Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects and Ethical Approval
2.2. Genomic DNA Extraction and Targeted NGS
2.3. NGS Data Processing and Classification of Variants
2.4. Variants’ Validation
3. Results
3.1. Clinical and Molecular Analysis
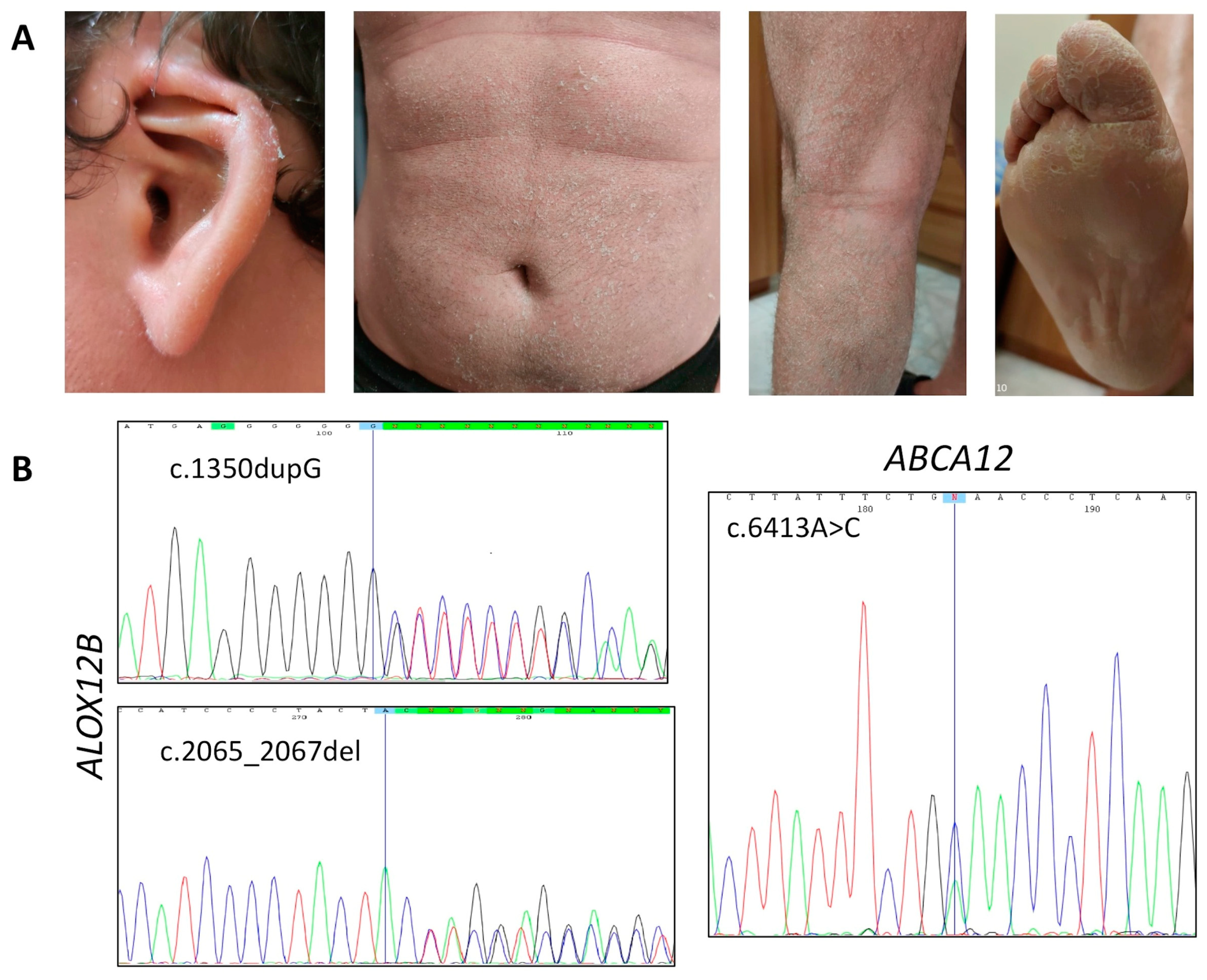
3.1.1. Affected Individual 1
