
Identification of the First Single GSDME Exon 8 Structural Variants Associated with Autosomal Dominant Hearing Loss

 , , , ,
, , , ,  and
and 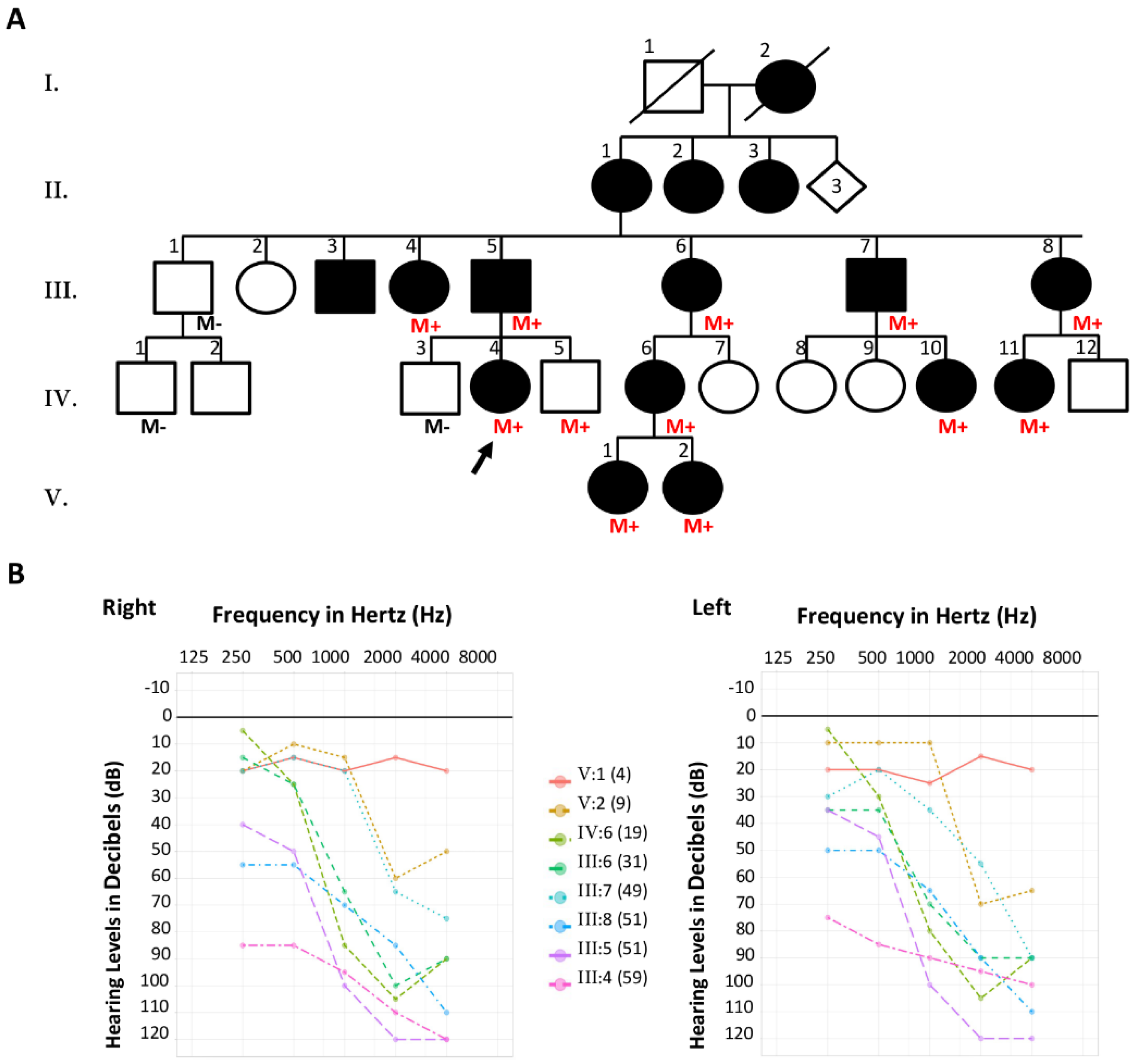{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Report of the Families
2.1.1. Family S2426
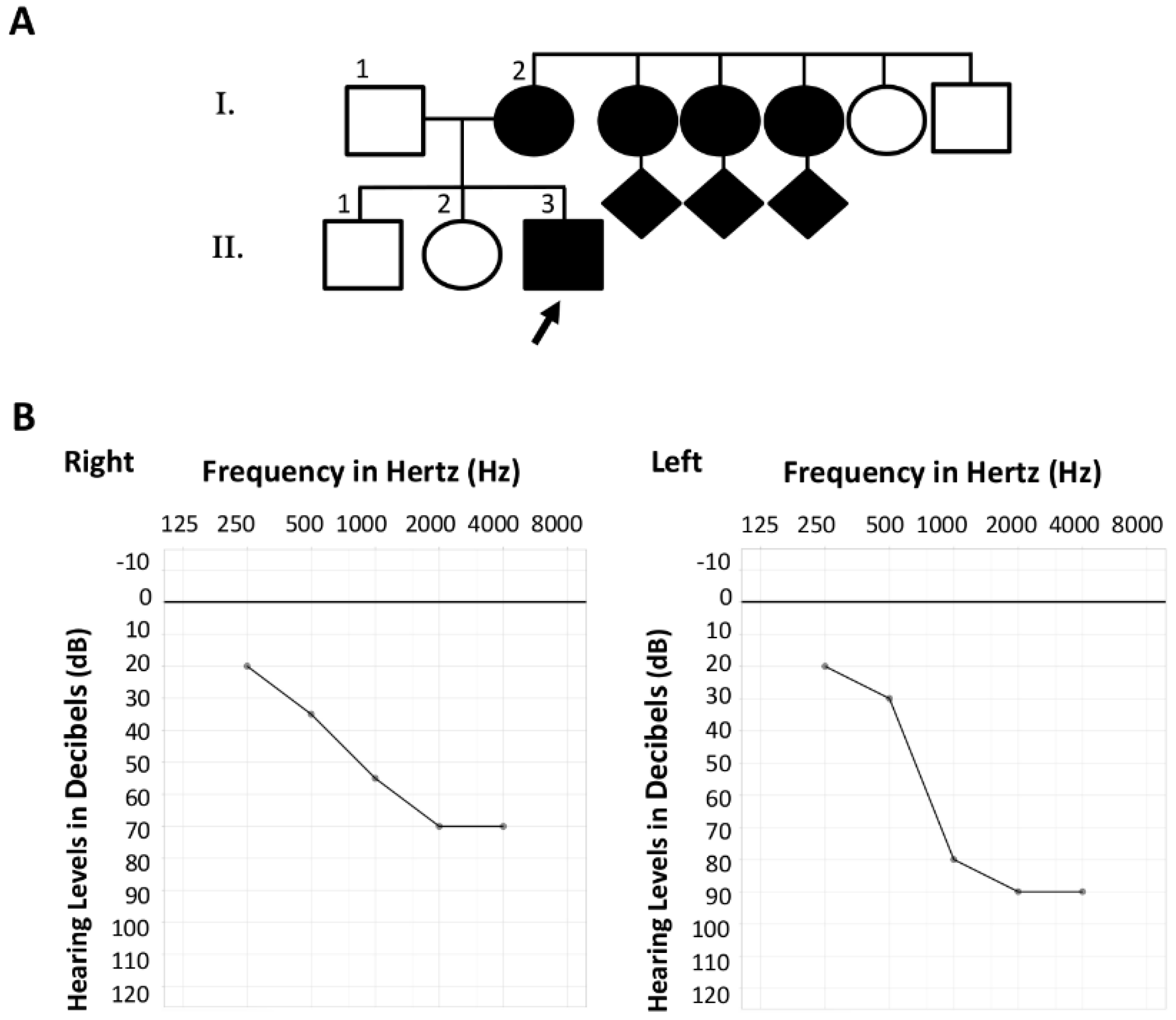
2.1.2. Family S2106
2.2. DNA Analysis
2.3. RNA Analysis
2.4. Variant Description
3. Results
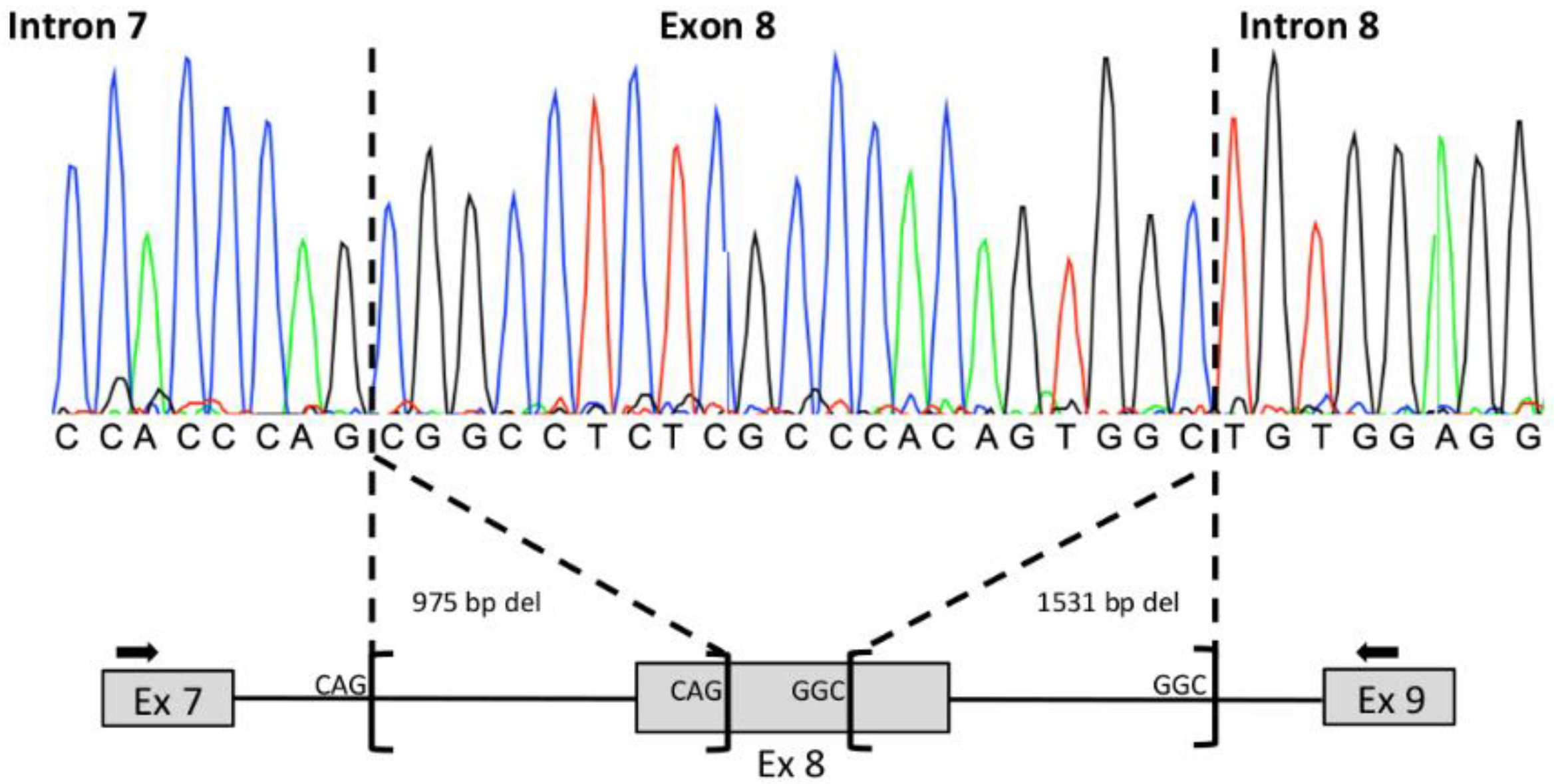
3.1. Family S2426
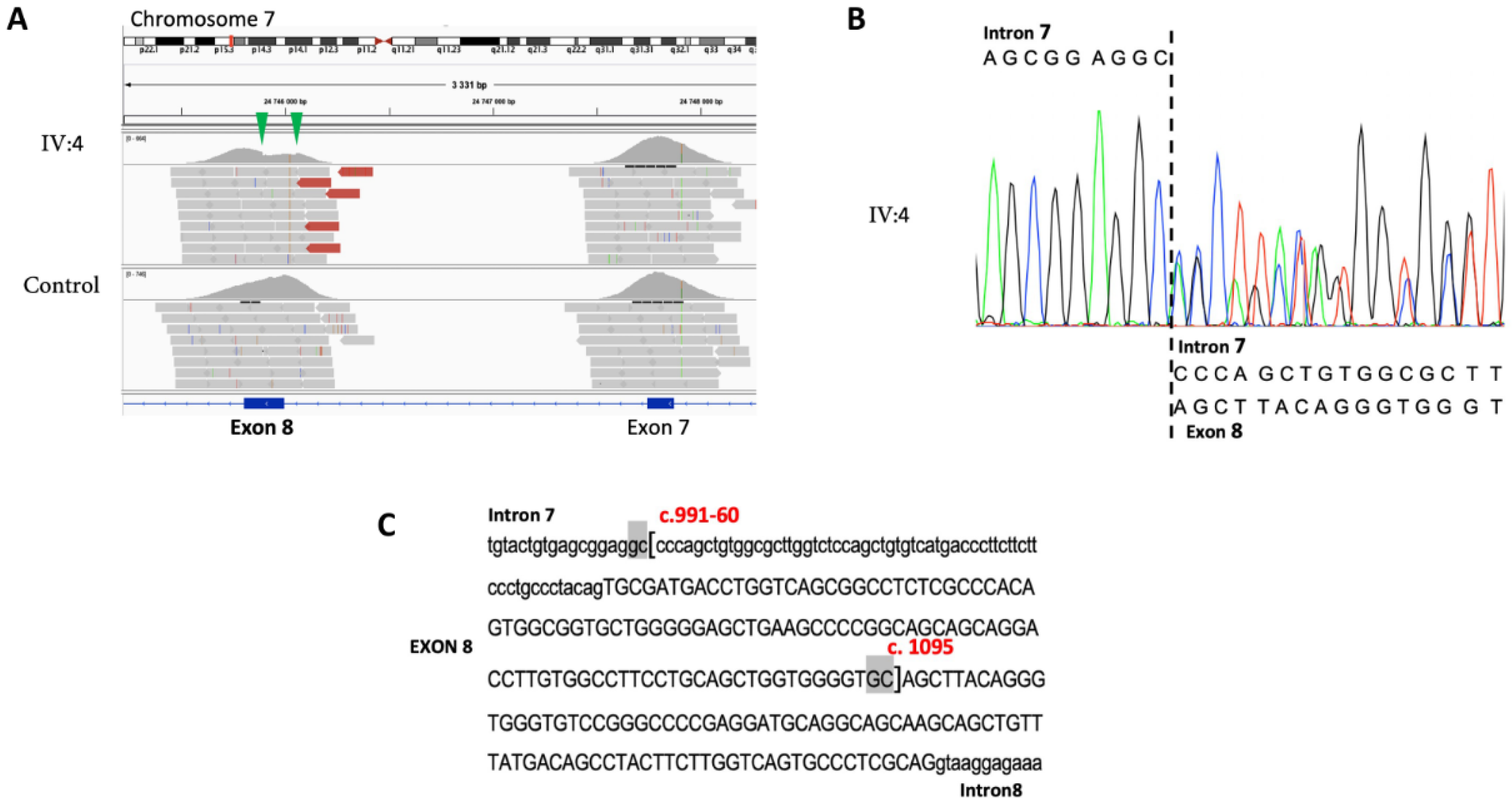
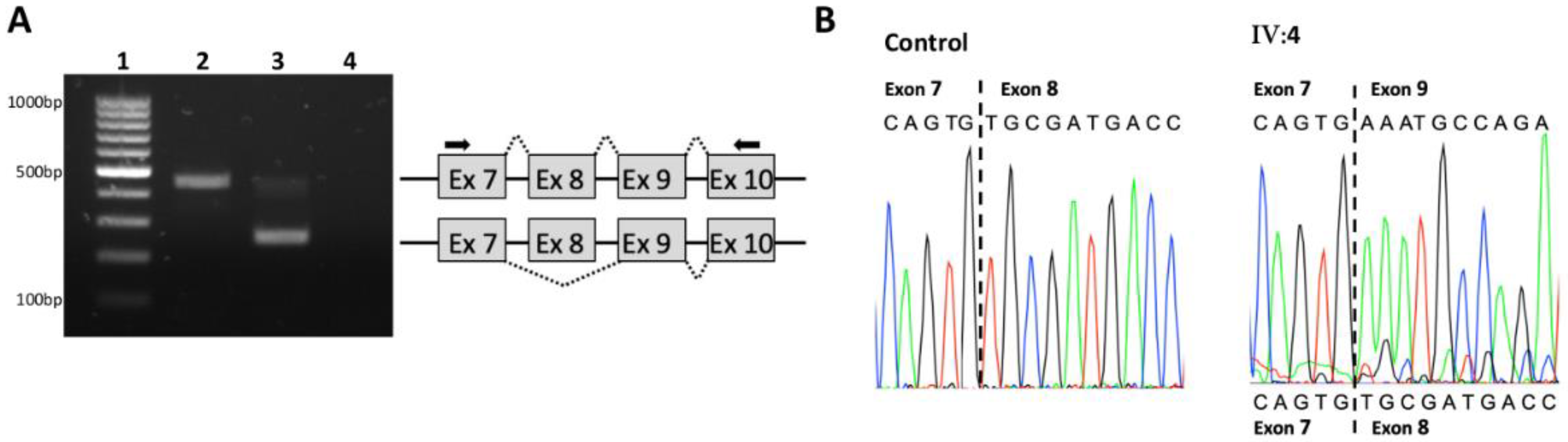
3.2. Family S2106
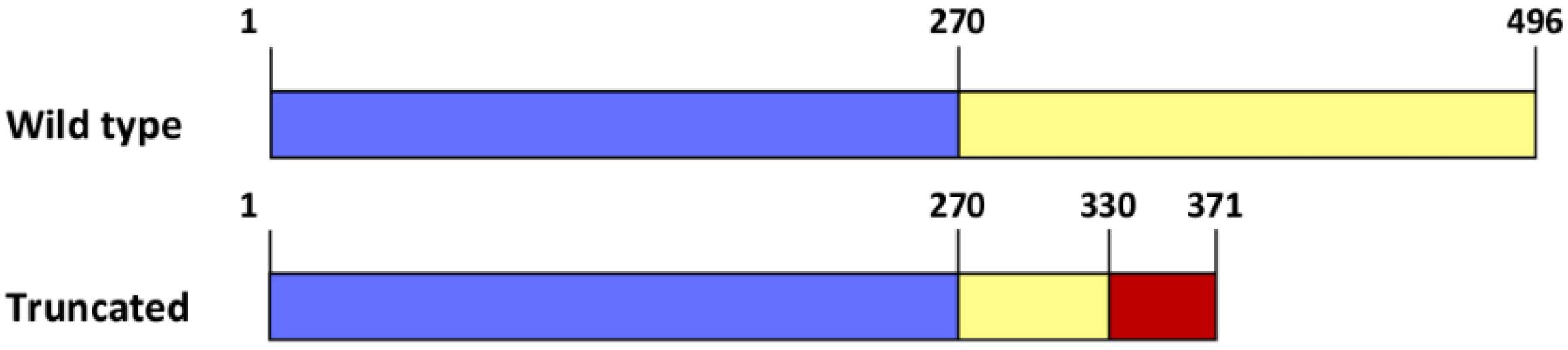
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Op de Beeck, K.; Van Camp, G.; Thys, S.; Cools, N.; Callebaut, I.; Vrijens, K.; Van Nassauw, L.; Van Tendeloo, V.F.I.; Timmermans, J.P.; Van Laer, L. The DFNA5 Gene, Responsible for Hearing Loss and Involved in Cancer, Encodes a Novel Apoptosis-Inducing Protein. Eur. J. Hum. Genet. 