
Diagnostic and Prognostic Value of Circulating Cell-Free DNA for Cholangiocarcinoma

 , , , ,
, , , ,  ,
,
Abstract
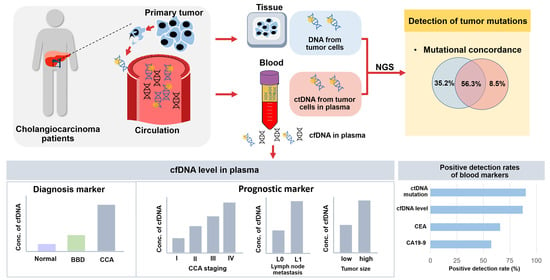
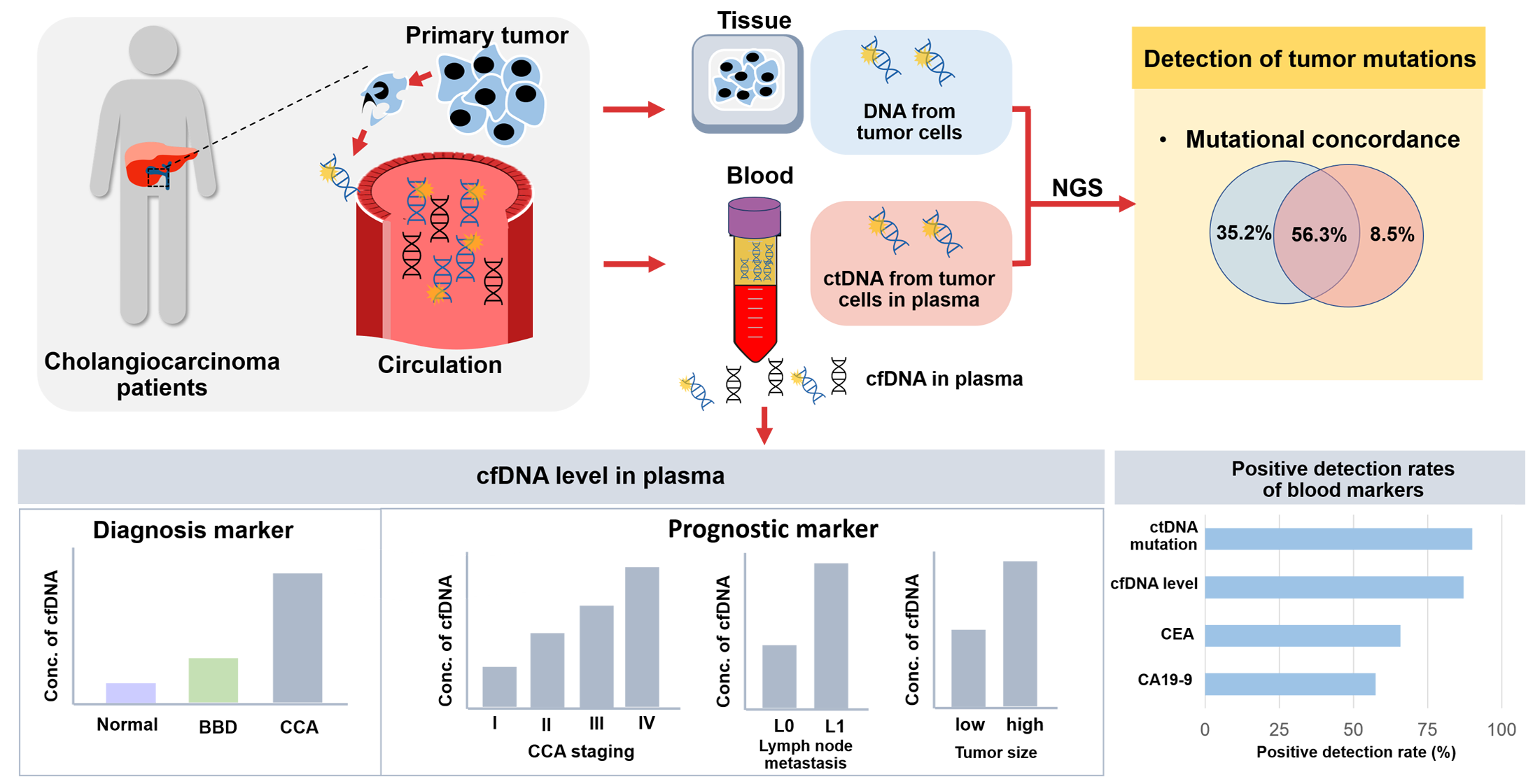
:
1. Introduction
2. Materials and Methods
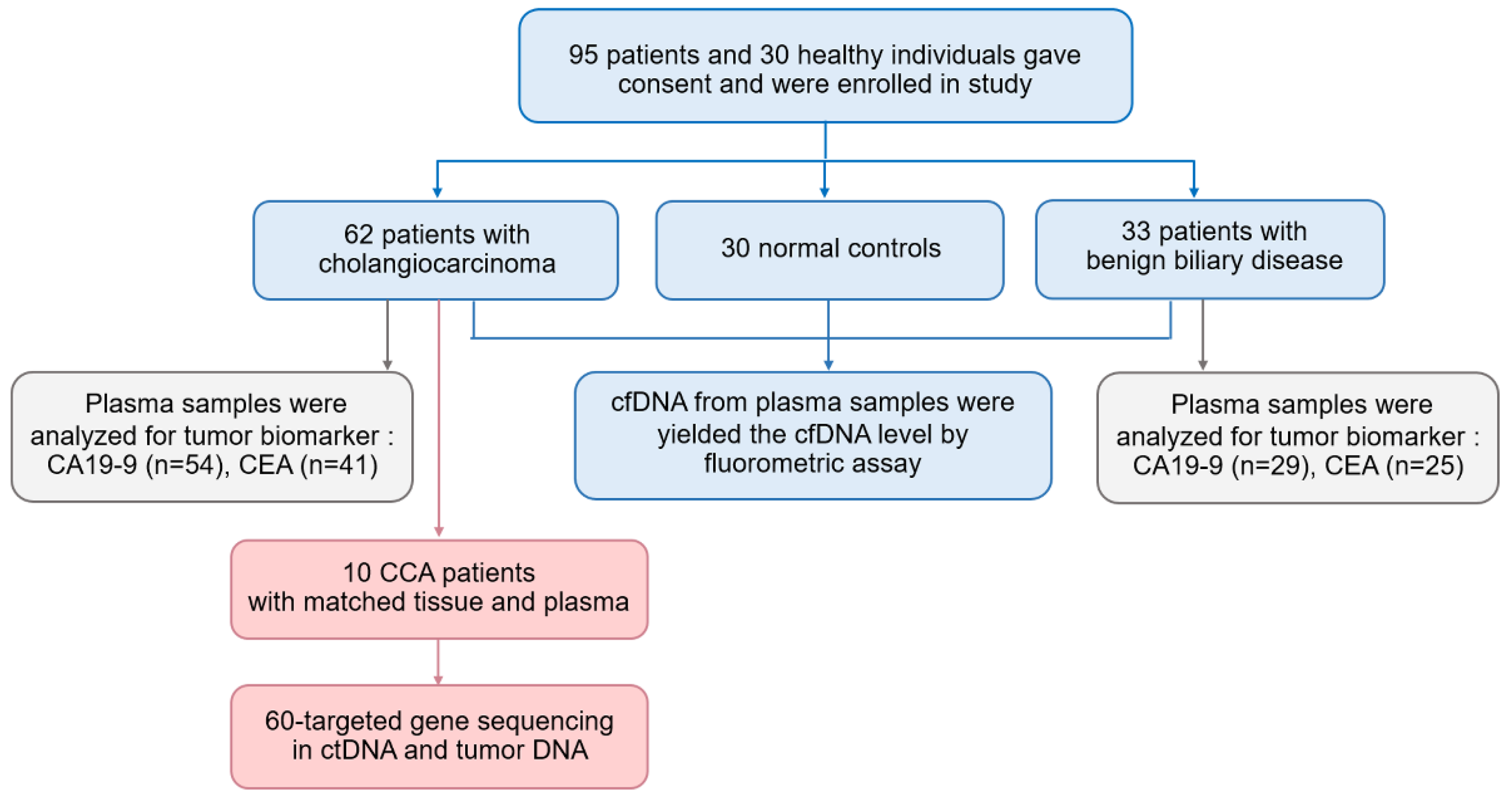
2.1. Patients and Samples
2.2. Plasma, cfDNA and Genomic DNA Isolation
2.3. Isolation of Tumor DNA from Fresh and FFPE CCA Tissues
2.4. Determination of cfDNA Level
2.5. Customization of Targeted Next-Generation Sequencing Panel
2.6. Library Preparation and Sequencing
- (i)
- Variants supported with reads from both strands, with ≥2 supporting reads, a sequencing depth of ≥200 and a variant allele frequency of ≥0.1 were reported.
- (ii)
- The p-value of the candidate somatic mutation to be <0.05.
- (iii)
- (iv)
- The variants present in more than 30% of patients and absent from the COSMIC cancer mutation database were discarded from the somatic analysis too. The final filtering step was manual examination of variant-supporting alignments with the Integrative Genomics Viewer (IGV) software [34].
2.7. Concordance between ctDNA and Tumor DNA Analysis
2.8. Statistical Analysis
3. Results
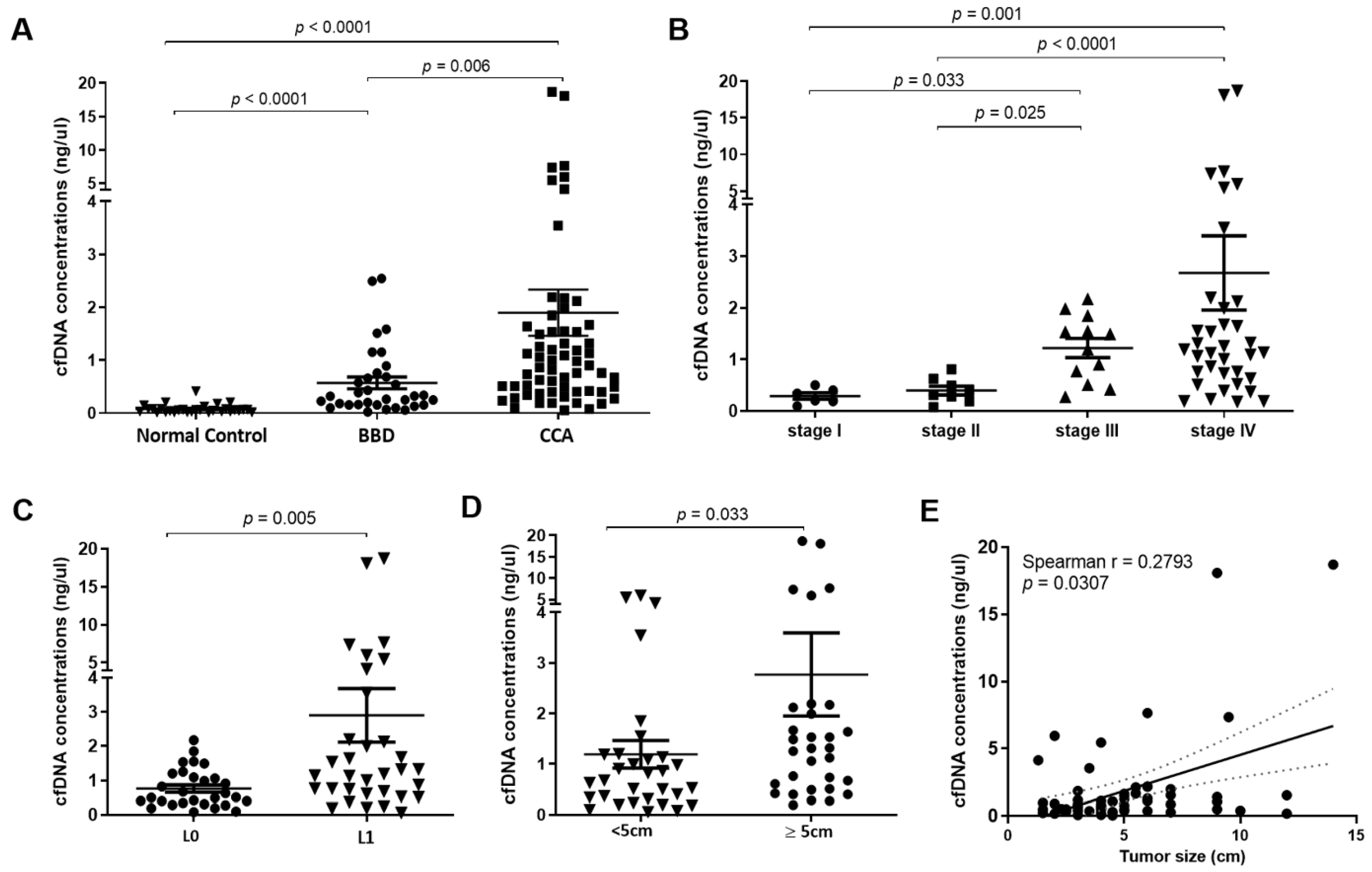
3.1. Levels of Plasma cfDNA Increased in Cholangiocarcinoma (CCA) Patients
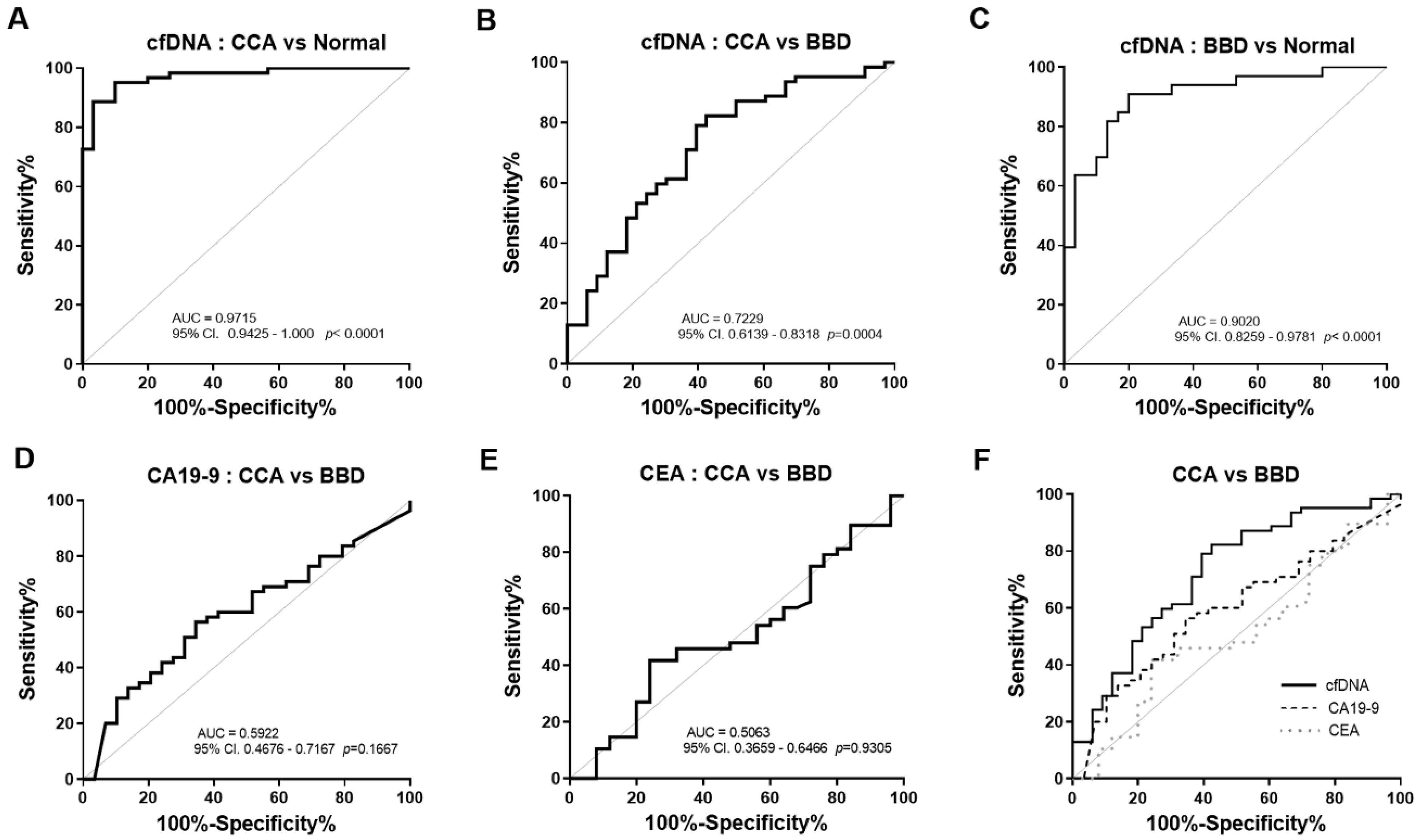
3.2. The Diagnostic Efficacy and Predictive Value of Plasma cfDNA Levels
3.3. The Diagnostic Performance of cfDNA Levels was Superior to Serum CA19-9 and CEA
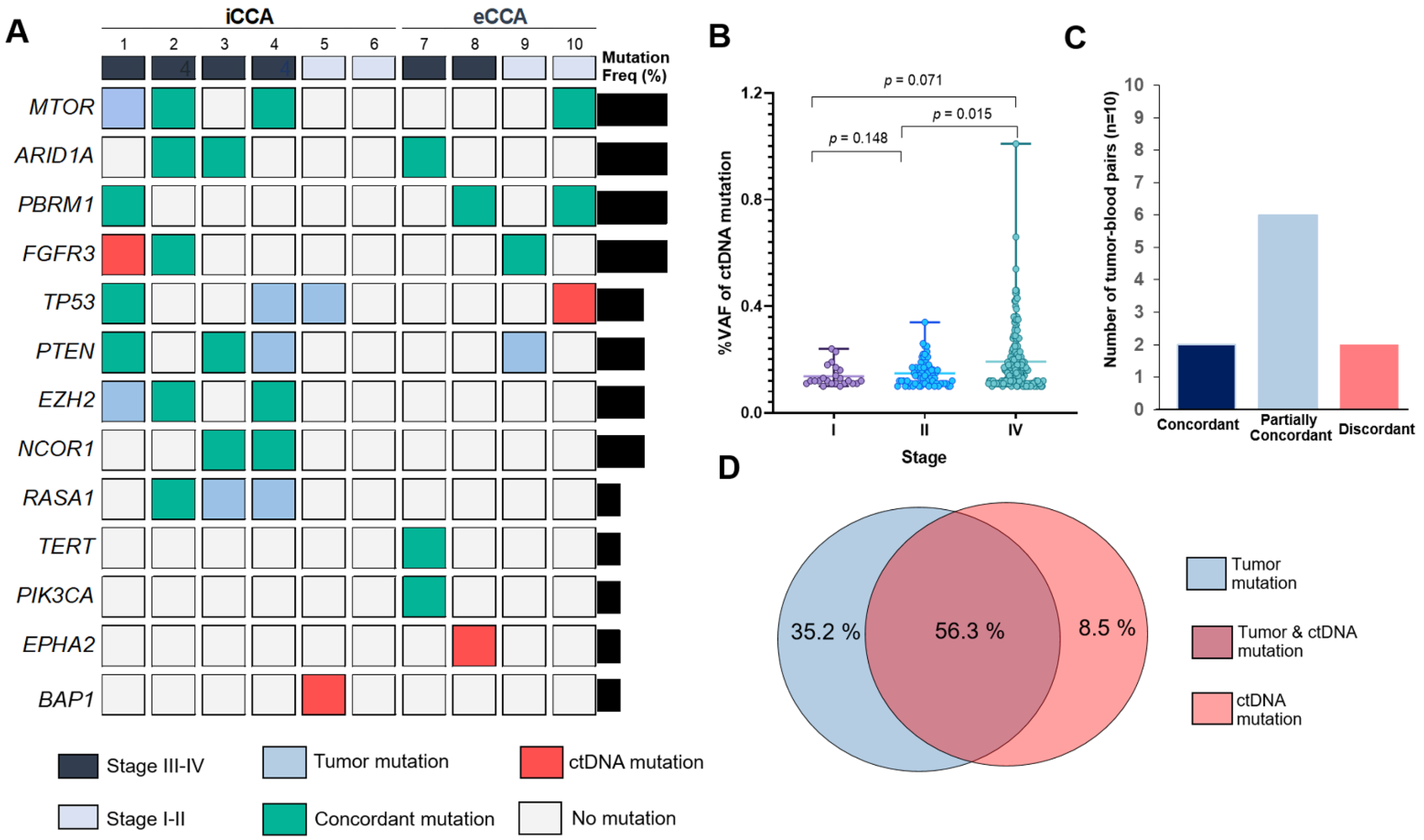
3.4. Profiling of Somatic Mutation in ctDNA and Diagnostic Sensitivity, Specificity and Accuracy for ctDNA Sequencing
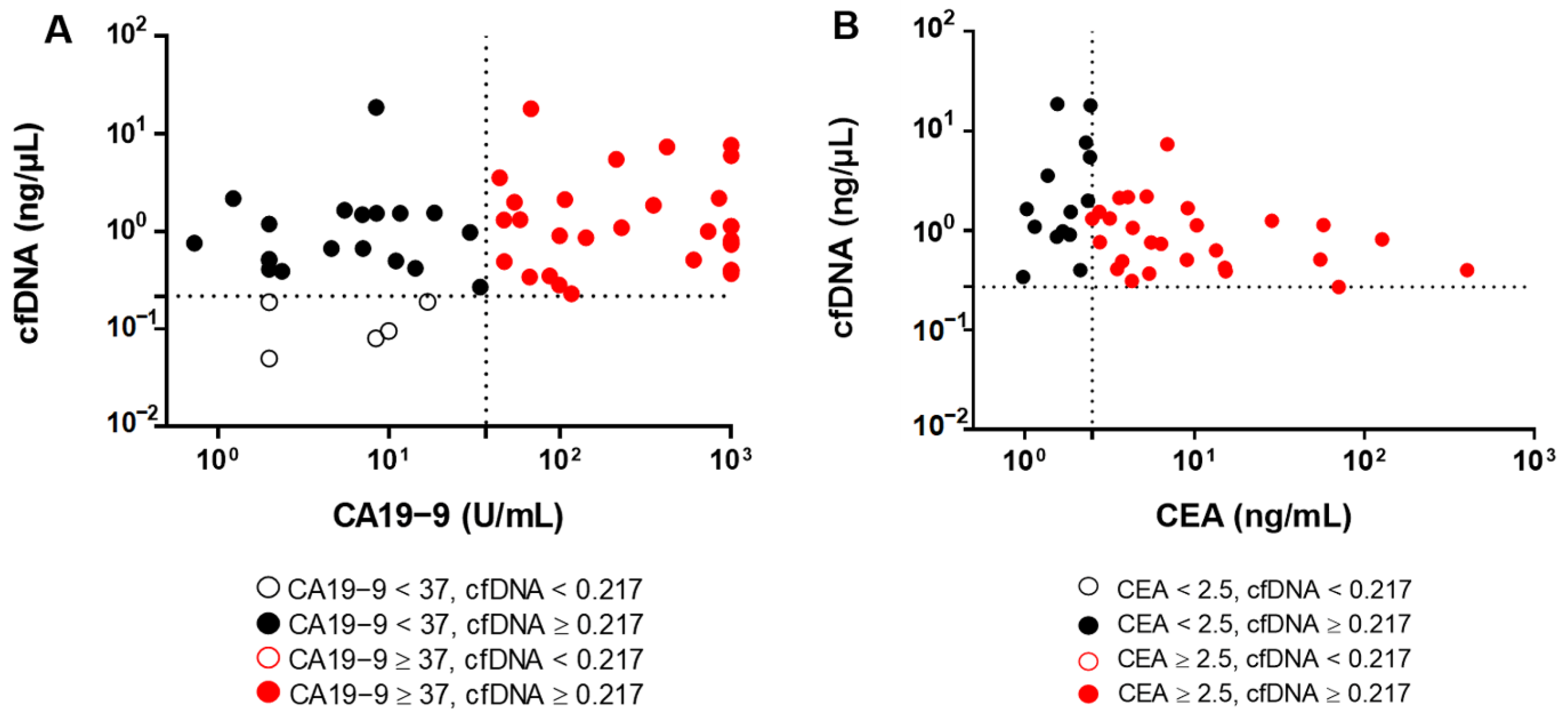
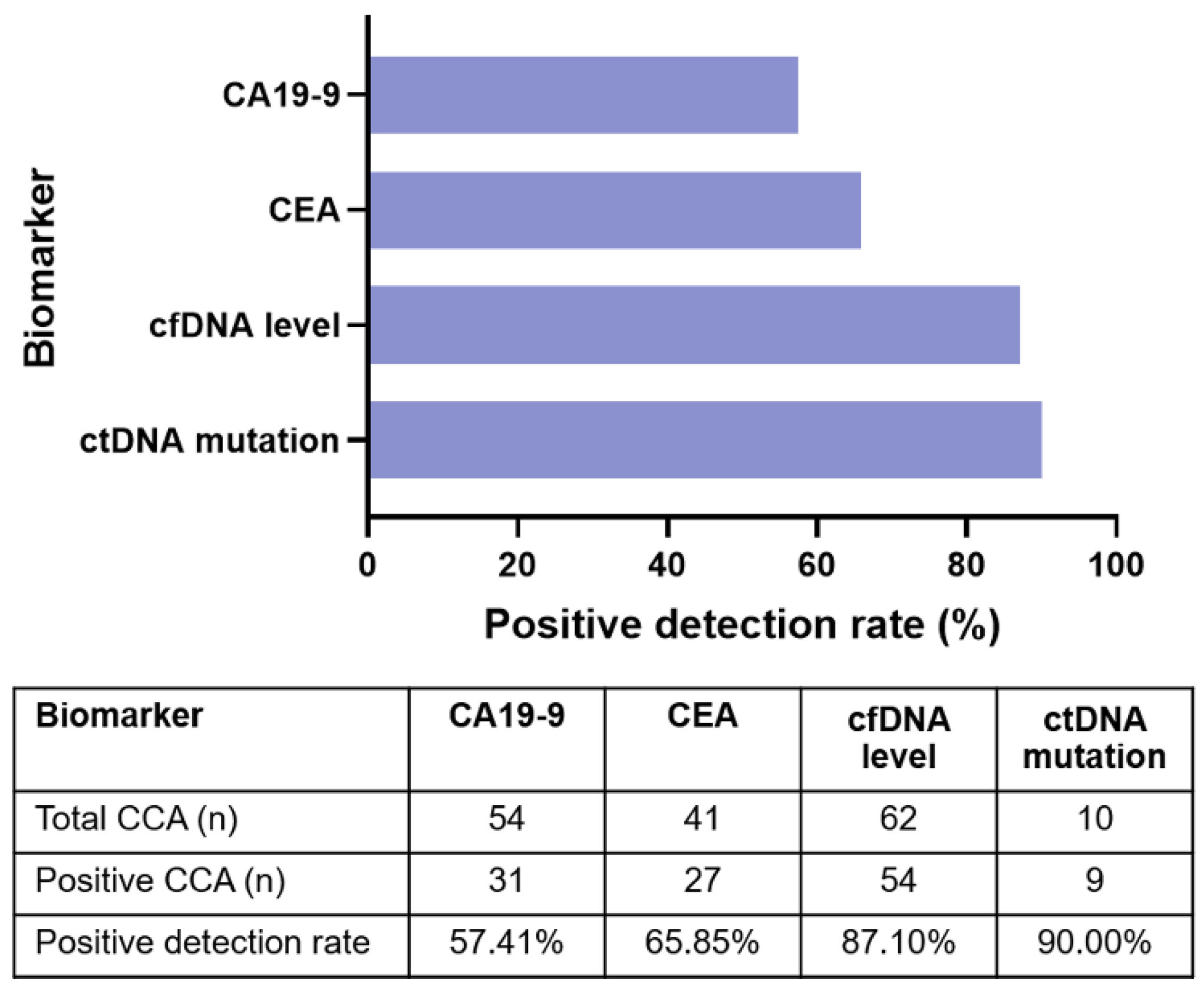
3.5. Plasma ctDNA Detection Versus Tumor Biomarkers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the ROC curve |
| CA 19-9 | Carbohydrate antigen 19-9 |
| CCA | Cholangiocarcinoma |
| cfDNA | Cell-free DNA |
| CI | Confidence interval |
| BBD | benign biliary disease |
| ctDNA | Circulating tumor DNA |
| LR | Likelihood ratio |
| NGS | Next-generation sequencing |
| NPV | Negative predictive value |
| OR | Odds ratios |
| PPV | Positive predictive value |
| ROC | Receiver operating characteristic curve |
| SD | Standard deviation |
| SN | Sensitivity |
| SP | Specificity |
| VAF | Variant allele frequency |
| VCF | Variant call format |
| YI | Youden index |
References