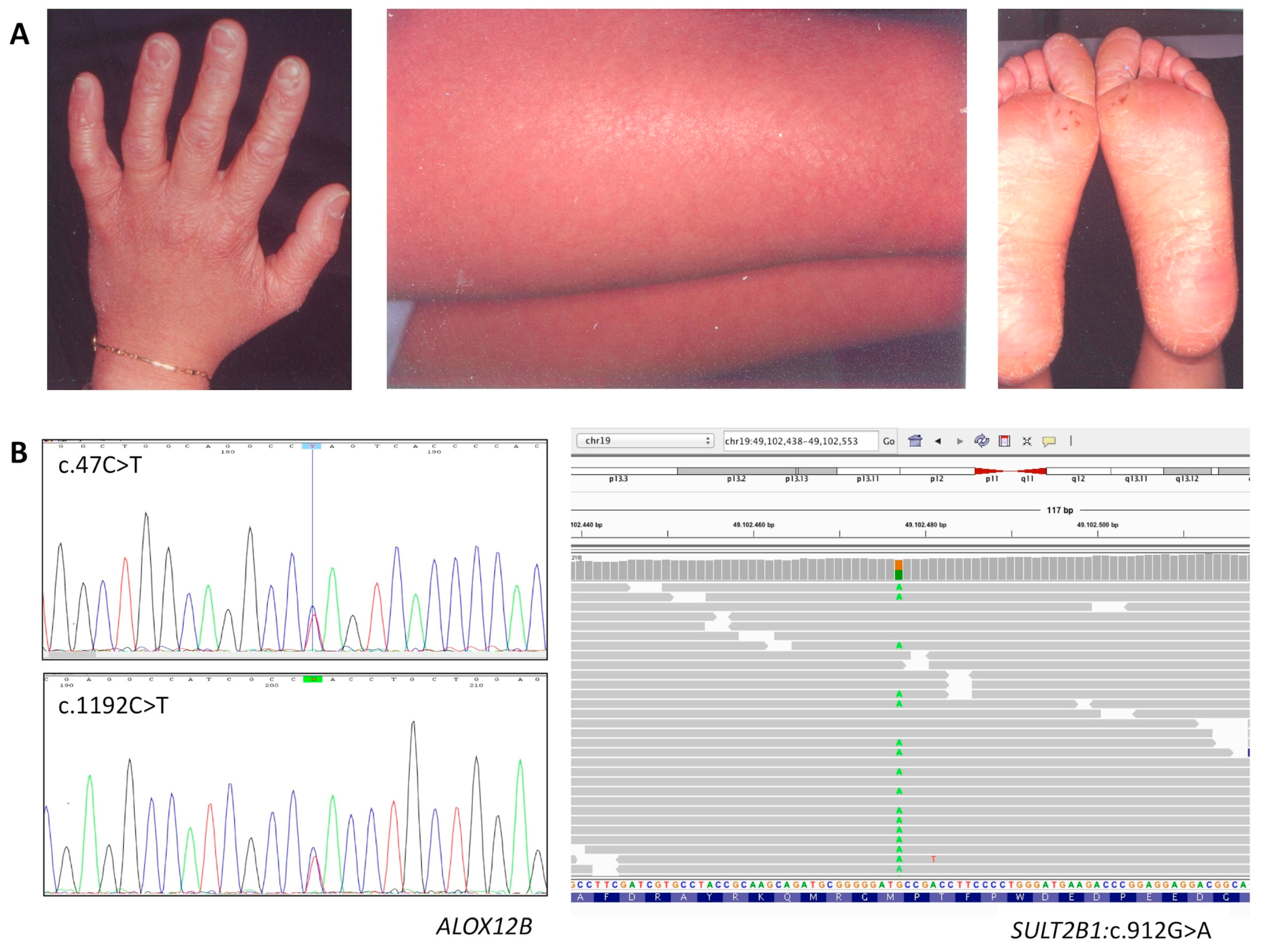
3.1.2. Affected Individual 2
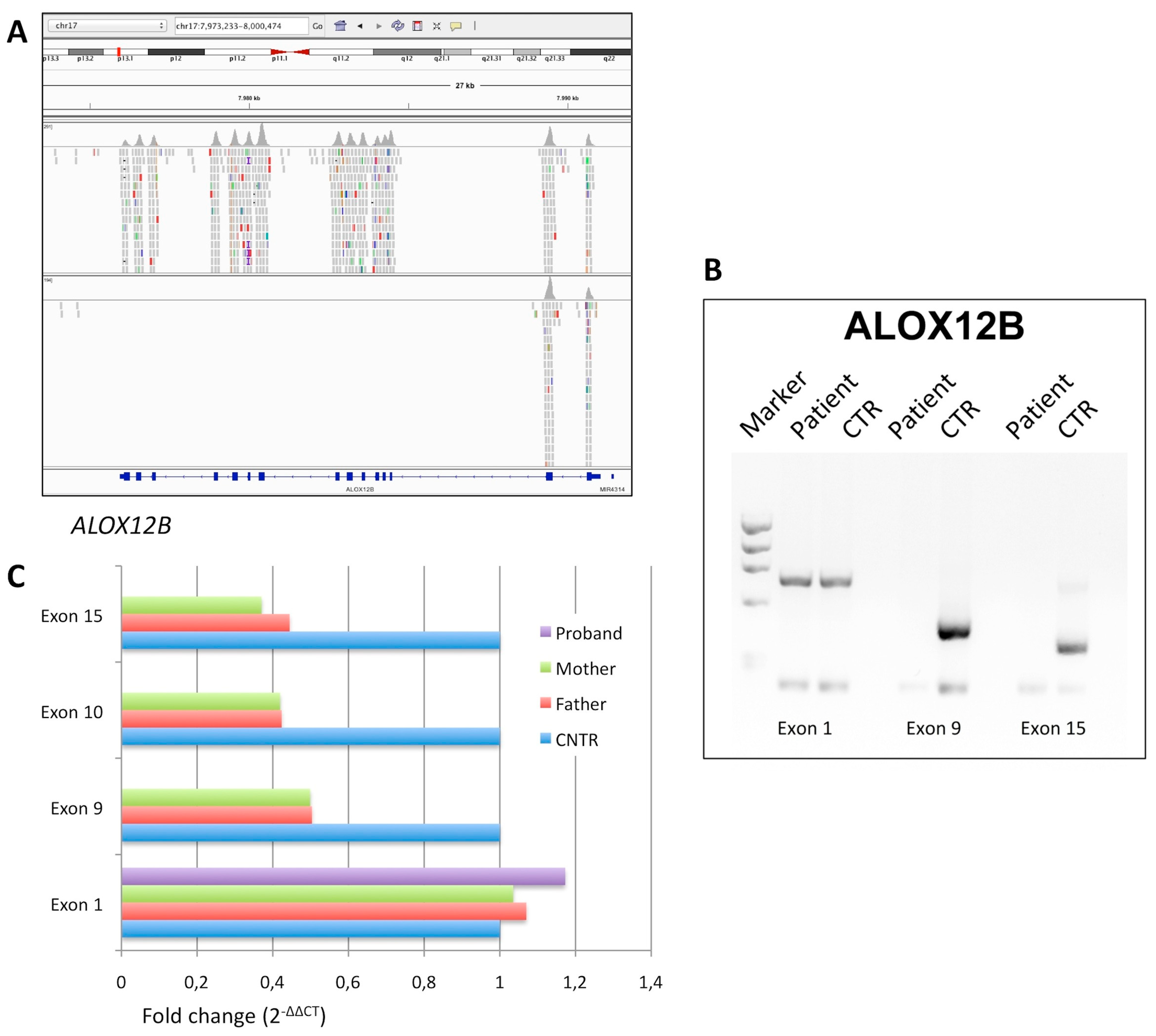
3.1.3. Affected Individual 3
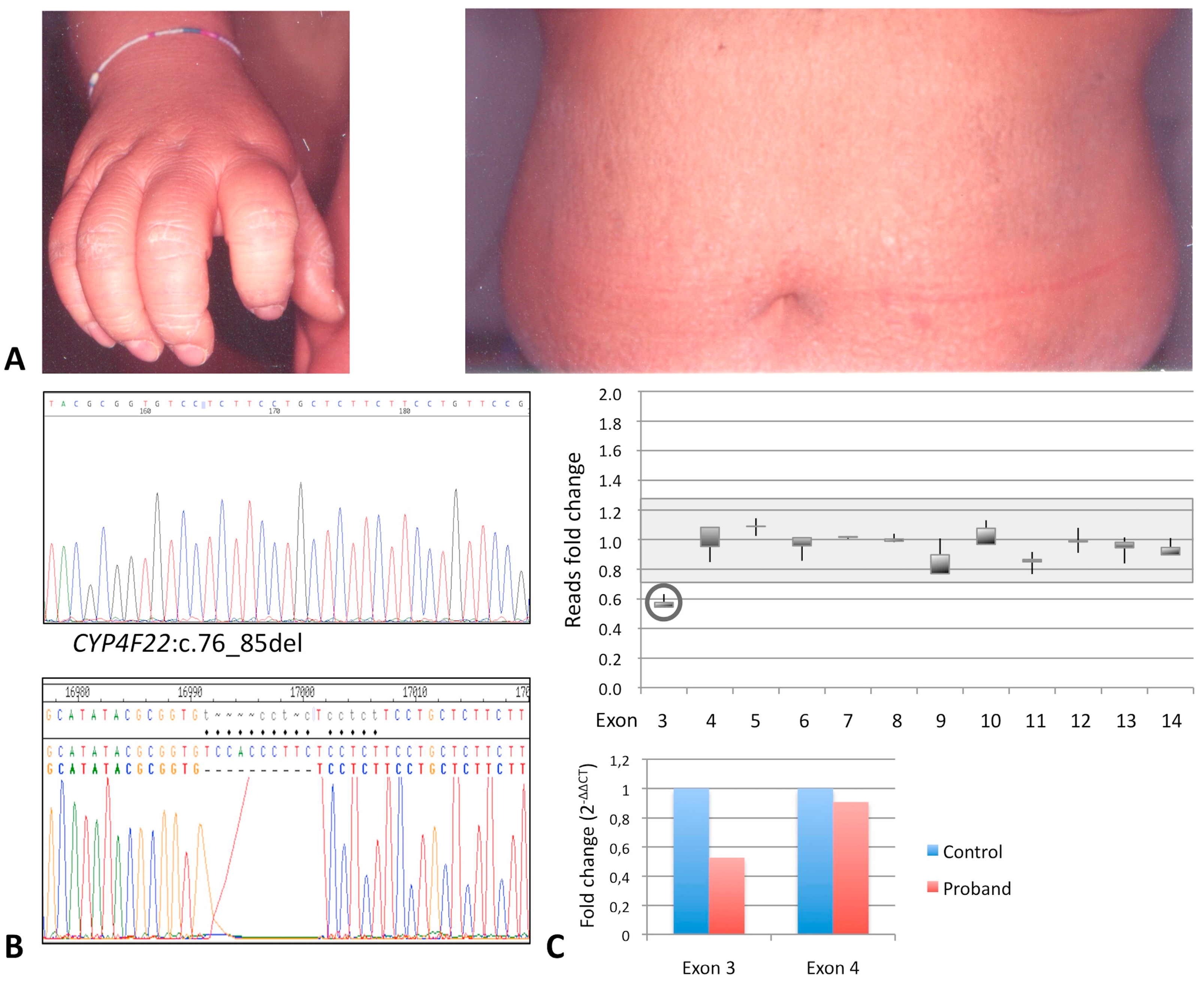
3.1.4. Affected Individual 4
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takeichi, T.; Akiyama, M. Inherited ichthyosis: Non-syndromic forms. J. Dermatol. 2016, 43, 242–251. [Google Scholar] [CrossRef]
- Uitto, J.; Youssefian, L.; Saeidian, A.H.; Vahidnezhad, H. Molecular Genetics of Keratinization Disorders—What’s New About Ichthyosis. Acta Derm.-Venereol. 2020, 100, adv00095. [Google Scholar] [CrossRef]
- Vahlquist, A.; Fischer, J.; Törmä, H. Inherited Nonsyndromic Ichthyoses: An Update on Pathophysiology, Diagnosis and Treatment. Am. J. Clin. Dermatol. 2018, 19, 51–66. [Google Scholar] [CrossRef]
- Richard, G. Autosomal Recessive Congenital Ichthyosis. 2001 Jan 10 [Updated 2017 May 18]. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1420/ (accessed on 23 November 2020).
