
Autophagy-Related Gene WD Repeat Domain 45B Promotes Tumor Proliferation and Migration of Hepatocellular Carcinoma through the Akt/mTOR Signaling Pathway
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformation Analysis
2.2. Immunohistochemistry (IHC)
2.3. Cell Culture and Lentivirus Infection
2.4. Quantitative Real-Time PCR
2.5. Western Blot
2.6. CCK-8 Assay
2.7. Wound-Healing Assay
2.8. Transwell Cell Migration and Invasion Assay
2.9. Statistics Analysis
3. Results
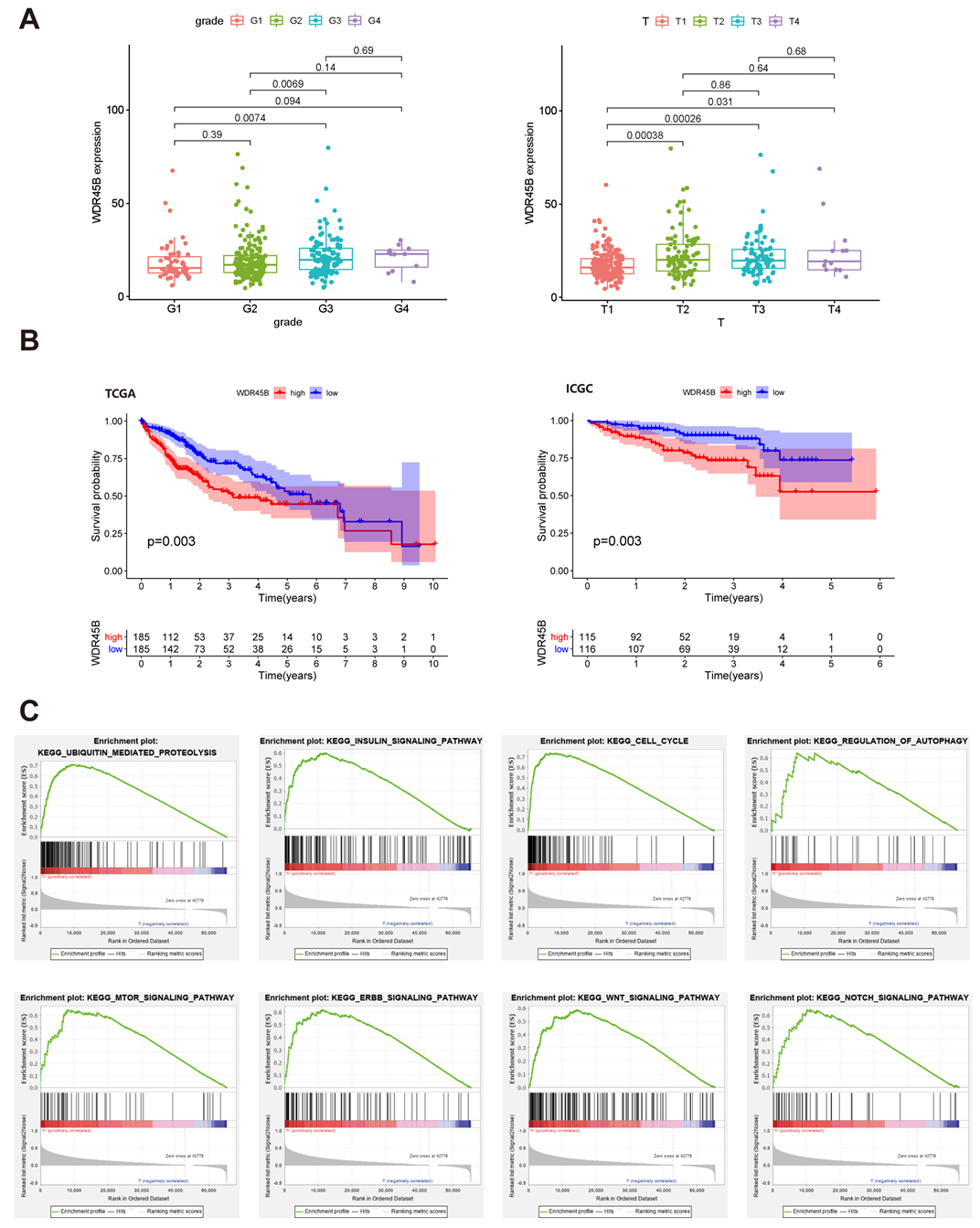
3.1. Identifying WDR45B as Prognostic Autophagy-Related Gene
3.2. Genomic Characteristics of WDR45B in HCC
3.3. Clinicopathological Characteristics and Enriched KEGG Pathways of Upregulated WDR45B in HCC
3.4. WDR45B Upregulated in FFPE Tissues of HCC Patients
3.5. WDR45B Upregulated in HCC Cell Lines
3.6. Knockdown of WDR45B Expression Inhibited Autophagy
3.7. Knockdown of WDR45B Expression Suppressed Akt/mTOR Signaling Pathway
3.8. Knockdown of WDR45B Expression Suppressed Proliferation and Migration in HCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA-Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.; Wangensteen, K.J. Incapacitated Capicua in Sorafenib-Resistant HCC. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 428–429. [Google Scholar] [CrossRef]
- Couri, T.; Pillai, A. Goals and targets for personalized therapy for HCC. Hepatol. Int. 2019, 13, 125–137. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Yang, S.; Park, D.; Manning, L.; Hill, S.E.; Cao, M.; Xuan, Z.; Gonzalez, I.; Dong, Y.; Clark, B.; Shao, L.; et al. Presynaptic autophagy is coupled to the synaptic vesicle cycle via ATG-9. Neuron 2022, 110, 824–840. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Nam, H.J. Autophagy Modulators in Cancer: Focus on Cancer Treatment. Life 2021, 11, 839. [Google Scholar] [CrossRef]
- Akkoc, Y.; Gozuacik, D. Autophagy and liver cancer. Turk. J. Gastroenterol. 2018, 29, 270–282. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, W.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Zhou, X.; Liang, C.; Li, X.; Ge, M.; Chen, Y.; Yin, J.; Zhu, J.; Zhong, C. Apatinib triggers autophagic and apoptotic cell death via VEGFR2/STAT3/PD-L1 and ROS/Nrf2/p62 signaling in lung cancer. J. Exp. Clin. Cancer Res. 2021, 40, 266. [Google Scholar] [CrossRef]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene 2020, 39, 1429–1444. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, R.; Chen, W.; Chen, Q.; Zhou, J. Construction of a prognosis-predicting model based on autophagy-related genes for hepatocellular carcinoma (HCC) patients. Aging 2020, 12, 14582–14592. [Google Scholar] [CrossRef]