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut. Liver 2017, 11, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Titapun, A.; Techasen, A.; Sa-Ngiamwibool, P.; Sithithaworn, P.; Luvira, V.; Srisuk, T.; Jareanrat, A.; Dokduang, H.; Loilome, W.; Thinkhamrop, B.; et al. Serum IgG as a marker for opisthorchis viverrini-associated cholangiocarcinoma correlated with HER2 overexpression. Int. J. Gen. Med. 2020, 13, 1271–1283. [Google Scholar] [CrossRef]
- Sriamporn, S.; Pisani, P.; Pipitgool, V.; Suwanrungruang, K.; Kamsa-ard, S.; Parkin, D.M. Prevalence of opisthorchis viverrini infection and incidence of cholangiocarcinoma in Khon Kaen, Northeast Thailand. Trop. Med. Int. Health 2004, 9, 588–594. [Google Scholar] [CrossRef]
- Cambridge, W.A.; Fairfield, C.; Powell, J.J.; Harrison, E.M.; Søreide, K.; Wigmore, S.J.; Guest, R.V. Meta-analysis and meta-regression of survival after liver transplantation for unresectable perihilar cholangiocarcinoma. Ann. Surg. 2021, 273, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Kamsa-ard, S.; Luvira, V.; Suwanrungruang, K.; Kamsa-ard, S.; Luvira, V.; Santong, C.; Srisuk, T.; Pugkhem, A.; Bhudhisawasdi, V.; Pairojkul, C. Cholangiocarcinoma trends, incidence, and relative survival in Khon Kaen, Thailand from 1989 through 2013: A population-based cancer registry study. J. Epidemiol. 2019, 29, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuntikeo, N.; Chamadol, N.; Yongvanit, P.; Loilome, W.; Namwat, N.; Sithithaworn, P.; Andrews, R.H.; Petney, T.N.; Promthet, S.; Thinkhamrop, K.; et al. Cohort profile: Cholangiocarcinoma Screening and Care Program (CASCAP). BMC Cancer 2015, 15, 459. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.; Peveling-Oberhag, J.; Schulze, F.; Bon, D.; Zeuzem, S.; Friedrich-Rust, M.; Albert, J.G. Intraductal biopsies in indeterminate biliary stricture: Evaluation of histopathological criteria in fluoroscopy- vs. cholangioscopy guided technique. Dig. Liver Dis. 2016, 48, 765–770. [Google Scholar] [CrossRef]
- Brandi, G.; Venturi, M.; Pantaleo, M.A.; Ercolani, G.; Fornaro, L.; Silvestris, N.; Leone, F.; Vasile, E.; Lonardi, S.; Cereda, S.; et al. Cholangiocarcinoma: Current opinion on clinical practice diagnostic and therapeutic algorithms. Dig. Liver Dis. 2016, 48, 231–241. [Google Scholar] [CrossRef]
- Deng, Y.; Zhong, R.; Xie, X.; Xiong, X.; He, J.; Peng, L.; Zeng, H.; Duan, C. Serum CEA, CA125, CA19-9, and CA724 levels for the diagnosis and staging of cholangiocarcinoma. Biomed. Res. 2017, 28, 1413–1418. [Google Scholar]
