Early Onset Ataxia with Comorbid Dystonia: Clinical, Anatomical and Biological Pathway Analysis Expose Shared Pathophysiology


 , ,
, ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Phenotypic Assessment of Dystonic Comorbidity in a Cohort of EOA Patients
2.1.1. EOA Database
2.1.2. Phenotypic Assessment
2.1.3. MRI Abnormalities in EOAD+ and EOAD− Subgroups
2.2. Network Analysis
2.2.1. Pathway and Network Analysis on the Study-Group (EOAD+) and Control-Group (EOAD−)
2.2.2. Pathway and Network Analysis in EOA, AOA and Dystonia Genes
2.2.3. Comparison of Shared Pathways between EOAD+ and EOA, AOA and Dystonia Gene Panels (2a versus 2b)
2.3. Statistics
3. Results
3.1. Prevalence of Comorbid Dystonia in 80 Patients with EOA
3.1.1. Clinical Characteristics of Included EOA-Patients
3.1.2. Evaluation of Comorbid Dystonia
3.1.3. Association between Phenotype and Underlying Etiology
3.1.4. Reliability of Agreement between the Observers
3.1.5. EOAD+ and EOAD− Groups and Associated MRI Abnormalities
3.2. Pathway and Network Analysis
3.2.1. EOAD+ Genotypes
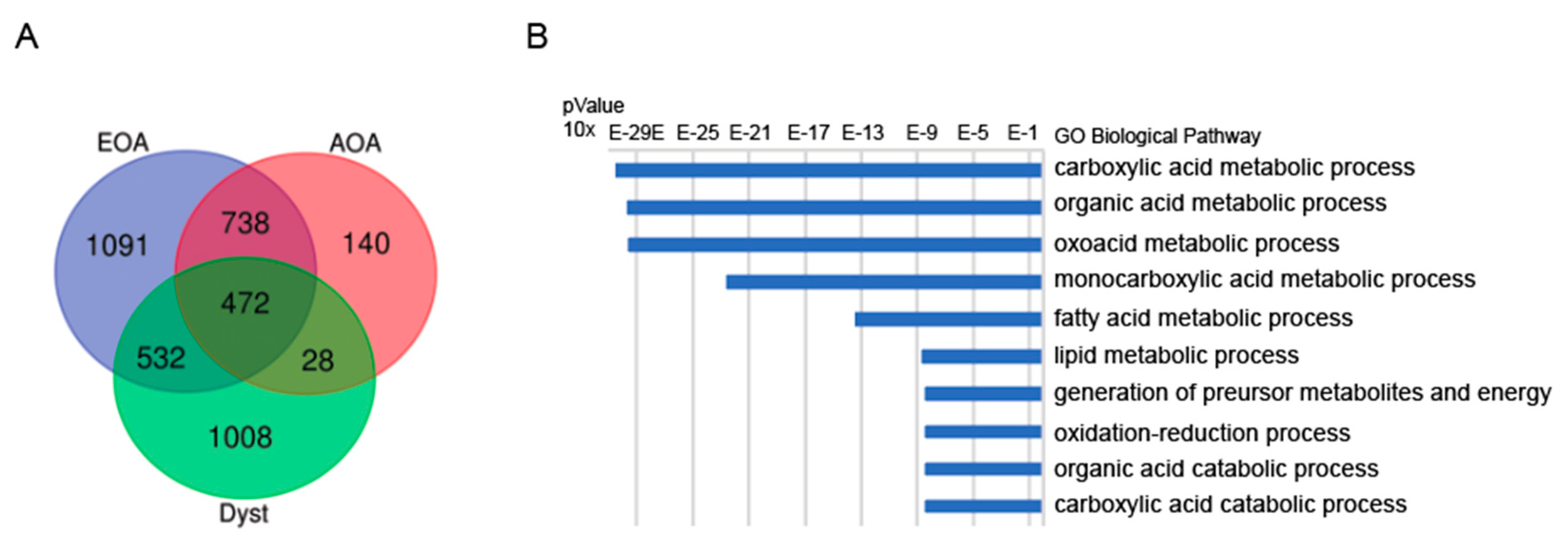
3.2.2. Shared Genes in EOA, AOA and Dystonia Gene-Lists (Panels)
3.2.3. Pathway Analysis in EOA, AOA and Dystonia Gene Lists (Panels)
3.2.4. Network Analysis in EOA, AOA, Dystonia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EOA | early onset ataxia |
| EOAD+ | early onset ataxia with dystonic comorbidity |
| EOAD− | early onset ataxia without dystonic comorbidity |
| TCA | tricarboxylic acid cycle |
| AOA | adult onset ataxia |
| AOAD+ | adult onset ataxia with dystonic comorbidity |
| SCA | spino-cerebellar ataxia |
References
- Lawerman, T.F.; Brandsma, R.; Maurits, N.M.; Martinez-Manzanera, O.; Verschuuren-Bemelmans, C.C.; Lunsing, R.J.; Brouwer, O.F.; Kremer, H.P.; Sival, D. Paediatric motor phenotypes in early-onset ataxia, developmental coordination disorder, and central hypotonia. Dev. Med. Child Neurol. 2020, 62, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, A.E. Clinical features and classification of inherited ataxias. Adv. Neurol. 1993, 61, 1–14. [Google Scholar] [PubMed]
- Nibbeling, E.A.R.; Duarri, A.; Verschuuren-Bemelmans, C.C.; Fokkens, M.R.; Karjalainen, J.M.; Smeets, C.J.; De Boer-Bergsma, J.J.; Van Der Vries, G.; Dooijes, D.; Bampi, G.B.; et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain 2017, 140, 2860–2878. [Google Scholar] [CrossRef] [PubMed]
- Prudente, C.N.; Hess, E.J.; Jinnah, H.A. Dystonia as a network disorder: What is the role of the cerebellum? Neuroscience 2014, 260, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gaalen, J.; Giunti, P.; Van De Warrenburg, B.P. Movement disorders in spinocerebellar ataxias. Mov. Disord. 2011, 26, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Tewari, A.; Fremont, R.; Khodakhah, K. It’s not just the basal ganglia: Cerebellum as a target for dystonia therapeutics. Mov. Disord. 2017, 32, 1537–1545. [Google Scholar] [CrossRef]