2011, 19, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, E.; Croes, L.; Ibrahim, J.; Pauwels, P.; Op de Beeck, K.; Vandenabeele, P.; Van Camp, G. GSDME and Its Role in Cancer: From behind the Scenes to the Front of the Stage. Int. J. Cancer 2021, 148, 2872–2883. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, A.M.L.C.; Luijendijk, M.W.J.; Huygen, P.L.M.; van Duijnhoven, G.; De Leenheer, E.M.R.; Oudesluijs, G.G.; Van Laer, L.; Cremers, F.P.M.; Cremers, C.W.R.J.; Kremer, H. A Novel Mutation Identified in the DFNA5 Gene in a Dutch Family: A Clinical and Genetic Evaluation. Audiol. Neurootol. 2004, 9, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Han, D.Y.; Dai, P.; Sun, H.J.; Tao, R.; Sun, Q.; Yan, D.; Qin, W.; Wang, H.Y.; Ouyang, X.M.; et al. A Novel DFNA5 Mutation, IVS8+4 A>G, in the Splice Donor Site of Intron 8 Causes Late-Onset Non-Syndromic Hearing Loss in a Chinese Family. Clin. Genet. 2007, 72, 471–477. [Google Scholar] [CrossRef]
- Chai, Y.; Chen, D.; Wang, X.; Wu, H.; Yang, T. A Novel Splice Site Mutation in DFNA5 Causes Late-Onset Progressive Non-Syndromic Hearing Loss in a Chinese Family. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1265–1268. [Google Scholar] [CrossRef]