- Qin, F.; Zhang, J.; Gong, J.; Zhang, W. Identification and Validation of a Prognostic Model Based on Three Autophagy-Related Genes in Hepatocellular Carcinoma. Biomed Res. Int. 2021, 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Vincent, O.; Anton-Esteban, L.; Bueno-Arribas, M.; Tornero-Ecija, A.; Navas, M.A.; Escalante, R. The WIPI Gene Family and Neurodegenerative Diseases: Insights From Yeast and Dictyostelium Models. Front. Cell. Dev. Biol. 2021, 9, 737071. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Mueller, A.J.; Proikas-Cezanne, T. WIPI beta-propellers function as scaffolds for STK11/LKB1-AMPK and AMPK-related kinase signaling in autophagy. Autophagy 2018, 14, 1082–1083. [Google Scholar] [PubMed]
- D’Arcangelo, D.; Giampietri, C.; Muscio, M.; Scatozza, F.; Facchiano, F.; Facchiano, A. WIPI1, BAG1, and PEX3 Autophagy-Related Genes Are Relevant Melanoma Markers. Oxidative Med. Cell. Longev. 2018, 2018, 1471682. [Google Scholar] [CrossRef]
- De Leo, M.G.; Berger, P.; Mayer, A. WIPI1 promotes fission of endosomal transport carriers and formation of autophagosomes through distinct mechanisms. Autophagy 2021, 17, 3644–3670. [Google Scholar] [CrossRef]
- Ran, T.; Ke, S.; Song, X.; Ma, T.; Xu, Y.; Wang, M. WIPI1 promotes osteosarcoma cell proliferation by inhibiting CDKN1A. Gene 2021, 782, 145537. [Google Scholar] [CrossRef]
- Liu, C.; Li, F.; Li, X.; Cao, M.; Feng, G.; Yuan, X.; Shi, X. WIPI2 depletion inhibits the growth of hepatocellular carcinoma cells through the AMPK signaling pathway. Oncol. Rep. 2020, 43, 1467–1478. [Google Scholar] [CrossRef]
- Wan, W.; You, Z.; Zhou, L.; Xu, Y.; Peng, C.; Zhou, T.; Yi, C.; Shi, Y.; Liu, W. mTORC1-Regulated and HUWE1-Mediated WIPI2 Degradation Controls Autophagy Flux. Mol. Cell 2018, 72, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Bakula, D.; Muller, A.J.; Zuleger, T.; Takacs, Z.; Franz-Wachtel, M.; Thost, A.K.; Brigger, D.; Tschan, M.P.; Frickey, T.; Robenek, H.; et al. WIPI3 and WIPI4 beta-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 2017, 8, 15637. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Honda, S.; Torii, S.; Shimizu, K.; Katoh, K.; Miyake, K.; Miyake, N.; Fujikake, N.; Sakurai, H.T.; Arakawa, S.; et al. Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration. Nat. Commun. 2020, 11, 5311. [Google Scholar] [CrossRef]
- Xun, Q.; Kuang, J.; Yang, Q.; Wang, W.; Zhu, G. GLCCI1 reduces collagen deposition and airway hyper-responsiveness in a mouse asthma model through binding with WD repeat domain 45B. J. Cell. Mol. Med. 2021, 25, 6573–6583. [Google Scholar] [CrossRef]
- Abe, H.; Ogawa, T.; Wang, L.; Kimura, M.; Tanaka, T.; Morita, R.; Yoshida, T.; Shibutani, M. Promoter-region hypermethylation and expression downregulation of Yy1 (Yin yang 1) in preneoplastic liver lesions in a thioacetamide rat hepatocarcinogenesis model. Toxicol. Appl. Pharmacol. 2014, 280, 467–474. [Google Scholar] [CrossRef]