- Muzaimi, M.B.; Wiles, C.M.; Robertson, N.P.; Ravine, D.; Compston, D.A.S. Task specific focal dystonia: A presentation of spinocerebellar ataxia type 6. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1444–1445. [Google Scholar] [CrossRef] [Green Version]
- Saunders-Pullman, R.; Raymond, D.; Stoessl, A.J.; Hobson, D.; Nakamura, T.; Pullman, S.; Lefton, D.; Okun, M.S.; Uitti, R.; Sachdev, R.; et al. Variant ataxia-telangiectasia presenting as primary-appearing dystonia in Canadian Mennonites. Neurology 2012, 78, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Vedolin, L.M.; González, G.; Souza, C.F.; Lourenço, C.; Barkovich, A.J. Inherited Cerebellar Ataxia in Childhood: A Pattern-Recognition Approach Using Brain MRI. Am. J. Neuroradiol. 2013, 34, 925–934. [Google Scholar] [CrossRef] [Green Version]
- Lawerman, T.F.; Brandsma, R.; Van Geffen, J.T.; Lunsing, R.J.; Burger, H.; Tijssen, M.A.J.; De Vries, J.J.; De Koning, T.J.; Sival, D. Reliability of phenotypic early-onset ataxia assessment: A pilot study. Dev. Med. Child Neurol. 2016, 58, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Hübsch, T.; Du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.-S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, P.; Takayanagi, T.; Hallett, M.; Currier, R.; Subramony, S.; Wessel, K.; Bryer, A.; Diener, H.; Massaquoi, S.; Gomez, C.; et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. J. Neurol. Sci. 1997, 145, 205–211. [Google Scholar] [CrossRef]
- Brandsma, R.R.; Spits, A.A.; Kuiper, M.M.; Lunsing, R.R.; Burger, H.; Kremer, H.H.; Sival, D. The Childhood Ataxia and Cerebellar Group Ataxia rating scales are age-dependent in healthy children. Dev. Med. Child Neurol. 2014, 56, 556–563. [Google Scholar] [CrossRef]
- Bürk, K.; Sival, D.A. Scales for the clinical evaluation of cerebellar disorders. Handb. Clin. Neurol. 2018, 154, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, R.; Lawerman, T.F.; Kuiper, M.J.; Lunsing, R.J.; Burger, H.; Sival, D.A. Reliability and discriminant validity of ataxia rating scales in early onset ataxia. Dev. Med. Child Neurol. 2017, 59, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Pers, T.H.; Karjalainen, J.M.; Chan, Y.; Westra, H.-J.; Wood, A.R.; Yang, J.; Lui, J.C.; Vedantam, S.; Gustafsson, S.; Esko, T.; et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The Measurement of Observer Agreement for Categorical Data. Biometrics 1977, 33, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweney, M.T.; Newcomb, T.M.; Swoboda, K.J. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, Alternating Hemiplegia of Childhood, Rapid-onset Dystonia-Parkinsonism, CAPOS and beyond. Pediatr. Neurol. 