- Loosen, S.H.; Roderburg, C.; Kauertz, K.L.; Koch, A.; Vucur, M.; Schneider, A.T.; Binnebösel, M.; Ulmer, T.F.; Lurje, G.; Schoening, W.; et al. CEA but Not CA19-9 is an independent prognostic factor in patients undergoing resection of cholangiocarcinoma. Sci. Rep. 2017, 7, 16975. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Ricci, A.D.; Tavolari, S.; Brandi, G. Circulating tumor DNA in biliary tract cancer: Current evidence and future perspectives. Cancer Genom. Proteom. 2020, 17, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative origins of cell-free DNA in humans: A review of active and passive nucleic acid release mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Chen, Y.; Zhou, J.; Zhao, H.; Zhang, H.; Wang, G. Diagnostic value of circulating cell-free DNA levels for hepatocellular carcinoma. Int. J. Infect. Dis. 2018, 67, 92–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizuka, N.; Sakaida, I.; Moribe, T.; Fujita, N.; Miura, T.; Stark, M.; Tamatsukuri, S.; Ishitsuka, H.; Uchida, K.; Terai, S.; et al. Elevated levels of circulating cell-free DNA in the blood of patients with hepatitis C virus-associated hepatocellular carcinoma. Anticancer Res. 2006, 26, 4713–4719. [Google Scholar]
- Guo, N.; Lou, F.; Ma, Y.; Li, J.; Yang, B.; Chen, W.; Ye, H.; Zhang, J.-B.; Zhao, M.-Y.; Wu, W.-J.; et al. Circulating tumor DNA detection in lung cancer patients before and after surgery. Sci. Rep. 2016, 6, 33519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zill, O.A.; Greene, C.; Sebisanovic, D.; Siew, L.M.; Leng, J.; Vu, M.; Hendifar, A.E.; Wang, Z.; Atreya, C.E.; Kelley, R.K.; et al. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov. 2015, 5, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jiang, W.; Wei, J.; Zhang, J.; Cai, L.; Luo, M.; Wang, Z.; Sun, W.; Wang, S.; Wang, C.; et al. Patient specific circulating tumor DNA fingerprints to monitor treatment response across multiple tumors. J. Transl. Med. 2020, 18, 293. [Google Scholar] [CrossRef] [PubMed]
- Ettrich, T.J.; Schwerdel, D.; Dolnik, A.; Beuter, F.; Blätte, T.J.; Schmidt, S.A.; Stanescu-Siegmund, N.; Steinacker, J.; Marienfeld, R.; Kleger, A.; et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci. Rep. 2019, 9, 13261. [Google Scholar] [CrossRef] [Green Version]
