Long-Term Outcomes of Early Enzyme Replacement Therapy for Mucopolysaccharidosis IV: Clinical Case Studies of Two Siblings

Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Subjects
2.2. Outcome Measures
- Safety and compliance—safety evaluations included continuous monitoring of adverse events. Absences from treatment sessions were recorded.
- Hospitalization and surgeries history—data regarding the surgical and hospitalization (inpatient and outpatient) history of P1 and P2 were retrieved from medical records.
- Growth—height and weight were measured in routine visitations to the clinic.
- Orthopedic and radiographic assessments and procedures—orthopedic and radiographic assessments were routinely conducted in order to assess structural changes. As recommended, the radiographic assessments mainly focused on the lower extremities (e.g., presence of progressive hip dysplasia, genu valgus, and ankle valgus), upper extremities, cervical spine, and thoracolumbar spine [31]. All orthopedic procedures were documented.
- Respiratory function and sleep test—the respiratory function evaluation consisted of a pediatric pulmonologist physical examination, spirometry test (forced vital capacity, forced expiratory volume in one second), and oxygen saturation and overnight sleep study.
- Ear, nose, and throat (ENT) manifestations—patients underwent a routine evaluation by an otorhinolaryngologist.
- Physical function—evaluation of physical function was routinely conducted by physical and occupational therapists. The evaluation focused on the patient’s impairment levels (e.g., muscle strength, range of motion), mobility ability (e.g., walking and stair climbing ability), activities of daily living (e.g., offing and doffing), and equilibrium and protective reactions.
3. Results
3.1. Safety and Compliance
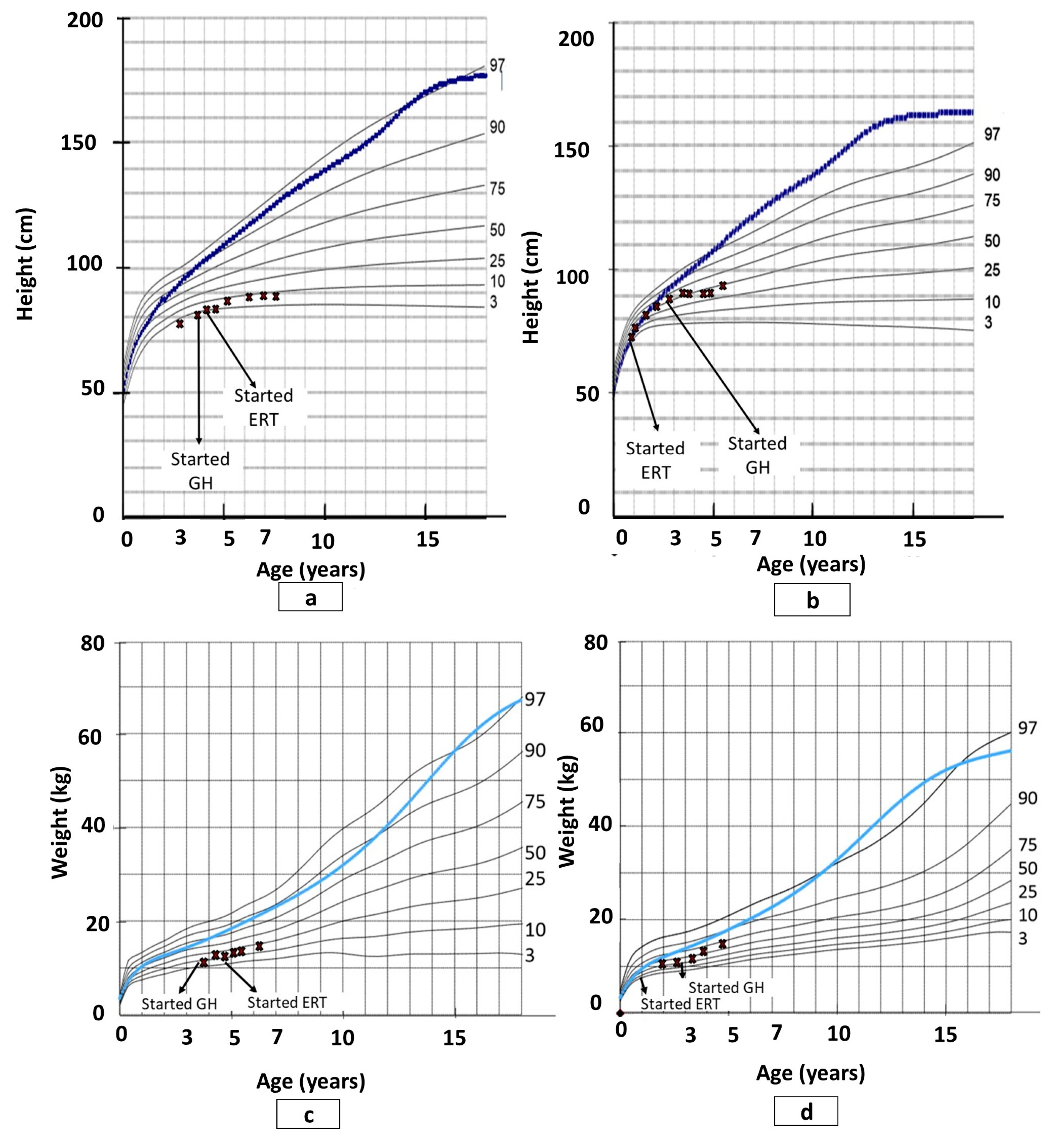
3.2. Growth
3.3. Clinical Course
3.4. Orthopedic and Radiographic Assessments and Procedures
3.5. Pulmonology and Ear, Nose, and Throat (ENT) Manifestations
3.6. Physical Function
3.6.1. Impairment Level
3.6.2. Mobility
3.6.3. Activities of Daily Living
4. Discussion
4.1. Safety and Compliance
4.2. Growth
4.3. Clinical Course, and Orthopedic and Radiographic Assessments
4.4. Respiratory Function and Sleep Test
4.5. ENT Manifestations
4.6. Physical and Functional Status
4.7. Study Limitations and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MPS | Mucopolysaccharidosis |
| GAG | Glycosaminoglycan |
| GALNS | N-acetylgalactosamine 6-sulfatase |
| ERT | Enzyme replacement therapy |
| GH | Growth hormone |
| P1 | Patient 1 |
| P2 | Patient 2 |
| ENT | Ear, nose, and throat |
References
- Neufeld, E.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, W., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume III, pp. 3421–3452. [Google Scholar]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Applegarth, D.A.; Toone, J.R.; Lowry, R.B. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics 2000, 105, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Fukuda, S.; Masue, M.; Sukegawa, K.; Fukao, T.; Yamagishi, A.; Hori, T.; Iwata, H.; Ogawa, T.; Nakashima, Y. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem. Biophys. Res. Commun. 1991, 181, 677–683. [Google Scholar] [CrossRef]
