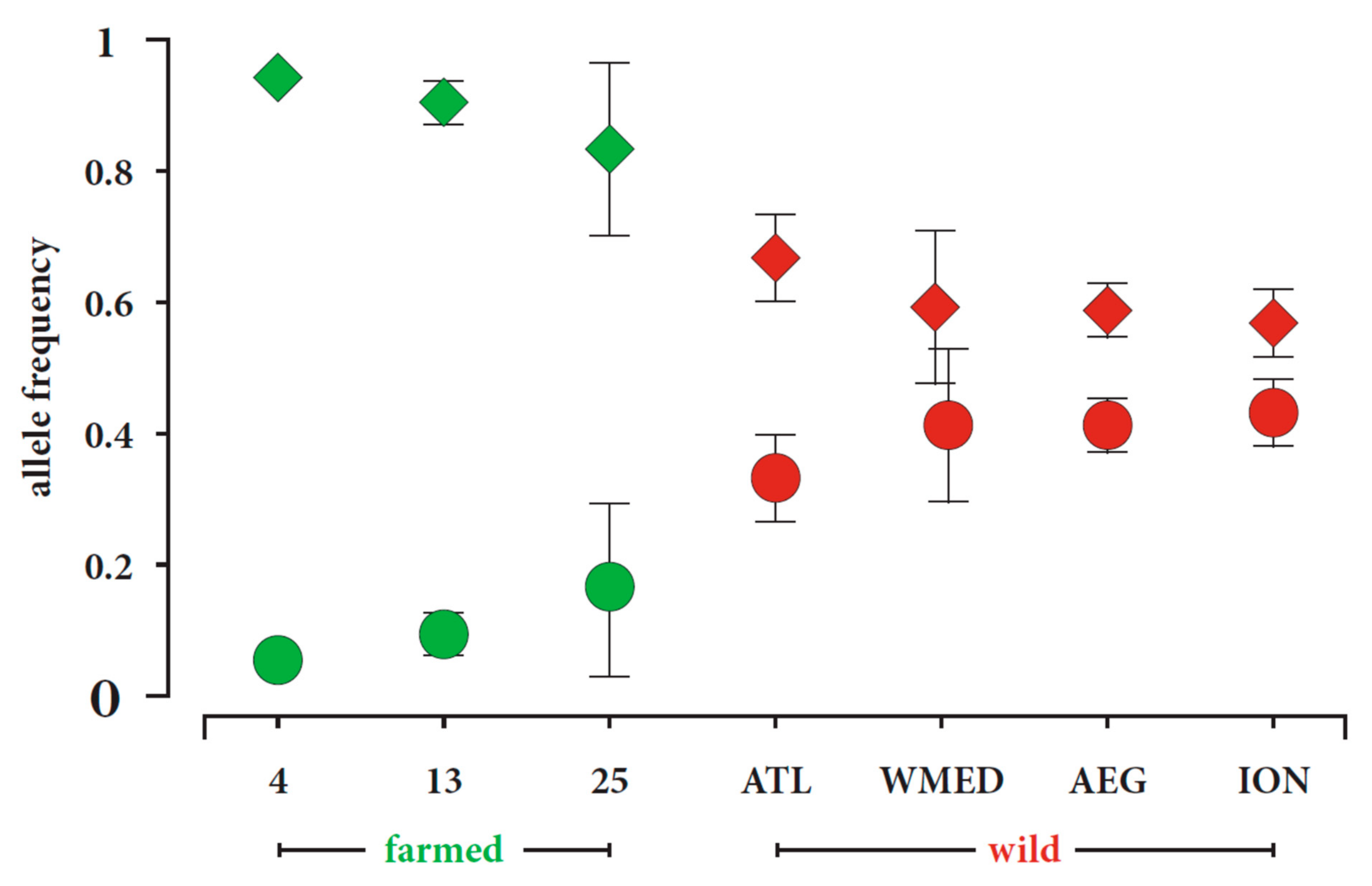
4.2.1. Genes Associated with the SNP 901_49
4.2.2. Genes Associated with SNP 8901_12 and SNP 12232_7
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerbault, P.; Allaby, R.G.; Boivin, N.; Rudzinski, A.; Grimaldi, I.M.; Pires, J.C.; Climer Vigueira, C.; Dobney, K.; Gremillion, K.J.; Barton, L.; et al. Storytelling and story testing in domestication. Proc. Natl. Acad. Sci. USA 2014, 111, 6159–6164. [Google Scholar] [CrossRef] [Green Version]
- Larson, G.; Piperno, D.R.; Allaby, R.G.; Purugganan, M.D.; Andersson, L.; Arroyo-Kalin, M.; Barton, L.; Climer Vigueira, C.; Denham, T.; Dobney, K.; et al. Current perspectives and the future of domestication studies. Proc. Natl. Acad. Sci. USA 2014, 111, 6139–6146. [Google Scholar] [CrossRef] [Green Version]
- Larson, G.; Fuller, D.Q. The Evolution of Animal Domestication. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 115–136. [Google Scholar] [CrossRef] [Green Version]
- Frantz, L.A.F.; Bradley, D.G.; Larson, G.; Orlando, L. Animal domestication in the era of ancient genomics. Nat. Rev. Genet. 2020, 21, 449–460. [Google Scholar] [CrossRef]
- Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; Couldrey, C.; et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, M.F.; Conceição, L.E.C.; Millot, S.; Rey, S.; Bégout, M.-L.; Damsgard, B.; Kristiansen, T.; Höglund, E.; Øverli, Ø.; Martins, C.I.M. Coping styles in farmed fish: Consequences for aquaculture. Rev. Aquac. 2017, 9, 23–41. [Google Scholar] [CrossRef] [Green Version]
- Milla, S.; Pasquet, A.; El Mohajer, L.; Fontaine, P. How domestication alters fish phenotypes. Rev. Aquac. 2021, 13, 388–405. [Google Scholar] [CrossRef]
- Zenger, K.R.; Khatkar, M.S.; Jones, D.B.; Khalilisamani, N.; Jerry, D.R.; Raadsma, H.W. Genomic Selection in Aquaculture: Application, Limitations and Opportunities with Special Reference to Marine Shrimp and Pearl Oysters. Front. Genet. 2019, 9, 693. [Google Scholar] [CrossRef]
- Purugganan, M.D. Evolutionary Insights into the Nature of Plant Domestication. Curr. Biol. 2019, 29, R705–R714. [Google Scholar] [CrossRef] [Green Version]
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M.; et al. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Southgate, P.C. Aquaculture; Lucas, J.S., Southgate, P.C., Eds.; Blackwell Publishing Ltd.: West Sussex, UK, 2012; ISBN 9781118687932. [Google Scholar]
- Teletchea, F. Fish domestication in aquaculture: 10 unanswered questions. Anim. Front. 2021, 11, 87–91. [Google Scholar] [CrossRef]
- Teletchea, F.; Fontaine, P. Levels of domestication in fish: Implications for the sustainable future of aquaculture. Fish Fish. 2014, 15, 181–195. [Google Scholar] [CrossRef]