- Li-Yang, M.-N.; Shen, X.-F.; Wei, Q.-J.; Yao, J.; Lu, Y.-J.; Cao, X.; Xing, G.-Q. IVS8+1 DelG, a Novel Splice Site Mutation Causing DFNA5 Deafness in a Chinese Family. Chin. Med. J. (Engl.) 2015, 128, 2510–2515. [Google Scholar] [CrossRef]
- Chen, S.; Dong, C.; Wang, Q.; Zhong, Z.; Qi, Y.; Ke, X.; Liu, Y. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients with Hereditary Hearing Loss. Genet. Test. Mol. Biomark. 2016, 20, 660–665. [Google Scholar] [CrossRef]
- Booth, K.T.; Azaiez, H.; Kahrizi, K.; Wang, D.; Zhang, Y.; Frees, K.; Nishimura, C.; Najmabadi, H.; Smith, R.J. Exonic Mutations and Exon Skipping: Lessons Learned from DFNA5. Hum. Mutat. 2018, 39, 433–440. [Google Scholar] [CrossRef]
- Wang, H.; Guan, J.; Guan, L.; Yang, J.; Wu, K.; Lin, Q.; Xiong, W.; Lan, L.; Zhao, C.; Xie, L.; et al. Further Evidence for “Gain-of-Function” Mechanism of DFNA5 Related Hearing Loss. Sci. Rep. 2018, 8, 8424. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, Q.; Su, Y.; Lin, Q.; Gao, X.; Liu, H.; Huang, S.; Kang, D.; Todd, N.W.; Mattox, D.; et al. Comprehensive Genetic Testing of Chinese SNHL Patients and Variants Interpretation Using ACMG Guidelines and Ethnically Matched Normal Controls. Eur. J. Hum. Genet. 2020, 28, 231–243. [Google Scholar] [CrossRef]
- Yu, C.; Meng, X.; Zhang, S.; Zhao, G.; Hu, L.; Kong, X. A 3-Nucleotide Deletion in the Polypyrimidine Tract of Intron 7 of the DFNA5 Gene Causes Nonsyndromic Hearing Impairment in a Chinese Family. Genomics 2003, 82, 575–579. [Google Scholar] [CrossRef]
- Van Laer, L.; Huizing, E.H.; Verstreken, M.; van Zuijlen, D.; Wauters, J.G.; Bossuyt, P.J.; Van de Heyning, P.; McGuirt, W.T.; Smith, R.J.; Willems, P.J.; et al. Nonsyndromic Hearing Impairment Is Associated with a Mutation in DFNA5. Nat. Genet. 1998, 20, 194–197. [Google Scholar] [CrossRef]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined Genetic Approaches Yield a 48% Diagnostic Rate in a Large Cohort of French Hearing-Impaired Patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert Specification of the ACMG/AMP Variant