- Oji, V.; Tadini, G.; Akiyama, M.; Blanchet Bardon, C.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; De Falco, F.; Neri, I.; Graziano, C.; Toschi, B.; Auricchio, L.; Gouveia, C.; Sousa, A.B.; Salvatore, F. Different TGM1 mutation spectra in Italian and Portuguese patients with autosomal recessive congenital ichthyosis: Evidence of founder effects in Portugal. Br. J. Dermatol. 2013, 168, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, I.; Pauwels, C.; Bourrat, E.; Bursztejn, A.C.; Maruani, A.; Chiaverini, C.; Maza, A.; Mallet, S.; Bessis, D.; Barbarot, S.; et al. Burden of inherited ichthyosis: A French national survey. Acta Derm. Venereol. 2015, 95, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hachem, M.; Abeni, D.; Diociaiuti, A.; Rotunno, R.; Gesualdo, F.; Zambruno, G.; Bodemer, C. Italian translation, cultural adaptation, and pilot testing of a questionnaire to assess family burden in inherited ichthyoses. Ital. J. Pediatr. 2019, 45, 26. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; De Falco, F.; Brazzelli, V.; Montanari, L.; Larizza, D.; Salvatore, F. Autosomal recessive congenital ichthyosis and congenital hypothyroidism in a Tunisian patient with a nonsense mutation in TGM1. J. Dermatol. Sci. 2009, 55, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.K.; Martinez-Queipo, M.; Onoufriadis, A.; Tso, S.; Glass, E.; Liu, L.; Higashino, T.; Scott, W.; Tierney, C.; Simpson, M.A.; et al. Genotype-phenotype correlation in a large English cohort of patients with autosomal recessive ichthyosis. Br. J. Dermatol. 2020, 182, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Tadini, G.; Paparo, F.; Viola, A.; Ieno, L.; Pennacchia, W.; Messina, F.; Giordano, L.; Piccirillo, A.; Auricchio, L. TGase1 deficiency and corneocyte collapse: An indication for targeted molecular screening in autosomal recessive congenital ichthyosis. Br. J. Dermatol. 2007, 157, 808–810. [Google Scholar] [CrossRef]
- Mazereeuw-Hautier, J.; Hernández-Martín, A.; O’Toole, E.A.; Bygum, A.; Amaro, C.; Aldwin, M.; Audouze, A.; Bodemer, C.; Bourrat, E.; Diociaiuti, A.; et al. Management of congenital ichthyoses: European guidelines of care, part two. Br. J. Dermatol. 2019, 180, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Mazereeuw-Hautier, J.U.; Vahlquist, A.; Traupe, H.; Bygum, A.; Amaro, C.; Aldwin, M.; Audouze, A.; Bodemer, C.; Bourrat, E.; Diociaiuti, A.; et al. Management of congenital ichthyoses: European guidelines of care, part one. Br. J. Dermatol. 2019, 180, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, L.; Roberts, R.; Tong, W. Toward Clinical Implementation of Next-Generation Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends Genet. 2019, 35, 852–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, G.; Ruggiero, R.; Savarese, M.; Savarese, G.; Tremolaterra, M.R.; Salvatore, F.; Carsana, A. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy, myotonic dystrophy type 1 and spinal muscular atrophy. Clin. Chem. Lab. Med. 2013, 51, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Vahlquist, A.; Törmä, H. Ichthyosis: A Road Model for Skin Research. Acta Derm. Venereol. 2020, 100, adv00097. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alström syndrome. Mol. Genet. Genomic Med. 2020, 8, e1260. [Google Scholar] [CrossRef]