- Araki, H.; Cooper, B.; Blouin, M.S. Genetic Effects of Captive Breeding Cause a Rapid, Cumulative Fitness Decline in the Wild. Science 2007, 318, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, M.R.; Marine, M.L.; French, R.A.; Blouin, M.S. Genetic adaptation to captivity can occur in a single generation. Proc. Natl. Acad. Sci. USA 2012, 109, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, G.; Kocour, M. Applications of next-generation sequencing in fisheries research: A review. Fish. Res. 2017, 186, 11–22. [Google Scholar] [CrossRef]
- Li, Y.-H.; Wang, H.-P. Advances of genotyping-by-sequencing in fisheries and aquaculture. Rev. Fish Biol. Fish. 2017, 27, 535–559. [Google Scholar] [CrossRef]
- Lu, G.; Luo, M. Genomes of major fishes in world fisheries and aquaculture: Status, application and perspective. Aquac. Fish. 2020, 5, 163–173. [Google Scholar] [CrossRef]
- Dittmar, E.L.; Oakley, C.G.; Conner, J.K.; Gould, B.A.; Schemske, D.W. Factors influencing the effect size distribution of adaptive substitutions. Proc. R. Soc. B Biol. Sci. 2016, 283, 20153065. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Yeaman, S. Local Adaptation by Alleles of Small Effect. Am. Nat. 2015, 186, S74–S89. [Google Scholar] [CrossRef] [PubMed]
- Gagnaire, P.-A.; Gaggiotti, O.E. Detecting polygenic selection in marine populations by combining population genomics and quantitative genetics approaches. Curr. Zool. 2016, 62, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellenreuther, M.; Hansson, B. Detecting Polygenic Evolution: Problems, Pitfalls, and Promises. Trends Genet. 2016, 32, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bomba, L.; Walter, K.; Soranzo, N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Darnaude, A.; Ferraton, F.; Guinand, B.; Bonhomme, F.; Bierne, N.; Gagnaire, P.-A. Within-Generation Polygenic Selection Shapes Fitness-Related Traits across Environments in Juvenile Sea Bream. Genes 2020, 11, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, L.; Moretti, A.; Crosetti, D.; Karaiskou, N.; Magoulas, A.; Rossi, A.R.; Rye, M.; Triantafyllidis, A.; Tsigenipoulos, C.S. Genetic effects of domestification, culture and breeding of fish and shellfish, and their impact on wild populations. Gilthead seabream-Sparus aurata. In Genetic Impact of Aquaculture Activities on Native Populations. Genetic Final Scientific Report (EU Contract no. RICA-CT-2005-022802); Svasand, T., Crosetti, D., Garcia-Vázquez, E., Verspoor, E., Eds.; 2007; pp. 47–54. Available online: https://www.researchgate.net/profile/Philip-Mcginnity/publication/268359165_Research_Priorities_for_Modelling/links/547459fb0cf2778985abd9ef/Research-Priorities-for-Modelling.pdf#page=47 (accessed on 26 March 2021).