- Mody, K.; Kasi, P.M.; Yang, J.; Surapaneni, P.K.; Bekaii-Saab, T.; Ahn, D.H.; Mahipal, A.; Sonbol, M.B.; Starr, J.S.; Roberts, A.; et al. Circulating tumor DNA profiling of advanced biliary tract cancers. JCO Precis. Oncol. 2019. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: Evidence to date and future perspectives. Expert Opin. Investig. Drugs 2021, 30, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Chan-on, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Perera-Bel, J.; Hutter, B.; Heining, C.; Bleckmann, A.; Fröhlich, M.; Fröhling, S.; Glimm, H.; Brors, B.; Beißbarth, T. From somatic variants towards precision oncology: Evidence-driven reporting of treatment options in molecular tumor boards. Genome Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce Framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Wang, K. WANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D. Biliary cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Mollica, V.; Ricci, A.D.; Maggio, I.; Massucci, M.; Rojas Limpe, F.L.; Fabio, F.D.; Ardizzoni, A. Third- and later-line treatment in advanced or metastatic gastric cancer: A systematic review and meta-analysis. Future Oncol. 2019, 16, 4409–4418. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Hua, D.; Hu, Y.; Cheng, Z.; Zhou, X.; Xie, Q.; Wang, Q.; Wang, F.; Du, X.; Zeng, Y. Quantitation of plasma circulating DNA using quantitative PCR for the detection of hepatocellular carcinoma. Pathol. Oncol. Res. 2012, 18, 271–276. [Google Scholar] [CrossRef]
- Okamura, R.; Kurzrock, R.; Mallory, R.J.; Fanta, P.T.; Burgoyne, A.M.; Clary, B.M.; Kato, S.; Sicklick, J.K. Comprehensive genomic landscape and precision therapeutic approach in biliary tract cancers. Int. J. Cancer. 2021, 148, 702–712. [Google Scholar] [CrossRef]
- Noguchi, T.; Iwahashi, N.; Sakai, K.; Matsuda, K.; Matsukawa, H.; Toujima, S.; Nishio, K.; Ino, K. Comprehensive gene mutation profiling of circulating tumor DNA in ovarian cancer: Its pathological and prognostic impact. Cancers 2020, 12, 3382. [Google Scholar] [CrossRef]
- Lapin, M.; Oltedal, S.; Tjensvoll, K.; Buhl, T.; Smaaland, R.; Garresori, H.; Javle, M.; Glenjen, N.I.; Abelseth, B.K.; Gilje, B.; et al. Fragment size and level of cell-free DNA provide prognostic information in patients with advanced pancreatic cancer. J. Transl. Med. 2018, 16, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvvuri, B.; Lood, C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. Front. Immunol. 2019, 10, 502. [Google Scholar] [CrossRef] [Green Version]