2015, 52, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirinzi, T.; Sciamanna, G.; Mercuri, N.B.; Pisani, A. Dystonia as a network disorder. Curr. Opin. Neurol. 2018, 31, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Calderon, D.P.; Fremont, R.; Kraenzlin, F.; Khodakhah, K. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat. Neurosci. 2011, 14, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Atai, N.A.; Ryan, S.D.; Kothary, R.; Breakefield, X.O.; Nery, F.C. Untethering the Nuclear Envelope and Cytoskeleton: Biologically Distinct Dystonias Arising from a Common Cellular Dysfunction. Int. J. Cell Biol. 2012, 2012, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibbeling, E.A.R.; Delnooz, C.C.S.; De Koning, T.J.; Sinke, R.J.; Jinnah, H.A.; Tijssen, M.A.J.; Verbeek, D.S. Using the shared genetics of dystonia and ataxia to unravel their pathogenesis. Neurosci. Biobehav. Rev. 2017, 75, 22–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreglmann, S.R.; Riederer, F.; Galovic, M.; Ganos, C.; Kägi, G.; Waldvogel, D.; Jaunmuktane, Z.; Schaller, A.; Hidding, U.; Krasemann, E.; et al. Movement disorders in genetically confirmed mitochondrial disease and the putative role of the cerebellum. Mov. Disord. 2018, 33, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neychev, V.K.; Gross, R.E.; Lehéricy, S.; Hess, E.J.; Jinnah, H.A. The functional neuroanatomy of dystonia. Neurobiol. Dis. 2011, 42, 185–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoons, E.; Tijssen, M. Pathologic changes in the brain in cervical dystonia pre- and post-mortem—A commentary with a special focus on the cerebellum. Exp. Neurol. 2013, 247, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Raethjen, J.; Deuschl, G. The oscillating central network of Essential tremor. Clin. Neurophysiol. 2012, 123, 61–64. [Google Scholar] [CrossRef]
- Teo, J.T.; Van De Warrenburg, B.; Schneider, S.; Rothwell, J.; Bhatia, K. Neurophysiological evidence for cerebellar dysfunction in primary focal dystonia. J. Neurol. Neurosurg. Psychiatry 2009, 80, 80–83. [Google Scholar] [CrossRef]
- Hubsch, C.; Vidailhet, M.; Rivaud-Péchoux, S.; Pouget, P.; Brochard, V.; Degos, B.; Pélisson, D.; Golmard, J.-L.; Gaymard, B.; Roze, E. Impaired saccadic adaptation in DYT11 dystonia. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1103–1106. [Google Scholar] [CrossRef]
- Mori, F.; Okada, K.-I.; Nomura, T.; Kobayashi, Y. The Pedunculopontine Tegmental Nucleus as a Motor and Cognitive Interface between the Cerebellum and Basal Ganglia. Front. Neuroanat. 2016, 10, 109. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Hubsch, T.; Coudert, M.; Bauer, P.; Giunti, P.; Globas, C.; Baliko, L.; Filla, A.; Mariotti, C.; Rakowicz, M.; Charles, P.; et al. Spinocerebellar ataxia types 1, 2, 3, and 6: Disease severity and nonataxia symptoms. Neurology 2008, 71, 982–989. [Google Scholar] [CrossRef]