- FAO Fishery and Aquaculture Statistics. Global Aquaculture Production 1950–2019 (FishstatJ). FAO Fisheries Division, FAO Fisheries and Aquaculture Department, FIPS—Statistics and Information: Rome, Italy, 2021. Updated 2021. 2.0.0. Available online: www.fao.org/fishery/statistics/software/fishstatj/en (accessed on 25 August 2021).
- Arabaci, M.; Yilmaz, Y.; Ceyhun, S.B.; Erdogan, O.; Dorlay, H.G.; Diler, I.; Akhan, S.; Kocabas, M.; Ozdemir, K.; Koyun, H.; et al. A Review on Population Characteristics of Gilthead Seabream (Sparus aurata). J. Anim. Vet. Adv. 2010, 9, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Gkagkavouzis, K.; Karaiskou, N.; Katopodi, T.; Leonardos, I.; Abatzopoulos, T.J.; Triantafyllidis, A. The genetic population structure and temporal genetic stability of gilthead sea bream Sparus aurata populations in the Aegean and Ionian Seas, using microsatellite DNA markers. J. Fish Biol. 2019, 94, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Thorland, I.; Papaioannou, N.; Kottaras, L.; Refstie, T.; Papasolomontos, S.; Rye, M. The Kego breeding programs for sea bream (Sparus aurata) and sea bass (Dicentrarchus labrax) in Greece. In Proceedings of the 8th Hellenic Symposium on Oceanography and Fisheries, Thessaloniki, Greece, 4–8 June 2006. [Google Scholar]
- Brown, R.C. Genetic Management and Selective Breeding in Farmed Populations of Gilthead Seabream (Sparus aurata). Ph.D. Thesis, University of Stirling, Stirling, Scotland, 2003. [Google Scholar]
- Janssen, K.; Chavanne, H.; Berentsen, P.; Komen, H. Gilthead Seabream (Sparus aurata)—Current status of selective breeding in Europe. In Proceedings of the the International Symposium on Genetics in Aquaculture XII, Santiago de Compostela, Spain, 21–27 June 2015. [Google Scholar]
- Knibb, W. Genetic improvement of marine fish—Which method for industry? Aquac. Res. 2000, 31, 11–23. [Google Scholar] [CrossRef]
- Gjedrem, T.; Baranski, M. Selective Breeding in Aquaculture: An Introduction; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010; Volume 10, ISBN 9048127734. [Google Scholar]
- Chavanne, H.; Parati, K.; Cambuli, C.; Capoferri, R.; Jiménez, C.A.; Galli, A. Microsatellites markers to depict the reproductive and genetic patterns of farmed gilthead seabream (Sparus aurata): Illustration by a case study on mass spawning. Aquac. Res. 2014, 45, 577–590. [Google Scholar] [CrossRef]
- Loughnan, S.R.; Domingos, J.A.; Smith-Keune, C.; Forrester, J.P.; Jerry, D.R.; Beheregaray, L.B.; Robinson, N.A. Broodstock contribution after mass spawning and size grading in barramundi (Lates calcarifer, Bloch). Aquaculture 2013, 404–405, 139–149. [Google Scholar] [CrossRef]
- Fessehaye, Y.; Bovenhuis, H.; Rezk, M.A.; Crooijmans, R.; van Arendonk, J.A.M.; Komen, H. Effects of relatedness and inbreeding on reproductive success of Nile tilapia (Oreochromis niloticus). Aquaculture 2009, 294, 180–186. [Google Scholar] [CrossRef]