- Zemmour, H.; Planer, D.; Magenheim, J.; Moss, J.; Neiman, D.; Gilon, D.; Korach, A.; Glaser, B.; Shemer, R.; Landesberg, G.; et al. Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nat. Commun. 2018, 9, 1443. [Google Scholar] [CrossRef] [PubMed]
- Neto-Neves, E.M. Cell-free DNA as a promising marker for risk stratification of pulmonary embolism. Crit. Care 2013, 17, 464. [Google Scholar] [CrossRef] [Green Version]
- Peled, M.; Agassi, R.; Czeiger, D.; Ariad, S.; Riff, R.; Rosenthal, M.; Lazarev, I.; Novack, V.; Yarza, S.; Mizrakli, Y.; et al. Cell-free DNA concentration in patients with clinical or mammographic suspicion of breast cancer. Sci. Rep. 2020, 10, 14601. [Google Scholar] [CrossRef]
- Kumari, S.; Tewari, S.; Husain, N.; Agarwal, A.; Pandey, A.; Singhal, A.; Lohani, M. Quantification of circulating free DNA as a diagnostic marker in gall bladder cancer. Pathol. Oncol. Res. 2017, 23, 91–97. [Google Scholar] [CrossRef] [PubMed]
- da Costa, W.H.; Fares, A.F.; Bezerra, S.M.; Morini, M.A.; de Toledo Benigno, L.A.; Clavijo, D.A.; Fornazieri, L.; Rocha, M.M.; da Cunha, I.W.; de Cassio Zequi, S. Loss of BAP1 expression in metastatic tumor tissue is an event of poor prognosis in patients with metastatic clear cell renal cell carcinoma. Urol. Oncol. 2019, 37, 78–85. [Google Scholar] [CrossRef]
- Namjan, A.; Techasen, A.; Loilome, W.; Sa-ngaimwibool, P.; Jusakul, A. ARID1A Alterations and their clinical significance in cholangiocarcinoma. PeerJ 2020, 8. [Google Scholar] [CrossRef]
- Luchini, C.; Robertson, S.A.; Hong, S.M.; Felsenstein, M.; Anders, R.A.; Pea, A.; Nottegar, A.; Veronese, N.; He, J.; Weiss, M.J.; et al. PBRM1 loss is a late event during the development of cholangiocarcinoma. Histopathology 2017, 71, 375–382. [Google Scholar] [CrossRef]
- Puetkasichonpasutha, J.; Namwat, N.; Sa-Ngiamwibool, P.; Titapun, A.; Suthiphongchai, T. Evaluation of P53 and its target gene expression as potential biomarkers of cholangiocarcinoma in Thai patients. Asian Pac. J. Cancer 2020, 21, 791–798. [Google Scholar] [CrossRef]
- Rizvi, S.; Borad, M.J.; Patel, T.; Gores, G.J. Cholangiocarcinoma: Molecular pathways and therapeutic opportunities. Semin. Liver Dis. 2014, 34, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Miao, D.; Margolis, C.; Martini, D.; Mullane, S.A.; Cullen, D.; Horak, C.; Wind-Rotolo, M.; Hellmann, M.D.; Voss, M.H.; Motzer, R.J.; et al. Loss-of-function of PBRM1 to predict response to anti-PD-1/PD-L1 therapy in metastatic renal cell carcinoma. JCO 2017, 35, 3016. [Google Scholar] [CrossRef]