- Iwata, T.; Sedukhina, A.S.; Kubota, M.; Oonuma, S.; Maeda, I.; Yoshiike, M.; Usuba, W.; Minagawa, K.; Hames, E.; Meguro, R.; et al. A new bioinformatics approach identifies overexpression of GRB2 as a poor prognostic biomarker for prostate cancer. Sci. Rep. 2021, 11, 5696. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Yan, H.; Cheng, Y.; Wang, Y.; Yang, Y.; Deng, M.; Che, X.; Hou, K.; Qu, X.; et al. Lymecycline reverses acquired EGFR-TKI resistance in non-small-cell lung cancer by targeting GRB2. Pharmacol. Res. 2020, 159, 105007. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, S.; Wu, H.; Lu, J.; Lu, Y.; Wang, F.; Zhao, W.; Zhan, P.; Lu, J.; Fang, Q.; et al. Deubiquitinase PSMD14 enhances hepatocellular carcinoma growth and metastasis by stabilizing GRB2. Cancer Lett. 2020, 469, 22–34. [Google Scholar] [CrossRef]
- Meitinger, F.; Ohta, M.; Lee, K.Y.; Watanabe, S.; Davis, R.L.; Anzola, J.V.; Kabeche, R.; Jenkins, D.A.; Shiau, A.K.; Desai, A.; et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 2020, 585, 440–446. [Google Scholar] [CrossRef]
- Nishibeppu, K.; Komatsu, S.; Kiuchi, J.; Kishimoto, T.; Takashima, Y.; Shoda, K.; Arita, T.; Kosuga, T.; Konishi, H.; Shiozaki, A.; et al. TRIM37 contributes to malignant outcomes and CDDP resistance in gastric cancer. J. Cancer 2021, 12, 316–325. [Google Scholar] [CrossRef]
- Yeow, Z.Y.; Lambrus, B.G.; Marlow, R.; Zhan, K.H.; Durin, M.A.; Evans, L.T.; Scott, P.M.; Phan, T.; Park, E.; Ruiz, L.A.; et al. Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature 2020, 585, 447–452. [Google Scholar] [CrossRef]
- Tan, G.; Xie, B.; Yu, N.; Huang, J.; Zhang, B.; Lin, F.; Li, H. TRIM37 overexpression is associated with chemoresistance in hepatocellular carcinoma via activating the AKT signaling pathway. Int. J. Clin. Oncol. 2021, 26, 532–542. [Google Scholar] [CrossRef]
- Fan, S.; Yue, L.; Wan, W.; Zhang, Y.; Zhang, B.; Otomo, C.; Li, Q.; Lin, T.; Hu, J.; Xu, P.; et al. Inhibition of Autophagy by a Small Molecule through Covalent Modification of the LC3 Protein. Angew. Chem.-Int. Edit. 2021, 60, 26105–26114. [Google Scholar] [CrossRef]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. Retraction Note: p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatr. 2021, 26, 3664. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Rick, J.; Yagnik, G.; Aghi, M.K. Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin. Cancer Biol. 2020, 66, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Liu, X.; Zhang, Y.; Li, A.; Xie, Y.; Zhou, S.; Song, L.; Xu, R.; Ma, Y.; Cai, S.; et al. BEZ235 Increases the Sensitivity of Hepatocellular Carcinoma to Sorafenib by Inhibiting PI3K/AKT/mTOR and Inducing Autophagy. Biomed Res. Int. 2021, 2021, 5556306. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, S.; Du, X.; Ji, J.; Xi, Y.; Zhai, G. Advances in autophagy as a target in the treatment of tumours. J. Drug Target. 2022, 30, 166–187. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total HCC Patients | WDR45B Expression in HCC Patients | χ2 | p | ||