Interpretation Guidelines for Genetic Hearing Loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Cho, H.J.; Baek, J.I.; Ben-Yosef, T.; Kwon, T.J.; Griffith, A.J.; Kim, U.-K. Evidence for a Founder Mutation Causing DFNA5 Hearing Loss in East Asians. J. Hum. Genet. 2010, 55, 59–62. [Google Scholar] [CrossRef]
- Cabanillas, R.; Diñeiro, M.; Cifuentes, G.A.; Castillo, D.; Pruneda, P.C.; Álvarez, R.; Sánchez-Durán, N.; Capín, R.; Plasencia, A.; Viejo-Díaz, M.; et al. Comprehensive Genomic Diagnosis of Non-Syndromic and Syndromic Hereditary Hearing Loss in Spanish Patients. BMC Med. Genom. 2018, 11, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butz, M.; McDonald, A.; Lundquist, P.A.; Meyer, M.; Harrington, S.; Kester, S.; Stein, M.I.; Mistry, N.A.; Zimmerman Zuckerman, E.; Niu, Z.; et al. Development and Validation of a Next-Generation Sequencing Panel for Syndromic and Nonsyndromic Hearing Loss. J. Appl. Lab. Med. 2020, 5, 467–479. [Google Scholar] [CrossRef]
- Abu Rayyan, A.; Kamal, L.; Casadei, S.; Brownstein, Z.; Zahdeh, F.; Shahin, H.; Canavati, C.; Dweik, D.; Jaraysa, T.; Rabie, G.; et al. Genomic Analysis of Inherited Hearing Loss in the Palestinian Population. Proc. Natl. Acad. Sci. USA 2020, 117, 20070–20076. [Google Scholar] [CrossRef] [PubMed]
- Tropitzsch, A.; Schade-Mann, T.; Gamerdinger, P.; Dofek, S.; Schulte, B.; Schulze, M.; Battke, F.; Fehr, S.; Biskup, S.; Heyd, A.; et al. Diagnostic Yield of Targeted Hearing Loss Gene Panel Sequencing in a Large German Cohort With a Balanced Age Distribution from a Single Diagnostic Center: An Eight-Year Study. Ear Hear. 2021. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base Editing: Precision Chemistry on the Genome and Transcriptome of Living Cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Grajcarek, J.; Monlong, J.; Nishinaka-Arai, Y.; Nakamura, M.; Nagai, M.; Matsuo, S.; Lougheed, D.; Sakurai, H.; Saito, M.K.; Bourque, G.; et al. Genome-Wide Microhomologies Enable Precise Template-Free Editing of Biologically Relevant Deletion Mutations. Nat. Commun. 2019, 10, 4856. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansard, L.; Vaché, C.; Bianchi, J.; Baudoin, C.; Perthus, I.; Isidor, B.; Blanchet, C.; Baux, D.; Koenig, M.; Kalatzis, V.; et al. Identification of the First Single GSDME Exon 8 Structural Variants Associated with Autosomal Dominant Hearing Loss. Diagnostics 2022, 12, 207. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12010207
Mansard L, Vaché C, Bianchi J, Baudoin C, Perthus I, Isidor B, Blanchet C, Baux D, Koenig M, Kalatzis V, et al. Identification of the First Single GSDME Exon 8 Structural Variants Associated with Autosomal Dominant Hearing Loss. Diagnostics. 2022; 12(1):207. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12010207
Chicago/Turabian StyleMansard, Luke, Christel Vaché, Julie Bianchi, Corinne Baudoin, Isabelle Perthus, Bertrand Isidor, Catherine Blanchet, David Baux, Michel Koenig, Vasiliki Kalatzis, and et al. 2022. "Identification of the First Single GSDME Exon 8 Structural Variants Associated with Autosomal Dominant Hearing Loss" Diagnostics 12, no. 1: 207. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12010207