- Cui, X.-D.; Lee, M.-J.; Kim, J.-H.; Hao, P.-P.; Liu, L.; Yu, G.-R.; Kim, D.-G. Activation of mammalian target of rapamycin complex 1 (MTORC1) and Raf/Pyk2 by growth factor-mediated Eph Receptor 2 (EphA2) is required for cholangiocarcinoma growth and metastasis. Hepatology 2013, 57, 2248–2260. [Google Scholar] [CrossRef]
- Sheng, Y.; Wei, J.; Zhang, Y.; Gao, X.; Wang, Z.; Yang, J.; Yan, S.; Zhu, Y.; Zhang, Z.; Xu, D.; et al. Mutated EPHA2 is a target for combating lymphatic metastasis in intrahepatic cholangiocarcinoma. Int. J. Cancer 2019, 144, 2440–2452. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-E.; Chen, M.-H.; Yeh, C.-N. MTOR inhibitors in advanced biliary tract cancers. Int. J. Mol. Sci. 2019, 20, 500. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Lira, M.; Deng, S.; Lee, S.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Mao, M.; Heo, J.S.; Kwon, W.; et al. PIK3CA mutation detection in metastatic biliary cancer using cell-free DNA. Oncotarget 2015, 6, 40026–40035. [Google Scholar] [CrossRef] [Green Version]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Xu, Y.; Gong, Y.; Zhang, Y.; Lu, Y.; Wang, C.; Yao, R.; Li, P.; Guan, Y.; Wang, J.; et al. Clinical factors associated with circulating tumor DNA (CtDNA) in primary breast cancer. Mol. Oncol. 2019, 13, 1033–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, I.S.; Elemento, O. Challenges in using CtDNA to achieve early detection of cancer. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | CCA | BBD | Normal Controls | p-Value |
|---|---|---|---|---|
| Gender (n) | 62 | 33 | 30 | 0.141 |
| male | 43 | 18 | 15 | |
| female | 19 | 15 | 15 | |
| Age (n) | 62 | 33 | 30 | <0.001 * |
| range | 51–81 | 34–79 | 22–61 | |
| mean ± SD (years) | 64 ± 8 | 60 ± 10 | 41 ± 11 | |
| WBC count (n) | 60 | 31 | - | 0.463 |
| range | 4.1–52.5 | 4.3–12.5 | - | |
| mean ± SD (× 103/µL) | 8.7 ± 6.5 | 7.8 ± 2.7 | - | |
| Neutrophils (n) | 60 | 31 | - | 0.152 |
| range | 39.0–85.6 | 33.6–89.9 | - | |
| mean ± SD (%) | 61.9 ± 10.9 | 58.1 ± 12.9 | - | |
| Hemoglobin (n) | 60 | 31 | - | 0.796 |
| range | 7.7–15.8 | 7.2–15.5 | - | |
| mean ± SD (g/dL) | 12.1 ± 1.6 | 12.2 ± 1.9 | - | |
| cfDNA levels (n) | 62 | 33 | 30 | 0.002 * |
| range | 0.05–18.69 | 0.02–2.54 | 0.01–0.41 | |
| mean ± SD (ng/mL) | 1.89 ± 3.43 | 0.57 ± 0.64 | 0.08 ± 0.09 | |
| CA19-9 levels (n) | 54 | 29 | - | 0.086 |
| range | 0.73–1000 | 2–1000 | - | |