- Alarcón, J.A.; Magoulas, A.; Georgakopoulos, T.; Zouros, E.; Alvarez, M.C. Genetic comparison of wild and cultivated European populations of the gilthead sea bream (Sparus aurata). Aquaculture 2004, 230, 65–80. [Google Scholar] [CrossRef]
- Šegvić-Bubić, T.; Talijančić, I.; Grubišić, L.; Izquierdo-Gomez, D.; Katavić, I. Morphological and molecular differentiation of wild and farmed gilthead sea bream Sparus aurata: Implications for management. Aquac. Environ. Interact. 2014, 6, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Maroso, F.; Gkagkavouzis, K.; De Innocentiis, S.; Hillen, J.; do Prado, F.; Karaiskou, N.; Taggart, J.B.; Carr, A.; Nielsen, E.; Triantafyllidis, A.; et al. Genome-wide analysis clarifies the population genetic structure of wild gilthead sea bream (Sparus aurata). PLoS ONE 2021, 16, e0236230. [Google Scholar] [CrossRef] [PubMed]
- Gkagkavouzis, K. Development and Evaluation of Methodologies for Genomic Analysis in Wild and Farmed Sea Bream (Sparus aurata) Populations; Aristotle University of Thessaloniki: Thessaloniki, Greece, 2019. [Google Scholar]
- Karlsson, S.; Moen, T.; Lien, S.; Glover, K.A.; Hindar, K. Generic genetic differences between farmed and wild Atlantic salmon identified from a 7K SNP-chip. Mol. Ecol. Resour. 2011, 11, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and genotyping Loci de novo from short-read sequences. G3 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-L.; Liu, J.-X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Antolin, M.F.; Bradburd, G.; Lowry, D.B.; Poss, M.L.; Reed, L.K.; Storfer, A.; Whitlock, M.C. Finding the Genomic Basis of Local Adaptation: Pitfalls, Practical Solutions, and Future Directions. Am. Nat. 2016, 188, 379–397. [Google Scholar] [CrossRef] [Green Version]
- Foll, M.; Gaggiotti, O. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Foll, M.; Fischer, M.C.; Heckel, G.; Excoffier, L. Estimating population structure from AFLP amplification intensity. Mol. Ecol. 2010, 19, 4638–4647. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.C.; Foll, M.; Excoffier, L.; Heckel, G. Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Mol. Ecol. 2011, 20, 1450–1462. [Google Scholar] [CrossRef]
- Villemereuil, P.; Gaggiotti, O.E. A new FST—Based method to uncover local adaptation using environmental variables. Methods Ecol. Evol. 2015, 6, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a F st -outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Rellstab, C.; Gugerli, F.; Eckert, A.J.; Hancock, A.M.; Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 2015, 24, 4348–4370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, J.D.; Bass, A.J.; Dabney, A.; Robinson, D. Qvalue: Q-Value Estimation for False Discovery Rate Control. R Package Version 2.24.0. 2021. Available online: https://www.bioconductor.org/packages/release/bioc/html/qvalue.html (accessed on 26 August 2021).