- Kresse, H.; von Figura, K.; Klein, U.; Glössl, J.; Paschke, E.; Pohlmann, R. Enzymic diagnosis of the genetic mucopolysaccharide storage disorders. Meth. Enzymol. 1982, 83, 559–572. [Google Scholar]
- Dorfman, A.; Arbogast, B.; Matalon, R. The enzymic defects in Morquio and Maroteaux-Lamy syndrome. Adv. Exp. Med. Biol. 1976, 68, 261–276. [Google Scholar] [PubMed]
- Wraith, J.E. The mucopolysaccharidoses: A clinical review and guide to management. Arch. Dis. Child. 1995, 72, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Montaño, A.M.; Oikawa, H.; Smith, M.; Barrera, L.; Chinen, Y.; Thacker, M.M.; Mackenzie, W.G.; Suzuki, Y.; Orii, T. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr. Pharm. Biotechnol. 2011, 12, 931–945. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.; Ohashi, A.; Gutierrez, M.; Oikawa, H.; Oguma, T.; Vu, D.; Takahashi, T.; Orii, T.; Sly, W. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum. Mol. Genet. 2008, 17, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Ohashi, A.; Oikawa, H.; Oguma, T.; Orii, T.; Barrera, L.; Sly, W.S. Enhancement of drug delivery: enzyme-replacement therapy for murine Morquio A syndrome. Mol. Ther. 2010, 18, 1094–1102. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oikawa, H.; Giugliani, R.; Harmatz, P.; Smith, M.; Suzuki, Y.; Orii, T. Impairment of Body Growth in Mucopolysaccharidoses. In Handbook of Growth and Growth Monitoring in Health and Disease; Preedy, V.R., Ed.; Springer: New York, NY, USA, 2012; pp. 2091–2117. ISBN 978-1-4419-1795-9. [Google Scholar]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef]
- Almassi, G.H. Algahim Current and emerging management options for patients with Morquio A syndrome. TCRM 2013, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.H.; Sawamoto, K.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; Tomatsu, S. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. J. Hum. Genet. 2019, 64, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Wraith, J.E.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Sidman, M.; Kakkis, E.D.; et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009, 123, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Ceravolo, F.; Mascaro, I.; Sestito, S.; Pascale, E.; Lauricella, A.; Dizione, E.; Concolino, D. Home treatment in paediatric patients with Hunter syndrome: The first Italian experience. Ital. J. Pediatr. 2013, 39, 53. [Google Scholar] [CrossRef] [Green Version]
- Cox-Brinkman, J.; Timmermans, R.G.M.; Wijburg, F.A.; Donker, W.E.; van de Ploeg, A.T.; Aerts, J.M.F.G.; Hollak, C.E.M. Home treatment with enzyme replacement therapy for mucopolysaccharidosis type I is feasible and safe. J. Inherit. Metab. Dis. 2007, 30, 984. [Google Scholar] [CrossRef]
- Sly, W.S. Enzyme replacement therapy: from concept to clinical practice. Acta Paediatr. Suppl. 2002, 91, 71–78. [Google Scholar] [CrossRef]