| mean ± SD (U/mL) | 271.4 ± 392.1 | 139.4 ± 290.7 | - | |
| CEA levels (n) | 41 | 25 | - | 0.361 |
| range | 1.0–402.0 | 0.3–1341 | - | |
| mean ± SD (ng/mL) | 22.0 ± 65.4 | 62.0 ± 266.8 | - | |
| CCA | n (%) | |||
| Primary tumor location | ||||
| Intrahepatic CCA | 31 (50%) | |||
| Extrahepatic CCA | 27 (43.5%) | |||
| Intrahepatic and Extrahepatic CCA | 4 (6.5%) | |||
| Lymph node metastatic | ||||
| Yes | 33 (53.2%) | |||
| No | 29 (46.8%) | |||
| Tumor size | ||||
| <5 cm | 31 (50.9%) | |||
| ≥5 cm | 30 (49.1%) | |||
| Tumor stage | ||||
| stage I | 6 (9.6%) | |||
| stage II | 8 (12.9%) | |||
| stage III | 12 (18.8%) | |||
| stage IV | 36 (58.1%) | |||
| Variables | Odds Ratio | p-Value | Confidence Interval (95%) | |
|---|---|---|---|---|
| Lower | Upper | |||
| CCA vs. normal | ||||
| cfDNA | 227.86 | <0.001 | 26.73 | 1942.52 |
| age | 5.32 | <0.001 | 2.08 | 13.57 |
| sex | 2.26 | 0.074 | 0.92 | 5.55 |
| BBD vs. normal | ||||
| cfDNA | 32.86 | <0.001 | 7.65 | 141.13 |
| age | 3.45 | 0.019 | 1.23 | 9.74 |
| sex | 1.20 | 0.718 | 0.45 | 3.23 |
| CCA vs. BBD | ||||
| cfDNA | 6.29 | <0.001 | 2.44 | 16.26 |
| age | 2.33 | 0.123 | 0.80 | 6.83 |
| sex | 1.96 | 0.177 | 0.74 | 5.20 |
| Group Comparisons | Biomarkers | Cut-Off | AUC (95% CI) | YI | SN (%) | SP (%) | LR | p-Value |
|---|---|---|---|---|---|---|---|---|
| Normal vs. CCA | cfDNA level (ng/µL) | >0.2175 | 0.9715 (0.943–1.000) | 0.85 | 88.71 | 96.67 | 26.61 | <0.0001 |
| Normal vs. BBD | cfDNA level (ng/µL) | >0.0897 | 0.9020 (0.826–0.978) | 0.71 | 90.91 | 80.00 | 4.55 | <0.0001 |
| BBD vs. CCA | cfDNA level (ng/µL) | >0.3388 | 0.7229 (0.614–0.832) | 0.40 | 82.26 | 57.58 | 1.94 | 0.0004 |
| BBD vs. CCA | CA19-9 (U/mL) | >39.90 | 0.5922 (0.468–0.717) | 0.22 | 56.36 | 65.52 | 1.64 | 0.1667 |
| BBD vs. CCA | CEA (ng/mL) | >2.53 | 0.5063 (0.366–0.647) | 0.18 | 41.67 | 67.00 | 1.74 | 0.9305 |
| Comparative Diagnosis | Biomarkers | Crude | Adjusted | ||
|---|---|---|---|---|---|
| OR (95% CI) | p-Value | OR * (95% CI) | p-Value | ||
| Normal vs. CCA | cfDNA < 0.2175 vs. ≥ 0.2175 ng/µL | 227.86 (26.73–1942.52) | <0.0001 | 227.05 (25.93–1988.27) | <0.0001 |
| Normal vs. BBD | cfDNA < 0.0897 vs. ≥ 0.0897 ng/µL | 32.86 (7.65–141.13) | <0.0001 | 101.46 (10.70–961.76) | <0.0001 |
| BBD vs. CCA | cfDNA < 0.3388 vs. ≥ 0.3388 ng/µL | 6.29 (2.44–16.26) | <0.0001 | 7.649 (2.72–21.50) | <0.0001 |
| BBD vs. CCA | CA19-9 < 39.90 vs. ≥ 39.90 U/mL | 2.30 (0.94–5.62) | 0.068 | 2.18 (0.86–5.46) | 0.099 |