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Stephan, W. Joint Effects of Genetic Hitchhiking and Background Selection on Neutral Variation. Genetics 2000, 155, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How far from the SNP may the causative genes be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef]
- Sherry, S.T. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoof, N.; Iles, M.M.; Bishop, D.T.; Newton-Bishop, J.A.; Barrett, J.H.; Consortium, G. Pathway-Based Analysis of a Melanoma Genome-Wide Association Study: Analysis of Genes Related to Tumour-Immunosuppression. PLoS ONE 2011, 6, e29451. [Google Scholar] [CrossRef] [Green Version]
- Pesquita, C.; Faria, D.; Falcão, A.O.; Lord, P.; Couto, F.M. Semantic Similarity in Biomedical Ontologies. PLoS Comput. Biol. 2009, 5, e1000443. [Google Scholar] [CrossRef]
- Papakostas, S.; Vøllestad, L.A.; Bruneaux, M.; Aykanat, T.; Vanoverbeke, J.; Ning, M.; Primmer, C.R.; Leder, E.H. Gene pleiotropy constrains gene expression changes in fish adapted to different thermal conditions. Nat. Commun. 2014, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Papakostas, S.; Vasemägi, A.; Himberg, M.; Primmer, C.R. Proteome variance differences within populations of European whitefish (Coregonus lavaretus) originating from contrasting salinity environments. J. Proteom. 2014, 105, 144–150. [Google Scholar] [CrossRef]
- Primmer, C.R.; Papakostas, S.; Leder, E.H.; Davis, M.J.; Ragan, M.A. Annotated genes and nonannotated genomes: Cross-species use of Gene Ontology in ecology and evolution research. Mol. Ecol. 2013, 22, 3216–3241. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Excoffier, L.; Hofer, T.; Foll, M. Detecting loci under selection in a hierarchically structured population. Heredity 2009, 103, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narum, S.R.; Hess, J.E. Comparison of FST outlier tests for SNP loci under selection. Mol. Ecol. Resour. 2011, 11, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, C.W.; Rymer, P.D.; Stow, A.; Bragg, J.; Dillon, S.; Umbers, K.D.L.; Dudaniec, R.Y. The search for loci under selection: Trends, biases and progress. Mol. Ecol. 2018, 27, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Peñaloza, C.; Manousaki, T.; Franch, R.; Tsakogiannis, A.; Sonesson, A.K.; Aslam, M.L.; Allal, F.; Bargelloni, L.; Houston, R.D.; Tsigenopoulos, C.S. Development and testing of a combined species SNP array for the European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata). Genomics 2021, 113, 2096–2107. [Google Scholar] [CrossRef] [PubMed]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, P.F.; Nielsen, E.E.; Williams, T.D.; Loeschcke, V. Intraspecific variation in expression of candidate genes for osmoregulation, heme biosynthesis and stress resistance suggests local adaptation in European flounder (Platichthys flesus). Heredity 2008, 101, 247–259. [Google Scholar] [CrossRef]
- Valenzuela-Muñoz, V.; Gallardo-Escárate, C. Iron metabolism modulation in Atlantic salmon infested with the sea lice Lepeophtheirus salmonis and Caligus rogercresseyi: A matter of nutritional immunity? Fish Shellfish Immunol. 2017, 60, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Kapetanović, D.; Kurtović, B.; Vardić, I.; Teskeredžić, E.; Teskeredžić, Z. Gill disease in a gilthead sea bream (Sparus aurata L.). Med. Weter. 2006, 62, 1239–1241. [Google Scholar]
- Ibarz, A.; Padrós, F.; Gallardo, M.Á.; Fernández-Borràs, J.; Blasco, J.; Tort, L. Low-temperature challenges to gilthead sea bream culture: Review of cold-induced alterations and ‘Winter Syndrome’. Rev. Fish Biol. Fish. 2010, 20, 539–556. [Google Scholar] [CrossRef]
- Fazio, F.; Ferrantelli, V.; Piccione, G.; Saoca, C.; Levanti, M.; Mucciardi, M. Biochemical and hematological parameters in European sea bass (Dicentrarchus labrax Linnaeus, 1758) and Gilthead sea bream (Sparus aurata Linnaeus, 1758) in relation to temperature. Vet. Arh. 2018, 88, 397–411. [Google Scholar] [CrossRef]