- Mariotti, C.; Alpini, D.; Fancellu, R.; Soliveri, P.; Grisoli, M.; Ravaglia, S.; Lovati, C.; Fetoni, V.; Giaccone, G.; Castucci, A.; et al. Spinocerebellar ataxia type 17 (SCA17): Oculomotor phenotype and clinical characterization of 15 Italian patients. J. Neurol. 2007, 254, 1538–1546. [Google Scholar] [CrossRef]
- Hagenah, J.M.; Zühlke, C.; Hellenbroich, Y.; Heide, W.; Klein, C. Focal dystonia as a presenting sign of spinocerebellar ataxia 17. Mov. Disord. 2004, 19, 217–220. [Google Scholar] [CrossRef]
- Estrada, R.; Galarraga, J.; Orozco, G.; Nodarse, A.; Auburger, G. Spinocerebellar ataxia 2 (SCA2): Morphometric analyses in 11 autopsies. Acta Neuropathol. 1999, 97, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, M.J.; Vrijenhoek, L.; Brandsma, R.; Lunsing, R.J.; Burger, H.; Eggink, H.; Peall, K.J.; Contarino, M.F.; Speelman, J.D.; Tijssen, M.A.J.; et al. The Burke-Fahn-Marsden Dystonia Rating Scale is Age-Dependent in Healthy Children. Mov. Disord. Clin. Pr. 2016, 3, 580–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, M.; Brandsma, R.; Vrijenhoek, L.; Tijssen, M.; Burger, H.; Dan, B.; Sival, D.A. Physiological movement disorder-like features during typical motor development. Eur. J. Paediatr. Neurol. 2018, 22, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Matilla-Dueñas, A.; Ashizawa, T.; Brice, A.; Magri, S.; McFarland, K.N.; Pandolfo, M.; Pulst, S.M.; Riess, O.; Rubinsztein, D.C.; Schmidt, T.H.; et al. Consensus Paper: Pathological Mechanisms Underlying Neurodegeneration in Spinocerebellar Ataxias. Cerebellum 2014, 13, 269–302. [Google Scholar] [CrossRef] [Green Version]
- Smeets, C.; Verbeek, D.S. Cerebellar ataxia and functional genomics: Identifying the routes to cerebellar neurodegeneration. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2030–2038. [Google Scholar] [CrossRef] [Green Version]
- Jinnah, H.A.; Sun, Y.V. Dystonia genes and their biological pathways. Neurobiol. Dis. 2019, 129, 159–168. [Google Scholar] [CrossRef]
- Casper, C.; Kalliolia, E.; Warner, T.T. Recent advances in the molecular pathogenesis of dystonia-plus syndromes and heredodegenerative dystonias. Curr. Neuropharmacol. 2013, 11, 30–40. [Google Scholar]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Synofzik, M.; Helbig, K.L.; Harmuth, F.; Deconinck, T.; Tanpaiboon, P.; Sun, B.; Guo, W.; Wang, R.; Palmaer, E.; Tang, S.; et al. De novo ITPR1 variants are a recurrent cause of early-onset ataxia, acting via loss of channel function. Eur. J. Hum. Genet. 2018, 26, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, R.; Zhong, H.; Li, X.; Ma, Y.; Zhang, R.; Wang, L.; Zang, Z.; Fan, X. Abnormal Cerebellar Development Is Involved in Dystonia-Like Behaviors and Motor Dysfunction of Autistic BTBR Mice. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.D.; Pollard, R.T.; Shelton, E.; Karki, R.; Smith-Jones, P.M.; Miao, Y. GABAA Receptor Availability Changes Underlie Symptoms in Isolated Cervical Dystonia. Front. Neurol. 2018, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Tomelleri, G.; Tonin, P.; Scarpelli, M.; Vattemi, G.; Rizzuto, N.; Padovani, A.; Simonati, A. Neuropathology of mitochondrial diseases. Biosci. Rep. 2007, 27, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Lax, N.Z.; Reeve, A.K.; Ohno, N.; Zambonin, J.L.; Blakely, E.L.; Taylor, R.W.; Bonilla, E.; Tanji, K.; DiMauro, S.; et al. Loss of Myelin-Associated Glycoprotein in Kearns-Sayre Syndrome. Arch. Neurol. 2012, 69, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Nowaczyk, M.J.M.; Wassif, C.A. Smith-lemli-opitz syndrome. In GeneReviews is a Registered Trademark of the University of Washington, Seattle; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Nóbrega, C.; Mendonça, L.; Marcelo, A.; Lamazière, A.; Tomé, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; Dunnen, W.D.; et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019, 138, 837–858. [Google Scholar] [CrossRef]
- Chen, C.H.; Fremont, R.; Arteaga-Bracho, E.E.; Khodakhah, K. Short latency cerebellar modulation of the basal ganglia. Nat. Neurosci. 2014, 17, 1767–1775. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Bourque, P.R.; Chardon, J.W.; Lelli, D.A.; Laberge, L.; Kirshen, C.; Bradshaw, S.H.; Hartley, T.; Boycott, K.M. Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol. Genet. 2018, 4, e263. [Google Scholar] [CrossRef] [Green Version]
- Deák, F.; Anderson, R.E.; Fessler, J.L.; Sherry, D.M. Novel Cellular Functions of Very Long Chain-Fatty Acids: Insight from ELOVL4 Mutations. Front. Cell. Neurosci. 2019, 13, 428. [Google Scholar] [CrossRef] [PubMed]
- Gazulla, J.; Orduna-Hospital, E.; Benavente, I.; Rodríguez-Valle, A.; Osorio-Caicedo, P.; Andrés, S.A.-D.; García-González, E.; Fraile-Rodrigo, J.; Fernández-Tirado, F.J.; Berciano, J. Contributions to the study of spinocerebellar ataxia type 38 (SCA38). J. Neurol. 2020, 267, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2011, 1811, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Heimer, G.; Kerätär, J.M.; Riley, L.G.; Balasubramaniam, S.; Eyal, E.; Pietikäinen, L.P.; Hiltunen, J.K.; Marek-Yagel, D.; Hamada, J.; Gregory, A.; et al. MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder. Am. J. Hum. Genet. 2016, 99, 1229–1244. [Google Scholar] [CrossRef]


{kind=link}
| Case | Age of Onset (Year) | Duration (Full Years) | Age at Assessment (Year) | Gene Name * | Mutation Type | Neurological Diagnosis |
|---|---|---|---|---|---|---|
| 1 | 0 | 14 | 14 | RELN | VUS | cerebel cort dyspl, hypopl pons |
| 2 | 0 | 17 | 17 | LAMA1A | MM | Poretti Boltzhausen syndrome |
