
An Atypical Case of Head Tremor and Extensive White Matter in an Adult Female Caused by 3-Hydroxy-3-methylglutaryl-CoA Lyase Deficiency

 , , and
, , and
Abstract
:1. Introduction
2. Patient and Methods
2.1. Case Description
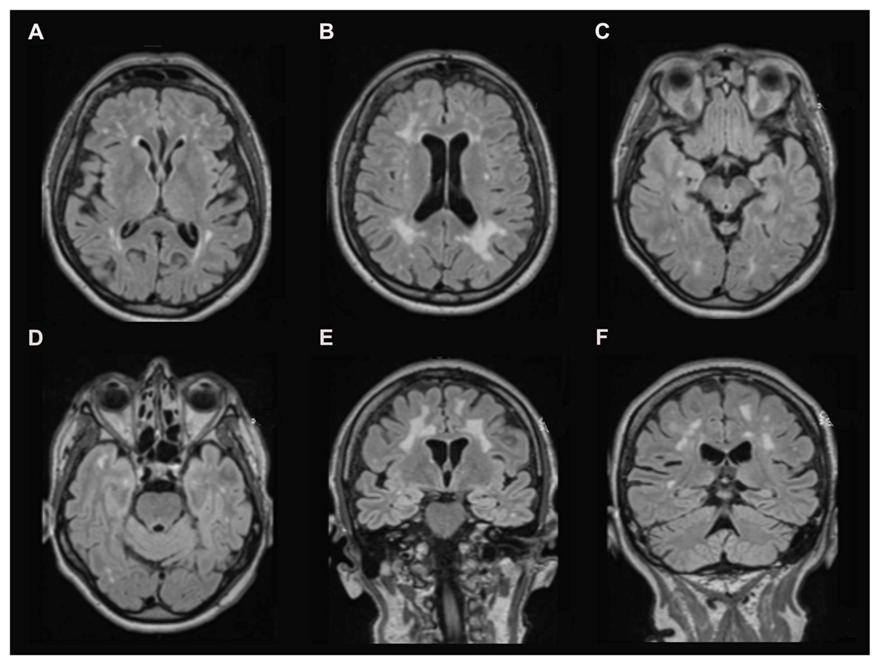
2.2. Brain Magnetic Resonance Imaging (MRI)
2.3. Biochemical Investigations
2.4. Molecular Analysis
3. Results
3.1. Brain MRI
3.2. Biochemical Investigations
3.3. Molecular Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gibson, K.M.; Breuer, J.; Nyhan, W.L. 3-hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: Review of 18 reported patients. Eur. J. Pediatr. 1988, 148, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C.; Schlatter, S.M.; Schmitt, R.N.; Gemperle-Britschgi, C.; Mrázová, L.; Balcı, M.C.; Bischof, F.; Çoker, M.; Das, A.M.; Demirkol, M.; et al. 3-hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: Clinical presentation and outcome in a series of 37 patients. Mol. Genet. Metab. 2017, 121, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.; Ketting, D.; Wadman, S.K.; Jakobs, C.; Schutgens, R.B.; Veder, H.A. Organic acid excretion in a patient with 3-hydroxy-3-methylglutaryl-coa lyase deficiency: Facts and artefacts. Clin. Chim. Acta Int. J. Clin. Chem. 1978, 90, 187–193. [Google Scholar] [CrossRef]
- Dos Santos Mello, M.; Ribas, G.S.; Wayhs, C.A.; Hammerschmidt, T.; Guerreiro, G.B.; Favenzani, J.L.; Sitta, Â.; de Moura Coelho, D.; Wajner, M.; Vargas, C.R. Increased oxidative stress in patients with 3-hydroxy-3-methylglutaric aciduria. Mol. Cell Biochem. 2015, 402, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Leipnitz, G.; Vargas, C.R.; Wajner, M. Disturbance of redox homeostasis as a contributing underlying pathomechanism of brain and liver alterations in 3-hydroxy-3-methylglutaryl-coa lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Leipnitz, G.; Seminotti, B.; Haubrich, J.; Dalcin, M.B.; Dalcin, K.B.; Solano, A.; de Bortoli, G.; Rosa, R.B.; Amaral, A.U.; Dutra-Filho, C.S.; et al. Evidence that 3-hydroxy-3-methylglutaric acid promotes lipid and protein oxidative damage and reduces the nonenzymatic antioxidant defenses in rat cerebral cortex. J. Neurosci. Res. 2008, 86, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Delgado, C.A.; Balbueno Guerreiro, G.B.; Diaz Jacques, C.E.; de Moura Coelho, D.; Sitta, A.; Manfredini, V.; Wajner, M.; Vargas, C.R. Prevention by l-carnitine of DNA damage induced by 3-hydroxy-3-methylglutaric and 3-methylglutaric acids and experimental evidence of lipid and DNA damage in patients with 3-hydroxy-3-methylglutaric aciduria. Arch. Biochem. Biophys. 2019, 668, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Matar, W.; Tolun, A.A.; Devanapalli, B.; Thompson, S.; Dalkeith, T.; Lichkus, K.; Tchan, M. The use of sodium dl-3-hydroxybutyrate in severe acute neuro-metabolic compromise in patients with inherited ketone body synthetic disorders. Orphanet. J. Rare Dis. 2020, 15, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierron, S.; Giudicelli, H.; Moreigne, M.; Khalfi, A.; Touati, G.; Caruba, C.; Rolland, M.O.; Acquaviva, C. Late onset 3-hmg-coa lyase deficiency: A rare but treatable disorder. Arch. Pediatr. 2010, 17, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Kitchen, S.; Pinto, A.; Daly, A.; Gerrard, A.; Hoban, R.; Santra, S.; Sreekantam, S.; Frost, K.; Pigott, A.; et al. 3-hydroxy-3-methylglutaryl-coa lyase deficiency: A case report and literature review. Nutr. Hosp. 2018, 35, 237–244. [Google Scholar] [PubMed]