- Anson, D.S.; McIntyre, C.; Byers, S. Therapies for neurological disease in the mucopolysaccharidoses. Curr. Gene. 2011, 11, 132–143. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Burton, B.; Fleming, T.R.; Harmatz, P.; Hughes, D.; Jones, S.A.; Lin, S.-P.; Mengel, E.; Scarpa, M.; Valayannopoulos, V.; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): A phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014, 37, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Harmatz, P.; Whitley, C.B.; Waber, L.; Pais, R.; Steiner, R.; Plecko, B.; Kaplan, P.; Simon, J.; Butensky, E.; Hopwood, J.J. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J. Pediatr. 2004, 144, 574–580. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the mucopolysaccharidoses. Rheumatology 2011, 50 Suppl. 5, v49–v59. [Google Scholar] [CrossRef] [Green Version]
- Furujo, M.; Kosuga, M.; Okuyama, T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI: 10-Year follow up. Mol Genet Metab Rep 2017, 13, 69–75. [Google Scholar] [CrossRef] [PubMed]
- McGill, J.J.; Inwood, A.C.; Coman, D.J.; Lipke, M.L.; de Lore, D.; Swiedler, S.J.; Hopwood, J.J. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age--a sibling control study. Clin. Genet. 2010, 77, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.F.; Soares, D.C.; Torres, L.C.; Leal, G.N.; Cunha, M.T.; Honjo, R.S.; Bertola, D.R.; Kim, C.A. Short Communication Impact of early enzyme-replacement therapy for mucopolysaccharidosis VI: results of a long-term follow-up of Brazilian siblings. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Horovitz, D.; Acosta, A.; Giuliani, L.; Ribeiro, E. Mucopolysaccharidosis type VI on enzyme replacement therapy since infancy: Six years follow-up of four children. Mol. Genet. Metab. Rep. 2015, 5, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Do Cao, J.; Wiedemann, A.; Quinaux, T.; Battaglia-Hsu, S.F.; Mainard, L.; Froissart, R.; Bonnemains, C.; Ragot, S.; Leheup, B.; Journeau, P.; et al. 30 months follow-up of an early enzyme replacement therapy in a severe Morquio A patient: About one case. Mol. Genet. Metab. Rep. 2016, 9, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.; Stapleton, M.; Piechnik, M.; Mason, R.W.; Mackenzie, W.G.; Yamaguchi, S.; Kobayashi, H.; Suzuki, Y.; Tomatsu, S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [Google Scholar] [CrossRef]
- Akyol, M.U.; Alden, T.D.; Amartino, H.; Ashworth, J.; Belani, K.; Berger, K.I.; Borgo, A.; Braunlin, E.; Eto, Y.; Gold, J.I.; et al. Recommendations for the management of MPS IVA: systematic evidence- and consensus-based guidance. Orphanet J. Rare Dis. 2019, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Muenzer, J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 63–72. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Berger, K.I.; Giugliani, R.; Harmatz, P.; Kampmann, C.; Mackenzie, W.G.; Raiman, J.; Villarreal, M.S.; Savarirayan, R. International guidelines for the management and treatment of Morquio A syndrome. Am. J. Med. Genet. A 2015, 167A, 11–25. [Google Scholar] [CrossRef]
- Montaño, A.M.; Tomatsu, S.; Brusius, A.; Smith, M.; Orii, T. Growth charts for patients affected with Morquio A disease. Am. J. Med. Genet. A 2008, 146A, 1286–1295. [Google Scholar] [CrossRef]