| 3 | 0 | 13 | 13 | - | - | Dandy Walker malformation |
| 4 | 0 | 14 | 14 | Unknown | - | Unknown |
| 5 | 0 | 9 | 9 | SOX 10 | MM | Shah-Waardenburg syndrome |
| 6 | 0 | 22 | 22 | CHD7 | MM | CHARGE Syndrome |
| 7 | 7 | 6 | 14 | Unknown | - | Unknown |
| 8 | 0 | 7 | 7 | KIAA0586 | MM | Joubert Syndrome 23 |
| 9 | 11 | 0 | 11 | - | - | Cediak Higashi |
| 10 | 0 | 12 | 12 | SPTBN2 | Del, MM | SCA5 |
| 11 | 3 | 7 | 11 | Unknown | - | Unknown |
| 12 | 2 | 8 | 10 | FXN | GAArepeat | Friedreich’s ataxia |
| 13 | 0 | 10 | 10 | CTNNB1-gen | MM | AD MR 19 |
| 14 | 0 | 8 | 9 | KCNC3 | MM | SCA13 |
| 15 | 0 | 9 | 10 | Unknown | - | Unknown |
| 16 | 1 | 7 | 8 | HSD17B10 | MM | MHBD-deficiency |
| 17 | 4 | 5 | 9 | FXN | GAA repeat | Friedreich’s ataxia |
| 18 | 3 | 4 | 7 | EBF3 mutation | MM | HADDS syndrome |
| 19 | 4 | 1 | 5 | FXN | GAA repeat | Friedreich’s ataxia |
| 20 | 0 | 0 | 1 | INPPE5 | MM | Joubert syndrome type 1 |
| 21 | 14 | 1 | 16 | Unknown | - | Unknown |
| 22 | 5 | 6 | 11 | GOSR2 | MM | Northsea progr myocl |
| 23 | 2 | 4 | 6 | Unknown | - | Unknown |
| 24 | 0 | 5 | 5 | Unknown | - | Unknown |
| 25 | 2 | 8 | 10 | Unknown | - | Unknown |
| 26 | 13 | 2 | 15 | CACNA1A | MM | Episodic Ataxia type 2 |
| 27 | 4 | 7 | 11 | FXN | GAA repeat | Friedreich’s ataxia |
| 28 | 2 | 5 | 7 | KCND3 | MM | SCA19 |
| 29 | 6 | 8 | 14 | CACNA1A | MM | Episodic Ataxia type 2 |
| 30 | 1 | 2 | 3 | CAMTA1 | MM | CAMTA1 |
| 31 | 0 | 13 | 13 | Unknown | - | Unknown |
| 32 | 2 | 7 | 9 | TITF1 | MM | Benign Hereditary Chorea |
| 33 | 4 | 2 | 6 | ZMYND11 | MM | AD, MR type 30 |
| 34 | 1 | 12 | 13 | ITPR1 | MM | SCA 29 |
| 35 | 3 | 9 | 12 | ITPR1 | MM | SCA29 |
| 36 | 4 | 11 | 15 | ITPR1 | MM | SCA29 |
| 37 | 12 | 0 | 12 | Unknown | - | Unknown |
| 38 | 1 | 1 | 2 | Unknown | - | Unknown |
| 39 | 6 | 3 | 9 | SPTBN2 | Del, MM | SCA5 |
| 40 | 1 | 7 | 8 | ATP1A3 | MM | RDP-AHC-Atax |
| 41 | 2 | 5 | 8 | ATP1A3 | MM | AHC |
| 42 | 9 | 23 | 32 | TTPA | MM | AVED |
| 43 | 4 | 11 | 15 | FXN | GAA repeat | Friedreich’s Ataxia |
| 44 | 12 | 3 | 15 | NPC | MM | Niemann Pick |
| 45 | 12 | 22 | 34 | TTPA | MM | AVED |
| 46 | 1 | 25 | 26 | T8993G | MM | NARP |
| 47 | 16 | 11 | 28 | TTPA | MM | AVED |
| 48 | 5 | 11 | 16 | HTT | CAG repeat | Juvenile Huntington |
| 49 | 1 | 18 | 19 | ATM | MM | Ataxia Telangiectasia |
| 50 | 5 | 3 | 8 | FXN | GAA repeat | Friedreich’s Ataxia |
| 51 | 0 | 5 | 5 | - | - | cong malf fossa pos |
| 52 | 11 | 6 | 18 | mtDNA | MM | Kearns Sayre Syndrome |
| 53 | 10 | 2 | 13 | FXN | GAA repeat | Friedreich’s ataxia |
| 54 | 14 | 3 | 18 | SPG-11 | MM | HSP |
| 55 | 1 | 17 | 18 | GOSR2 | MM | Northsea progr myocl |
| 56 | 2 | 23 | 25 | GOSR2 | MM | Northsea progr myocl |
| 57 | 0 | 6 | 6 | Unknown | - | Unknown |
| 58 | 3 | 13 | 16 | CACNA1A | CAG repeat | Episodic Ataxia type 1 |