- Xu, K.; Liu, B.; Ma, Y.; Xu, B.; Xing, X. A novel SWIM domain protein ZSWIM5 inhibits the malignant progression of non-small-cell lung cancer. Cancer Manag. Res. 2018, 10, 3245–3254. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Aravind, L.; Koonin, E.V. SWIM, a novel Zn-chelating domain present in bacteria, archaea and eukaryotes. Trends Biochem. Sci. 2002, 27, 384–386. [Google Scholar] [CrossRef]
- Chang, C.; Kuo, H.; Chen, S.; Lin, W.; Lu, K.; Saito, T.; Liu, F. Developmental characterization of Zswim5 expression in the progenitor domains and tangential migration pathways of cortical interneurons in the mouse forebrain. J. Comp. Neurol. 2020, 528, 2404–2419. [Google Scholar] [CrossRef]
- Micheletti, S.J.; Narum, S.R. Utility of pooled sequencing for association mapping in nonmodel organisms. Mol. Ecol. Resour. 2018, 18, 825–837. [Google Scholar] [CrossRef]
- Lowell, C.A. Src-family kinases: Rheostats of immune cell signaling. Mol. Immunol. 2004, 41, 631–643. [Google Scholar] [CrossRef]
- Corey, S.J.; Anderson, S.M. Src-Related Protein Tyrosine Kinases in Hematopoiesis. Blood 1999, 93, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ijiri, T.W.; Mahbub Hasan, A.K.M.; Sato, K. Protein-Tyrosine Kinase Signaling in the Biological Functions Associated with Sperm. J. Signal Transduct. 2012, 2012, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Weichselbaum, R.; Kharbanda, S.; Kufe, D. Role for Lyn Tyrosine Kinase as a Regulator of Stress-Activated Protein Kinase Activity in Response to DNA Damage. Mol. Cell. Biol. 2000, 20, 5370–5380. [Google Scholar] [CrossRef] [Green Version]
- Dunand-Sauthier, I.; Walker, C.A.; Narasimhan, J.; Pearce, A.K.; Wek, R.C.; Humphrey, T.C. Stress-Activated Protein Kinase Pathway Functions To Support Protein Synthesis and Translational Adaptation in Response to Environmental Stress in Fission Yeast. Eukaryot. Cell 2005, 4, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhao, Y.; Wang, J.; Liu, B.; Sun, X.; Guo, S.; Feng, J. Transcriptome profiling of developing spleen tissue and discovery of immune-related genes in grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. 2017, 60, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.G.; Somero, G.N. A microarray-based transcriptomic time-course of hyper- and hypo-osmotic stress signaling events in the euryhaline fish Gillichthys mirabilis:osmosensors to effectors. J. Exp. Biol. 2008, 211, 3636–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortuño, J.; Esteban, M.A.; Meseguer, J. Effects of short-term crowding stress on the gilthead seabream (Sparus aurata L.) innate immune response. Fish Shellfish Immunol. 2001, 11, 187–197. [Google Scholar] [CrossRef]
- Segner, H.; Sundh, H.; Buchmann, K.; Douxfils, J.; Sundell, K.S.; Mathieu, C.; Ruane, N.; Jutfelt, F.; Toften, H.; Vaughan, L. Health of farmed fish: Its relation to fish welfare and its utility as welfare indicator. Fish Physiol. Biochem. 2012, 38, 85–105. [Google Scholar] [CrossRef] [Green Version]
- Melamed, P.; Rosenfeld, H.; Elizur, A.; Yaron, Z. Endocrine regulation of gonadotropin and growth hormone gene transcription in fish. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1998, 119, 325–338. [Google Scholar] [CrossRef]
- Barbaro, A.; Francescon, A.; Bozzato, G.; Merlin, A.; Belvedere, P.; Colombo, L. Induction of spawning in gilthead seabream, Sparus aurata L., by a long-acting GnRH agonist and its effects on egg quality and daily timing of spawning. Aquaculture 1997, 154, 349–359. [Google Scholar] [CrossRef]
- Zohar, Y.; Mylonas, C.C. Endocrine manipulations of spawning in cultured fish: From hormones to genes. Aquaculture 2001, 197, 99–136. [Google Scholar] [CrossRef]
- Muñoz-Cueto, J.A.; Zmora, N.; Paullada-Salmerón, J.A.; Marvel, M.; Mañanos, E.; Zohar, Y. The gonadotropin-releasing hormones: Lessons from fish. Gen. Comp. Endocrinol. 2020, 291, 113422. [Google Scholar] [CrossRef]
- Deane, E.E.; Woo, N.Y.S. Modulation of fish growth hormone levels by salinity, temperature, pollutants and aquaculture related stress: A review. Rev. Fish Biol. Fish. 2009, 19, 97–120. [Google Scholar] [CrossRef]
- Webb, A.E. Characterisation of Human Peptidyl Prolyl ISOMERASE-Like Protein 1 (PPIL1) Mutations. 2019. Available online: https://etheses.whiterose.ac.uk/25250/ (accessed on 26 March 2021).