- Harmatz, P.; Giugliani, R.; Schwartz, I.V.D.; Guffon, N.; Teles, E.L.; Miranda, M.C.S.; Wraith, J.E.; Beck, M.; Arash, L.; Scarpa, M.; et al. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol. Genet. Metab. 2008, 94, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Furujo, M.; Kubo, T.; Kosuga, M.; Okuyama, T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol. Genet. Metab. 2011, 104, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Giugliani, R.; Harmatz, P.; Mengel, E.; Guffon, N.; Valayannopoulos, V.; Parini, R.; Hughes, D.; Pastores, G.M.; Lau, H.A.; et al. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Mol. Genet. Metab. 2015, 114, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Bialer, M.; Parini, R.; Martin, K.; Wang, H.; Yang, K.; Shaywitz, A.J.; Harmatz, P. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr Res. 2015, 78, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Dullenkopf, A.; Holzmann, D.; Feurer, R.; Gerber, A.; Weiss, M. Tracheal intubation in children with Morquio syndrome using the angulated video-intubation laryngoscope. Can. J. Anaesth. 2002, 49, 198–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaño, A.M.; Tomatsu, S.; Gottesman, G.S.; Smith, M.; Orii, T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J. Inherit. Metab. Dis. 2007, 30, 165–174. [Google Scholar] [CrossRef]
- Tomatsu, S.; Mackenzie, W.G.; Theroux, M.C.; Mason, R.W.; Thacker, M.M.; Shaffer, T.H.; Montano, A.M.; Rowan, D.; Sly, W.; Almeciga-Diaz, C.J.; et al. Current and emerging treatments and surgical interventions for Morquio A syndrome: A review. RRED 2012, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.G.; Chadderton, R.D.; Cowie, R.A.; Wraith, J.E.; Jenkins, J.P.R. MRI of the brain and craniocervical junction in Morquio’s disease. Neuroradiology 1997, 39, 381–385. [Google Scholar] [CrossRef]
- Hendriksz, C.; Vellodi, A.; Jones, S.; Takkele, H.; Lee, S.; Chesler, S.; Decker, C. Long Term Outcomes of a Phase 1/2, Multicenter, Open-Label, Dose-Escalation Study to Evaluate the Safety, Tolerability, and Efficacy of BMN 110 in Patients with Mucopolysaccharidosis IVA (Morquio A Syndrome). Mol. Genet. Metab. 2012, 105, S35. [Google Scholar] [CrossRef]
- Kenth, J.J.; Thompson, G.; Fullwood, C.; Wilkinson, S.; Jones, S.; Bruce, I.A. The characterisation of pulmonary function in patients with mucopolysaccharidoses IVA: A longitudinal analysis. Mol. Genet. Metab. Rep. 2019, 20, 100487. [Google Scholar] [CrossRef]
- Luzak, A.; Karrasch, S.; Thorand, B.; Nowak, D.; Holle, R.; Peters, A.; Schulz, H. Association of physical activity with lung function in lung-healthy German adults: Results from the KORA FF4 study. BMC Pulm. Med. 2017, 17, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parini, R.; Rigoldi, M.; Tedesco, L.; Boffi, L.; Brambilla, A.; Bertoletti, S.; Boncimino, A.; Del Longo, A.; De Lorenzo, P.; Gaini, R.; et al. Enzymatic replacement therapy for Hunter disease: Up to 9years experience with 17 patients. Mol. Genet. Metab. Rep. 2015, 3, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Brands, M.M.M.G.; Oussoren, E.; Ruijter, G.J.G.; Vollebregt, A.A.M.; van den Hout, H.M.P.; Joosten, K.F.M.; Hop, W.C.J.; Plug, I.; van der Ploeg, A.T. Up to five years experience with 11 mucopolysaccharidosis type VI patients. Mol. Genet. Metab. 2013, 109, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Horovitz, D.D.G.; Magalhães, T.S.P.C.; Acosta, A.; Ribeiro, E.M.; Giuliani, L.R.; Palhares, D.B.; Kim, C.A.; de Paula, A.C.; Kerstenestzy, M.; Pianovski, M.A.D.; et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol. Genet. Metab. 