| 59 | 0 | 15 | 15 | KIAA0586 | MM | Joubert Syndrome 23 |
| 60 | 3 | 3 | 6 | GOSR2 | MM | Northsea progr myocl |
| 61 | 3 | 13 | 16 | CACNA1A | CAG repeat | Episodic Ataxia type 1 |
| 62 | 13 | 9 | 22 | TTPA | MM | AVED |
| 63 | 2 | 0 | 3 | GOSR2 | MM | Northsea progr myocl |
| 64 | 2 | 18 | 20 | GOSR2 | MM | Northsea progr myocl |
| 65 | 2 | 1 | 3 | ALDH3A2 | MM | SjogrenLarsson |
| 66 | 8 | 5 | 13 | SPG11 | MM | Spastic paraplegia 11 |
| 67 | 6 | 13 | 19 | FXN | GAA repeat | Friedreich’s Ataxia |
| 68 | 7 | 14 | 21 | FXN | GAA repeat | Friedreich’s Ataxia |
| 69 | 9 | 13 | 22 | FXN | GAA repeat | Friedreich’s Ataxia |
| 70 | 6 | 10 | 17 | FXN | GAA repeat | Friedreich’s Ataxia |
| 71 | 4 | 10 | 14 | FXN | GAA repeat | Friedreich’s Ataxia |
| 72 | 5 | 7 | 12 | FXN | GAA repeat | Friedreich’s Ataxia |
| 73 | 2 | 9 | 11 | TUBB2A | MM | CDCBM5 |
| 74 | 1 | 1 | 3 | ATP1A3 | MM | FIPWE |
| 75 | 1 | 11 | 12 | CACNA1A | CAG repeat | Episodic Ataxia type 2 |
| 76 | 7 | 5 | 12 | ATXN7 | CAG repeat | SCA7 |
| 77 | 15 | 0 | 16 | SLC2A1 gen | MM | Glut-1 def |
| 78 | 0 | 3 | 3 | Unknown | - | Unknown |
| 79 | 5 | 4 | 9 | FXN | GAA repeat | Friedreich’s ataxia |
| 80 | 6 | 10 | 16 | FXN | GAA repeat | Friedreich’s Ataxia |
| a. with comorbid dystonia | |||
| Gene mutation | |||
| TUBB2A (n = 1) | ATXN7 (n = 1) | LAMA1A (n = 1) | |
| FTX (n = 9) | KCNC3 (n = 1) | CHD7 (n = 1) | |
| INPPE5 (n = 1) | ATM (n = 1) | LYST (n = 1) | |
| ATP1A3 (n = 3) | CAMTA1 (n = 1) | HSD17B10 (n = 1) | |
| TTPA (n = 3) | NARP (n = 1) | HADDS (n = 1) | |
| CACNA1A (n = 3) | ZMYND11 (n = 1) | CTNNB1 (n = 1) | |
| GOSR2 (n = 2) | ALDH3A2 (n = 1) | HTT (n = 1) | |
| SPTBN2 (n = 2) | TITF1 (n = 1) | SPG11 (n = 1) | |
| KIAA0586 (n = 2) | NPC (n = 1) | * unknown (n = 12) | |
| b. without comorbid dystonia | |||
| Gene Mutation | |||
| KCND3 (n = 1) | CACNA1A (n = 2) | ||
| FTX (n = 3) | SPG11 (n = 1) | ||
| GOSR2 (n = 3) | ** unknown (n = 3) | ||
| ITPR1 (n = 3) | |||
| Subgroup | Most Significant Pathways | p-Value |
|---|---|---|
| EOA, Dystonia + (EOAD+) | 1. organelle organization | 8.853 × 10−17 |
| 2. cellular component organization or biogenesis | 2.315 × 10−12 | |
| 3. cellular component organization | 1.767 × 10−11 | |
| 4. chromosome organization | 7.158 × 10−8 | |
| 5. cytoskeleton organization | 3.441 × 10−7 | |
| EOA, Dystonia − (EOAD−) | 1. small molecule metabolic process | 1.091 × 10−17 |
| 2. cellular lipid metabolic process | 2.773 × 10−15 | |
| 3. lipid metabolic process | 2.866 × 10−15 | |
| 4. cellular lipid catabolic process | 6.840 × 10−15 | |
| 5. carboxylic acid metabolic process | 1.376 × 10−14 | |
| EOA, White Matter damage + (EOAW+) | 1. organelle organization | 4.102 × 10−8 |
| 2. cellular component organization | 6.759 × 10−8 | |
| 3. cellular component organization or biogenesis | 8.330 × 10−8 | |