- Di Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. EJIFCC 2018, 29, 4–14. [Google Scholar]
- Esposito, G.; Testa, F.; Zacchia, M.; Crispo, A.A.; Di Iorio, V.; Capolongo, G.; Rinaldi, L.; D’Antonio, M.; Fioretti, T.; Iadicicco, P.; et al. Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: Identification of eleven novel pathogenic sequence variants. BMC Med. Genet. 2017, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Tumienė, B.; Maver, A.; Writzl, K.; Hodžić, A.; Čuturilo, G.; Kuzmanić-Šamija, R.; Čulić, V.; Peterlin, B. Diagnostic exome sequencing of syndromic epilepsy patients in clinical practice. Clin. Genet. 2018, 93, 1057–1062. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxid. Med. Cell. Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.; et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. ACMG Laboratory Quality Assurance Committee. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Imperato, M.R.; Ieno, L.; Sorvillo, R.; Benigno, V.; Parenti, G.; Parini, R.; Vitagliano, L.; Zagari, A.; Salvatore, F. Hereditary fructose intolerance: Functional study of two novel ALDOB natural variants, and characterization of a partial gene deletion. Hum. Mutat. 2010, 31, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diociaiuti, A.; El Hachem, M.; Pisaneschi, E.; Giancristoforo, S.; Genovese, S.; Sirleto, P.; Boldrini, R.; Angioni, A. Role of molecular testing in the multidisciplinary diagnostic approach of ichthyosis. Orphanet J. Rare Dis. 2016, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Sugiura, K.; Suzuki, A.; Ichiki, T.; Akiyama, M. Apparent homozygosity due to compound heterozygosity of one point mutation and an overlapping exon deletion mutation in ABCA12: A genetic diagnostic pitfall. J. Dermatol. Sci. 2015, 80, 196–202. [Google Scholar] [CrossRef]
- Zebisch, A.; Schulz, E.; Grosso, M.; Lombardo, B.; Acierno, G.; Sill, H.; Iolascon, A. Identification of a novel variant of epsilon-gamma-delta-beta thalassemia highlights limitations of next generation sequencing. Am. J. Hematol. 2015, 90, E52–E54. [Google Scholar] [CrossRef]
- Esposito, G.; Auricchio, L.; Rescigno, G.; Paparo, F.; Salvatore, F. Transglutaminase 1 Gene Mutations in Italian Patients with Autosomal Recessive Lamellar Ichthyosis. J. Investig. Dermatol. 2001, 116, 809–812. [Google Scholar] [CrossRef]
- De Leonibus, C.; Lembo, C.; Santantonio, A.; Fioretti, T.; Rojo, S.; Salvatore, F.; De Vivo, M.; Esposito, G.; Giliberti, P. Photoletter to the editor: Lamellar ichthyosis and arthrogryposis in a premature neonate. J. Dermatol. Case Rep. 2015, 9, 49–51. [Google Scholar] [CrossRef] [Green Version]