- Bischof, F.; Nägele, T.; Wanders, R.J.; Trefz, F.K.; Melms, A. 3-hydroxy-3-methylglutaryl-coa lyase deficiency in an adult with leukoencephalopathy. Ann. Neurol. 2004, 56, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Reimão, S.; Morgado, C.; Almeida, I.T.; Silva, M.; Corte Real, H.; Campos, J. 3-hydroxy-3-methylglutaryl-coenzyme a lyase deficiency: Initial presentation in a young adult. J. Inherit. Metab. Dis. 2009, 32, S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Hajnal, J.V.; Bryant, D.J.; Kasuboski, L.; Pattany, P.M.; De Coene, B.; Lewis, P.D.; Pennock, J.M.; Oatridge, A.; Young, I.R.; Bydder, G.M. Use of fluid attenuated inversion recovery (flair) pulse sequences in mri of the brain. J. Comput. Assist Tomogr. 1992, 16, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, S.L.; Goh, T.D.; Heffernan, T.; Rowan, D. Hyperintensity in the subarachnoid space on flair mri. AJR Am. J. Roentgenol. 2007, 189, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, P.; Menao, S.; López-Viñas, E.; Santpere, G.; Clotet, J.; Sierra, A.Y.; Gratacós, E.; Puisac, B.; Gómez-Puertas, P.; Hegardt, F.G.; et al. C-terminal end and aminoacid lys48 in hmg-coa lyase are involved in substrate binding and enzyme activity. Mol. Genet. Metab. 2007, 91, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, D.; Vojinovic, S.; Aracki-Trenkic, A.; Tasic, A.; Benedeto-Stojanov, D.; Ljubisavljevic, S.; Vujnovic, S. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (cadasil). Bosn. J. Basic Med. Sci. 2015, 15, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Investigation | Metabolite | Concentration | Reference Range |
|---|---|---|---|
| Urinary Organic Acids(mmol/mol of Creatinine) | 3-Hydroxy-3-MethylGlutaric acid | 304 | <200 |
| 3-hydroxyIsovaleric acid | 143 | <50 | |
| 3-methylGlutaric acid | 54 | <5 | |
| 3-methylGlutaconic acid | 399 | <25 | |
| Blood Carnitines(µmol/L) | Carnitine | 20 | 15–35 |
| 3-hydroxyIsovalerylcarnitine | 3.73 | <0.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutouchent, N.; Bourilhon, J.; Sudrié-Arnaud, B.; Bonnevalle, A.; Guyant-Maréchal, L.; Acquaviva, C.; Dujardin-Ippolito, L.; Bekri, S.; Dabaj, I.; Tebani, A. An Atypical Case of Head Tremor and Extensive White Matter in an Adult Female Caused by 3-Hydroxy-3-methylglutaryl-CoA Lyase Deficiency. Diagnostics 2021, 11, 1561. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11091561
Boutouchent N, Bourilhon J, Sudrié-Arnaud B, Bonnevalle A, Guyant-Maréchal L, Acquaviva C, Dujardin-Ippolito L, Bekri S, Dabaj I, Tebani A. An Atypical Case of Head Tremor and Extensive White Matter in an Adult Female Caused by 3-Hydroxy-3-methylglutaryl-CoA Lyase Deficiency. Diagnostics. 2021; 11(9):1561. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11091561
Chicago/Turabian StyleBoutouchent, Nassim, Julie Bourilhon, Bénédicte Sudrié-Arnaud, Antoine Bonnevalle, Lucie Guyant-Maréchal, Cécile Acquaviva, Loréna Dujardin-Ippolito, Soumeya Bekri, Ivana Dabaj, and Abdellah Tebani. 2021. "An Atypical Case of Head Tremor and Extensive White Matter in an Adult Female Caused by 3-Hydroxy-3-methylglutaryl-CoA Lyase Deficiency" Diagnostics 11, no. 9: 1561. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11091561