- Thapar, R. Roles of Prolyl Isomerases in RNA-Mediated Gene Expression. Biomolecules 2015, 5, 974–999. [Google Scholar] [CrossRef]
- Mesa, A.; Somarelli, J.A.; Herrera, R.J. Spliceosomal immunophilins. FEBS Lett. 2008, 582, 2345–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.D.; Singh, P. Comparative studies on drought-induced changes in peptidyl prolyl cis–trans isomerase activity in drought-tolerant and susceptible cultivars of Sorghum bicolor. Curr. Sci. 2003, 84, 911–918. [Google Scholar]
- Kaur, G.; Singh, S.; Dutta, T.; Kaur, H.; Singh, B.; Pareek, A.; Singh, P. The peptidyl-prolyl cis-trans isomerase activity of the wheat cyclophilin, TaCypA-1, is essential for inducing thermotolerance in Escherichia coli. Biochim. Open 2016, 2, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin Anduaga, A.; Evantal, N.; Patop, I.L.; Bartok, O.; Weiss, R.; Kadener, S. Thermosensitive alternative splicing senses and mediates temperature adaptation in Drosophila. eLife 2019, 8, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Yi, S.-K.; Yao, H.; Chi, W.; Guo, Y.; Ma, X.-F.; Wang, H.-P. Comparative transcriptome analysis reveals potential evolutionary differences in adaptation of temperature and body shape among four Percidae species. PLoS ONE 2019, 14, e0215933. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Rai, A.K.; Kanwar, S.S.; Sharma, T.R. Comparative Analysis of Zinc Finger Proteins Involved in Plant Disease Resistance. PLoS ONE 2012, 7, e42578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.; Lu, C.; Guo, J.; Qiao, Z.; Sui, N.; Qiu, N.; Wang, B. C2H2 Zinc Finger Proteins: Master Regulators of Abiotic Stress Responses in Plants. Front. Plant Sci. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vij, S.; Tyagi, A.K. A20/AN1 zinc-finger domain-containing proteins in plants and animals represent common elements in stress response. Funct. Integr. Genom. 2008, 8, 301–307. [Google Scholar] [CrossRef]
- Aceituno-Valenzuela, U.; Micol-Ponce, R.; Ponce, M.R. Genome-wide analysis of CCHC-type zinc finger (ZCCHC) proteins in yeast, Arabidopsis, and humans. Cell. Mol. Life Sci. 2020, 77, 3991–4014. [Google Scholar] [CrossRef]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simicek, M.; Lievens, S.; Laga, M.; Guzenko, D.; Aushev, V.N.; Kalev, P.; Baietti, M.F.; Strelkov, S.V.; Gevaert, K.; Tavernier, J.; et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol. 2013, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-G.; Yeo, M.-K. Comparison of gene expression changes induced by exposure to Ag, Cu-TiO2, and TiO2 nanoparticles in zebrafish embryos. Mol. Cell. Toxicol. 2013, 9, 129–139. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, Z.; Pan, Y.; Zhao, J.; Bai, H.; Chen, B.; Zhang, X.; Pu, F.; Chen, J.; Xu, P. GWAS identified candidate variants and genes associated with acute heat tolerance of large yellow croaker. Aquaculture 2021, 540, 736696. [Google Scholar] [CrossRef]
- Lahnsteiner, F.; Mansour, N. The effect of temperature on sperm motility and enzymatic activity in brown trout Salmo trutta, burbot Lota lota and grayling Thymallus thymallus. J. Fish Biol. 2012, 81, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Blier, P.U.; Guderley, H.E. Mitochondrial Activity in Rainbow Trout Red Muscle: The Effect of Temperature on the Adp-Dependence of Atp Synthesis. J. Exp. Biol. 1993, 176, 145–158. [Google Scholar] [CrossRef]
- Arai, K.; Inoue, A.; Ojima, T. Isolation and characterization of an adenylate kinase from the lateral muscle of bastard halibut Paralichthys olivaceus. Fish. Sci. 2020, 86, 385–394. [Google Scholar] [CrossRef]