- Vahlquist, A.; Bygum, A.; Gånemo, A.; Virtanen, M.; Hellström-Pigg, M.; Strauss, G.; Brandrup, F.; Fischer, J. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J. Investig. Dermatol. 2010, 130, 438–443. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M. ABCA12 mutations and autosomal recessive congenital ichthyosis: A review of genotype/phenotype correlations and of pathogenetic concepts. Hum. Mutat. 2010, 31, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Sugiura, K.; Aoyama, Y.; Ogawa, Y.; Hitomi, K.; Iwatsuki, K.; Akiyama, M. Novel ABCA12 missense mutation p.Phe2144Ser underlies congenital ichthyosiform erythroderma. J. Dermatol. 2013, 40, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M. Corneocyte lipid envelope (CLE), the key structure for skin barrier function and ichthyosis pathogenesis. J. Dermatol. Sci. 2017, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Crumrine, D.; Khnykin, D.; Krieg, P.; Man, M.Q.; Celli, A.; Mauro, T.M.; Wakefield, J.S.; Menon, G.; Mauldin, E.; Miner, J.H.; et al. Mutations in Recessive Congenital Ichthyoses Illuminate the Origin and Functions of the Corneocyte Lipid Envelope. J. Investig. Dermatol. 2019, 139, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Heinz, L.; Kim, G.J.; Marrakchi, S.; Christiansen, J.; Turki, H.; Rauschendorf, M.A.; Lathrop, M.; Hausser, I.; Zimmer, A.D.; Fischer, J. Mutations in SULT2B1 Cause Autosomal-Recessive Congenital Ichthyosis in Humans. Am. J. Hum. Genet. 2017, 100, 926–939. [Google Scholar] [CrossRef] [Green Version]
- Ohno, Y.; Nakamichi, S.; Ohkuni, A.; Kamiyama, N.; Naoe, A.; Tsujimura, H.; Yokose, U.; Sugiura, K.; Ishikawa, J.; Akiyama, M.; et al. Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc. Natl. Acad. Sci. USA 2015, 112, 7707–7712. [Google Scholar] [CrossRef] [Green Version]
- Hotz, A.; Bourrat, E.; Küsel, J.; Oji, V.; Alter, S.; Hake, L.; Korbi, M.; Ott, H.; Hausser, I.; Zimmer, A.D.; et al. Mutation update for CYP4F22 variants associated with autosomal recessive congenital ichthyosis. Hum. Mutat. 2018, 39, 1305–1313. [Google Scholar] [CrossRef]
- Esperón-Moldes, U.; Ginarte-Val, M.; Rodríguez-Pazos, L.; Fachal, L.; Martín-Santiago, A.; Vicente, A.; Jiménez-Gallo, D.; Guillén-Navarro, E.; Sampol, L.M.; González-Enseñat, M.A.; et al. Novel CYP4F22 mutations associated with autosomal recessive congenital ichthyosis (ARCI). Study of the CYP4F22 c.1303C>T founder mutation. PLoS ONE 2020, 15, e0229025. [Google Scholar] [CrossRef]
- Süßmuth, K.; Traupe, H.; Metze, D.; Oji, V. Ichthyoses in everyday practice: Management of a rare group of diseases. J. Dtsch. Dermatol. Ges. 2020, 18, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Carsana, A.; Frisso, G.; Intrieri, M.; Tremolaterra, M.R.; Savarese, G.; Scapagnini, G.; Esposito, G.; Santoro, L.; Salvatore, F. A 15-year molecular analysis of DMD/BMD: Genetic features in a large cohort. Front. Biosci. 2010, 2, 547–558. [Google Scholar]
- Posey, J.E. Genome sequencing and implications for rare disorders. Orphanet J. Rare Dis. 2019, 14, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Marmiesse, A.; Gouveia, S.; Couce, M.L. NGS Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Exon | Sequence | Tm (°C) | Fragment Size |
|---|---|---|---|---|
| ALOX12B | 1 | 5′-CATCCACGGCATCTTCTATC-3′ | 60.5 | 200 bp |