| 4. regulation of organelle organization | 4.108 × 10−7 | |
| 5. regulation of cellular component organization | 6.354 × 10−6 | |
| EOA, White matter damage − (EOAW−) | 1. ribonucleoprotein complex biogenesis | 1.897 × 10−6 |
| 2. cellular nitrogen compound metabolic process | 3.662 × 10−6 | |
| 3. ribosome biogenesis | 8.758 × 10−6 | |
| 4. cellular component organization or biogenesis | 9.220 × 10−5 * | |
| 5. RNA processing | 1.389 × 10−4 * | |
| EOA, extracerebellar damage + (EOAX+) | 1. carboxylic acid metabolic process | 5.703 × 10−10 |
| 2. oxoacid metabolic process | 9.228 × 10−9 | |
| 3. organic acid metabolic process | 1.635 × 10−8 | |
| 4. cellular lipid catabolic process | 1.742 × 10−6 | |
| 5. vacuolar transport | 3.896 × 10−6 | |
| EOA, extracerebellar damage − (EOAX−) | 1. RNA metabolic process | 4.933 × 10−16 |
| 2. mRNA metabolic process | 1.716 × 10−14 | |
| 3. nucleic acid metabolic process | 2.453 × 10−13 | |
| 4. gene expression | 9.775 × 10−13 | |
| 5. mRNA processing | 5.158 × 10−12 | |
| EOA, cerebellar damage + (EOAC+) | 1. cellular component organization | 9.435 × 10−11 |
| 2. cellular component organization or biogenesis | 1.286 × 10−10 | |
| 3. organelle organization | 9.573 × 10−10 | |
| 4. cellular localization | 3.517 × 10−7 | |
| 5. vacuolar transport | 3.177 × 10−6 | |
| EOA, cerebellar damage − (EOAC−) | 1. No statistical significant pathways could be found. | |
| EOA, dystonia+, White matter damage + (EOAD+W+) | 1. organelle organization | 9.603 × 10−15 |
| 2. cellular component organization or biogenesis | 3.714 × 10−13 | |
| 3. cellular component organization | 1.062 × 10−12 | |
| 4. cellular localization | 2.485 × 10−8 | |
| 5. microtubule-based process | 1.631 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sival, D.A.; Garofalo, M.; Brandsma, R.; Bokkers, T.A.; van den Berg, M.; de Koning, T.J.; Tijssen, M.A.J.; Verbeek, D.S. Early Onset Ataxia with Comorbid Dystonia: Clinical, Anatomical and Biological Pathway Analysis Expose Shared Pathophysiology. Diagnostics 2020, 10, 997. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10120997
Sival DA, Garofalo M, Brandsma R, Bokkers TA, van den Berg M, de Koning TJ, Tijssen MAJ, Verbeek DS. Early Onset Ataxia with Comorbid Dystonia: Clinical, Anatomical and Biological Pathway Analysis Expose Shared Pathophysiology. Diagnostics. 2020; 10(12):997. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10120997
Chicago/Turabian StyleSival, Deborah A., Martinica Garofalo, Rick Brandsma, Tom A. Bokkers, Marloes van den Berg, Tom J. de Koning, Marina A. J. Tijssen, and Dineke S. Verbeek. 2020. "Early Onset Ataxia with Comorbid Dystonia: Clinical, Anatomical and Biological Pathway Analysis Expose Shared Pathophysiology" Diagnostics 10, no. 12: 997. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10120997