|---|---|---|---|---|---|---|
| High | Low | |||||
| Age, n (%) | >45 | 42 (75.0%) | 24 (85.7%) | 18 (64.3%) | 3.429 | 0.121 |
| ≤45 | 14 (25.0%) | 4 (14.3%) | 10 (35.7%) | |||
| Sex, n (%) | male | 55 (98.2%) | 27 (96.4%) | 28 (100%) | 1.018 | 1.000 |
| female | 1 (2.8%) | 1 (3.6%) | 0 (0%) | |||
| Grade, n (%) | G1-G2 | 39 (69.6%) | 15 (53.6%) | 24 (85.7%) | 6.842 | 0.019 * |
| G3 | 17 (30.4%) | 13 (46.4%) | 4 (14.3%) | |||
| T stage, n (%) | T1-T2 | 33 (58.9%) | 13 (46.4%) | 20 (71.4%) | 3.615 | 0.102 |
| T3-T4 | 23 (41.1%) | 15 (53.6%) | 8 (28.6%) | |||
| macroscope vein invasion, n (%) | visible | 6 (10.7%) | 4 (14.3%) | 2 (7.1%) | 0.747 | 0.669 |
| invisible | 50 (89.3%) | 24 (85.7%) | 26 (92.9%) | |||
| microvascular invasion, n (%) | visible | 36 (64.3%) | 20 (71.4%) | 16 (57.1%) | 1.244 | 0.403 |
| invisible | 20 (35.7%) | 8 (28.6%) | 12 (42.9%) | |||
| cirrhosis, n (%) | yes | 22 (39.3%) | 12 (42.9%) | 10 (35.7%) | 0.299 | 0.785 |
| no | 34 (60.7%) | 16 (57.1%) | 18 (64.3%) | |||
| tumor involvement, n (%) | restricted to liver | 47 (88.7%) | 23 (85.2%) | 24 (92.3%) | 0.669 | 0.669 |
| invade portal vein, gallbladder or visceral peritoneum | 6 (11.3%) | 4 (14.8%) | 2 (7.7%) | |||
| AFP (IHC), n (%) | positive (+) | 16 (61.5%) | 9 (75.0%) | 7 (50.0%) | 1.706 | 0.248 |
| negative (−) | 10 (38.5%) | 3 (25.0%) | 7 (50.0%) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chen, L.; Pang, J.; Yang, C.; Xie, W.; Shen, G.; Chen, H.; Li, X.; Xiao, S.-Y.; Li, Y. Autophagy-Related Gene WD Repeat Domain 45B Promotes Tumor Proliferation and Migration of Hepatocellular Carcinoma through the Akt/mTOR Signaling Pathway. Diagnostics 2023, 13, 906. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics13050906
Li J, Chen L, Pang J, Yang C, Xie W, Shen G, Chen H, Li X, Xiao S-Y, Li Y. Autophagy-Related Gene WD Repeat Domain 45B Promotes Tumor Proliferation and Migration of Hepatocellular Carcinoma through the Akt/mTOR Signaling Pathway. Diagnostics. 2023; 13(5):906. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics13050906
Chicago/Turabian StyleLi, Jiahao, Lansi Chen, Jingjing Pang, Chunxiu Yang, Wen Xie, Guoyan Shen, Hongshan Chen, Xiaoyi Li, Shu-Yuan Xiao, and Yueying Li. 2023. "Autophagy-Related Gene WD Repeat Domain 45B Promotes Tumor Proliferation and Migration of Hepatocellular Carcinoma through the Akt/mTOR Signaling Pathway" Diagnostics 13, no. 5: 906. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics13050906