| 1 | 5′-AGAGATCTGGGACATGGGCG-3′ | 67.1 | ||
| 9 | 5′-CCTTCTCATACTCCCTTCTG-3′ | 56.3 | 360 bp | |
| 9 | 5′-GAAGTCCCTATGCCAAGCCC-3′ | 65.3 | ||
| 10 | 5′-CTTCAGCCCTCTCTCTTCAT-3′ | 58.5 | 449 bp | |
| 10 | 5′-TCCTCCTCTTCATCTAACTG-3′ | 54.3 | ||
| 15 | 5′-GGGATGGGGGAGGATAACTA-3′ | 62.2 | 745 bp | |
| 15 | 5′-AGAATGGGGAGAGGAGAGAC-3′ | 59.6 | ||
| CYP4F22 | 3 | 5′-TGTGCTGGGAACCTTCTGTG-3′ | 64.7 | 383 bp |
| 3 | 5′-CCTACCTATTACCCTGCACA-3′ | 57.5 |
| ID | Gene | Nucleotide Change | Protein Effect | RefSNP Database | Variant Significance |
|---|---|---|---|---|---|
| P1 | ABCA12 | c.6413A>C (het) | p.Glu2138Ala | -- | VUS |
| ABCA12 | c.7093G>A (het) | p.Asp2365Asn | rs726070 MAF = 0.028 | benign | |
| ALOX12B | c.1350dupG (het) | p.Leu451Alafs* | -- | pathogenic | |
| ALOX12B | c.2065_2067del (het) | p.Tyr687del | rs1414018786 MAF = 0.000008 | pathogenic | |
| P2 | ALOX12B | c.47C>T (het) | p.Ser16Leu | rs147784568 MAF = 0.000012 | likely pathogenic |
| ALOX12B | c.1192C>T (het) | p.His398Tyr | rs752176414 MAF = 0.000016 | likely pathogenic | |
| SULT2B1 | c.912G>A (het) | p.Met304Ile | rs142168444 MAF = 0.001 | likely benign | |
| P3 | ALOX12B | delExon3_15 (homo) | null | -- | pathogenic |
| P4 | CYP4F22 | c.76_85del (het) | p.Thr26Serfs* | -- | pathogenic |
| CYP4F22 | delExon3 (het) | null | -- | pathogenic |
| ID | Gene | Variants | Sex/Age | Phenotype | Additional Signs |
|---|---|---|---|---|---|
| P1 | ABCA12 | c.6413A>C | M 32 | CB, diffused white-gray scaling and erythema, hyperkeratosis on legs, arms and scalp, PPH | Premature birth, deformed auricles, anhidrosis, burning sensation, conductive hearing loss |
| ALOX12B | c.1350dupG/2065_2067del | ||||
| P2 | ALOX12B | c.47C>T/1192C>T | F 40 | CB, fine white scaling and mild erythema, mild keratosis on legs and arms, PPH | Hypohidrosis, skin erythema in a hot environment, frequent itching |
| SULT2B1 | c.912G>A | ||||
| P3 | ALOX12B | exon3_15del/exon3_15del | M 35 | CB; mild face erythema, palmar hyperlinearity | None |
| P4 | CYP4F22 | c.76_85del/exon3del | F 43 | CB, diffused white-gray scaling and erythema, hyperkeratosis on legs and arms, PPH | Brownish scales on trunk and back, hypohidrosis, hyperkeratosis of elbows and knees |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fioretti, T.; Auricchio, L.; Piccirillo, A.; Vitiello, G.; Ambrosio, A.; Cattaneo, F.; Ammendola, R.; Esposito, G. Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients. Diagnostics 2020, 10, 995. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10120995
Fioretti T, Auricchio L, Piccirillo A, Vitiello G, Ambrosio A, Cattaneo F, Ammendola R, Esposito G. Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients. Diagnostics. 2020; 10(12):995. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10120995
Chicago/Turabian StyleFioretti, Tiziana, Luigi Auricchio, Angelo Piccirillo, Giuseppina Vitiello, Adelaide Ambrosio, Fabio Cattaneo, Rosario Ammendola, and Gabriella Esposito. 2020. "Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients" Diagnostics 10, no. 12: 995. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10120995