- Pickart, C.M. Ubiquitin and the Stress Response; Springer: Berlin/Heidelberg, Germany, 1999; pp. 133–152. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Loc_ID | Cluster Analysis_ID | Location | Latitude | Longitude | Year | N | Ho | He | FIS |
|---|---|---|---|---|---|---|---|---|---|
| FRA-1 | ATL | Noirmoutier | −2.170 | 46.989 | 2003 | 22 | 0.13 | 0.15 | 0.08 |
| SPA-1 | ATL | Vigo | −8.953 | 41.831 | 2009 | 13 | 0.14 | 0.15 | 0.04 |
| SPA-2 | ATL | Cadiz | −6.400 | 36.500 | 2001 | 17 | 0.14 | 0.15 | 0.04 |
| SPA-3 | WMED | Alicante | −0.317 | 38.286 | 2009 | 20 | 0.12 | 0.14 | 0.08 |
| SPA-4 | WMED | Valencia | −0.100 | 39.500 | 2014 | 24 | 0.14 | 0.15 | 0.07 |
| SPA-5 | WMED | Balearic | 2.681 | 39.403 | 2013 | 36 | 0.14 | 0.16 | 0.07 |
| ITA-1 | WMED | W Sardinia | 8.402 | 39.826 | 2002 | 28 | 0.14 | 0.16 | 0.07 |
| ITA-2 | WMED | Genova | 8.901 | 44.360 | 2005 | 33 | 0.13 | 0.15 | 0.09 |
| ITA-3 | WMED | Sabaudia | 12.624 | 41.406 | 2013 | 52 | 0.14 | 0.16 | 0.07 |
| ITA-4 | WMED | Tortoli | 9.756 | 39.924 | 2002 | 29 | 0.14 | 0.16 | 0.07 |
| ITA-5 | WMED | Trapani | 12.449 | 38.006 | 2007 | 22 | 0.14 | 0.16 | 0.06 |
| TUN-1 | WMED | Tunis | 10.602 | 36.932 | 2014 | 106 | 0.13 | 0.15 | 0.10 |
| ITA-6 | WMED | Otranto | 18.532 | 40.360 | 2001 | 20 | 0.15 | 0.15 | 0.03 |
| ITA-7 | ION | Venice | 12.409 | 45.322 | 2014 | 40 | 0.12 | 0.14 | 0.10 |
| GRE-1 | ION | Ionio | 20.360 | 38.983 | 2013 | 31 | 0.15 | 0.15 | 0.04 |
| GRE-2 | ION | Igoumenitsa | 20.163 | 39.486 | 2006, 2014 | 53 | 0.14 | 0.15 | 0.08 |
| GRE-3 | ION | Mesologgi | 21.315 | 38.303 | 2004, 2013 | 49 | 0.14 | 0.15 | 0.09 |
| GRE-4 | ION | Korinthiakos | 22.945 | 37.270 | 2014 | 32 | 0.14 | 0.16 | 0.08 |
| GRE-5 | AEG | Nayplio | 22.758 | 38.046 | 2005 | 33 | 0.14 | 0.16 | 0.08 |
| GRE-6 | AEG | Basova Kavalas | 24.495 | 40.846 | 2006, 2013 | 79 | 0.14 | 0.15 | 0.07 |
| GRE-7 | AEG | Thermaikos gulf | 22.846 | 40.263 | 2014 | 81 | 0.14 | 0.16 | 0.09 |
| GRE-8 | AEG | Agiasma | 24.419 | 40.644 | 2006 | 43 | 0.14 | 0.16 | 0.07 |
| GRE-9 | AEG | Alexandroupolis | 25.916 | 40.778 | 2013 | 93 | 0.14 | 0.15 | 0.09 |
| FA1 | 13 | Hatchery 1 | - | - | 2014 | 78 | 0.14 | 0.15 | 0.07 |
| FA2 | 25 | Hatchery 2 | - | - | 2014 | 56 | 0.15 | 0.15 | 0.05 |
| FA3 | 13 | Hatchery 3 | - | - | 2014 | 80 | 0.14 | 0.15 | 0.08 |
| FA4 | 4 | Hatchery 4 | - | - | 2014 | 60 | 0.14 | 0.15 | 0.06 |
| FA5 | 25 | Hatchery 5 | - | - | 2014 | 88 | 0.13 | 0.15 | 0.09 |
| Total | 1318 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkagkavouzis, K.; Papakostas, S.; Maroso, F.; Karaiskou, N.; Carr, A.; Nielsen, E.E.; Bargelloni, L.; Triantafyllidis, A. Investigating Genetic Diversity and Genomic Signatures of Hatchery-Induced Evolution in Gilthead Seabream (Sparus aurata) Populations. Diversity 2021, 13, 563. https://0-doi-org.brum.beds.ac.uk/10.3390/d13110563
Gkagkavouzis K, Papakostas S, Maroso F, Karaiskou N, Carr A, Nielsen EE, Bargelloni L, Triantafyllidis A. Investigating Genetic Diversity and Genomic Signatures of Hatchery-Induced Evolution in Gilthead Seabream (Sparus aurata) Populations. Diversity. 2021; 13(11):563. https://0-doi-org.brum.beds.ac.uk/10.3390/d13110563
Chicago/Turabian StyleGkagkavouzis, Konstantinos, Spiros Papakostas, Francesco Maroso, Nikoleta Karaiskou, Adrian Carr, Einar Eg Nielsen, Luca Bargelloni, and Alexandros Triantafyllidis. 2021. "Investigating Genetic Diversity and Genomic Signatures of Hatchery-Induced Evolution in Gilthead Seabream (Sparus aurata) Populations" Diversity 13, no. 11: 563. https://0-doi-org.brum.beds.ac.uk/10.3390/d13110563