| BBD vs. CCA | CEA < 2.53 vs. ≥ 2.53 ng/mL | 0.61 (0.26–1.42) | 0.251 | 0.56 (0.23–1.37) | 0.204 |
| Gene | ctDNA Mutations | Tumor Mutations | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | False Positive (%) | False Negative (%) | Diagnostic Accuracy (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| + | − | |||||||||
| ARID1A | + | 3 | 0 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | 0.00 | 100.00 |
| − | 0 | 7 | ||||||||
| PBRM1 | + | 3 | 0 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | 0.00 | 100.00 |
| − | 0 | 7 | ||||||||
| NCOR1 | + | 2 | 0 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | 0.00 | 100.00 |
| − | 0 | 8 | ||||||||
| MTOR | + | 3 | 0 | 100.00 | 75.00 | 100.00 | 85.71 | 0.00 | 14.29 | 90.00 |
| − | 1 | 6 | ||||||||
| EZH2 | + | 2 | 0 | 100.00 | 66.67 | 100.00 | 87.50 | 0.00 | 12.50 | 90.00 |
| − | 1 | 7 | ||||||||
| PTEN | + | 2 | 0 | 100.00 | 66.67 | 100.00 | 87.50 | 0.00 | 12.50 | 90.00 |
| − | 1 | 7 | ||||||||
| FGFR3 | + | 2 | 1 | 87.50 | 100.00 | 66.67 | 100.00 | 33.33 | 0.00 | 90.00 |
| − | 0 | 7 | ||||||||
| RASA1 | + | 1 | 0 | 100.00 | 33.33 | 100.00 | 77.78 | 0.00 | 22.22 | 80.00 |
| − | 2 | 7 | ||||||||
| TP53 | + | 1 | 1 | 85.71 | 33.33 | 50.00 | 75.00 | 50.00 | 25.00 | 70.00 |
| − | 2 | 6 | ||||||||
| Total | + | 19 | 2 | 96.88 | 73.08 | 90.48 | 89.86 | 9.52 | 10.14 | 90.00 |
| − | 7 | 62 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wintachai, P.; Lim, J.Q.; Techasen, A.; Lert-itthiporn, W.; Kongpetch, S.; Loilome, W.; Chindaprasirt, J.; Titapun, A.; Namwat, N.; Khuntikeo, N.; et al. Diagnostic and Prognostic Value of Circulating Cell-Free DNA for Cholangiocarcinoma. Diagnostics 2021, 11, 999. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11060999
Wintachai P, Lim JQ, Techasen A, Lert-itthiporn W, Kongpetch S, Loilome W, Chindaprasirt J, Titapun A, Namwat N, Khuntikeo N, et al. Diagnostic and Prognostic Value of Circulating Cell-Free DNA for Cholangiocarcinoma. Diagnostics. 2021; 11(6):999. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11060999
Chicago/Turabian StyleWintachai, Preawwalee, Jing Quan Lim, Anchalee Techasen, Worachart Lert-itthiporn, Sarinya Kongpetch, Watcharin Loilome, Jarin Chindaprasirt, Attapol Titapun, Nisana Namwat, Narong Khuntikeo, and et al. 2021. "Diagnostic and Prognostic Value of Circulating Cell-Free DNA for Cholangiocarcinoma" Diagnostics 11, no. 6: 999. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11060999