2013, 109, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.; Giugliani, R.; Guffon, N.; Jones, S.A.; Mengel, K.E.; Parini, R.; Matousek, R.; Hawley, S.M.; Quartel, A. Clinical outcomes in a subpopulation of adults with Morquio A syndrome: results from a long-term extension study of elosulfase alfa. Orphanet J. Rare Dis. 2017, 12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Oxygen Drop | Minimal Oxygen Saturation (%) | Apnea-Hypopnea Index | Obstructive Sleep Apnea | Forced Vital Capacity (%) | Forced Expiratory Volume in 1 Second (%) |
|---|---|---|---|---|---|---|
| 48 | 74 | 66 | 8.4 | Moderate to severe | 75 | 80 |
| 54 | 45 | 78 | 5.5 | Moderate | 80 | 85 |
| 102 | 16 | 90 | 2.3 | Normal | 82 | 89 |
| Function | Age (Months) | Patient 1 | Patient 2 |
|---|---|---|---|
| Range of motion | 30–78 | Neck and chest brace | |
| 48 | Elbow extension | Elbow extension | |
| Strength | 36 | Progressive weakness in four limbs and trunk | Pelvic girdle weakness; moderate trunk weakness |
| Protective reactions and balance | 36 | Partial protection reactions; impaired static and dynamic balance | Mild impairment in balance |
| Sitting | 84 | Difficulty in sitting upright | Independent |
| Walking | 36 | Independent for short distances on even surfaces/indoors; supervision for walking outdoors; poor endurance. | |
| 48 | Walking only indoors; outdoors—using toddler ride-on toy on even surfaces and for short distances | Independent with no assistance devices on even surfaces for short and long distances with postural compensations | |
| 66 | See aforementioned | Six-minute walk test—400 m (age-expected—573 m) | |
| 102 | Powered wheelchair | ||
| Standing up | 36 | Gower’s sign/external support | Independent, partial Gower’s sign; postural compensations |
| Stairs | 36–66 | Assistance, non-reciprocating | Handrail support |
| Offing and doffing | 48 | Partially independent | Independent |
| Drinking and eating | 66 | Partially independent | Partially independent |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barak, S.; Anikster, Y.; Sarouk, I.; Stern, E.; Eisenstein, E.; Yissar, T.; Sherr-Lurie, N.; Raas-Rothschild, A.; Guttman, D. Long-Term Outcomes of Early Enzyme Replacement Therapy for Mucopolysaccharidosis IV: Clinical Case Studies of Two Siblings. Diagnostics 2020, 10, 108. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10020108
Barak S, Anikster Y, Sarouk I, Stern E, Eisenstein E, Yissar T, Sherr-Lurie N, Raas-Rothschild A, Guttman D. Long-Term Outcomes of Early Enzyme Replacement Therapy for Mucopolysaccharidosis IV: Clinical Case Studies of Two Siblings. Diagnostics. 2020; 10(2):108. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10020108
Chicago/Turabian StyleBarak, Sharon, Yair Anikster, Ifat Sarouk, Eve Stern, Etzyona Eisenstein, Tamar Yissar, Nir Sherr-Lurie, Annick Raas-Rothschild, and Dafna Guttman. 2020. "Long-Term Outcomes of Early Enzyme Replacement Therapy for Mucopolysaccharidosis IV: Clinical Case Studies of Two Siblings" Diagnostics 10, no. 2: 108. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10020108