Primary Biliary Cholangitis and Bile Acid Farnesoid X Receptor Agonists

 ,
, 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
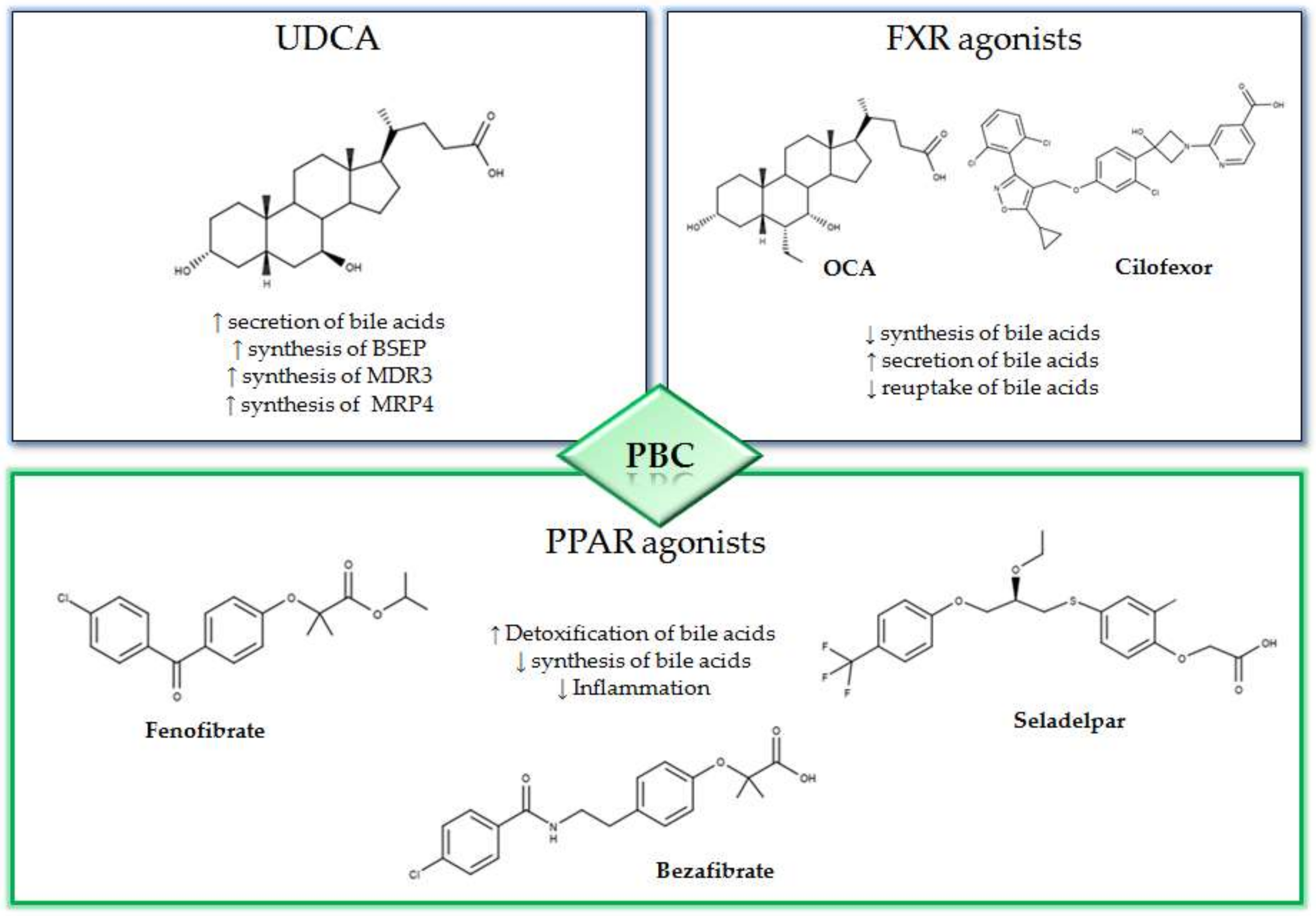
2. The First-and Second-Line Therapies for PBC
2.1. UDCA
2.2. OCA
3. New Compounds for the Treatment of PBC
3.1. NGM282
3.2. Non-Bile Acids FXR Agonists
3.3. Immunomodulatory Strategies
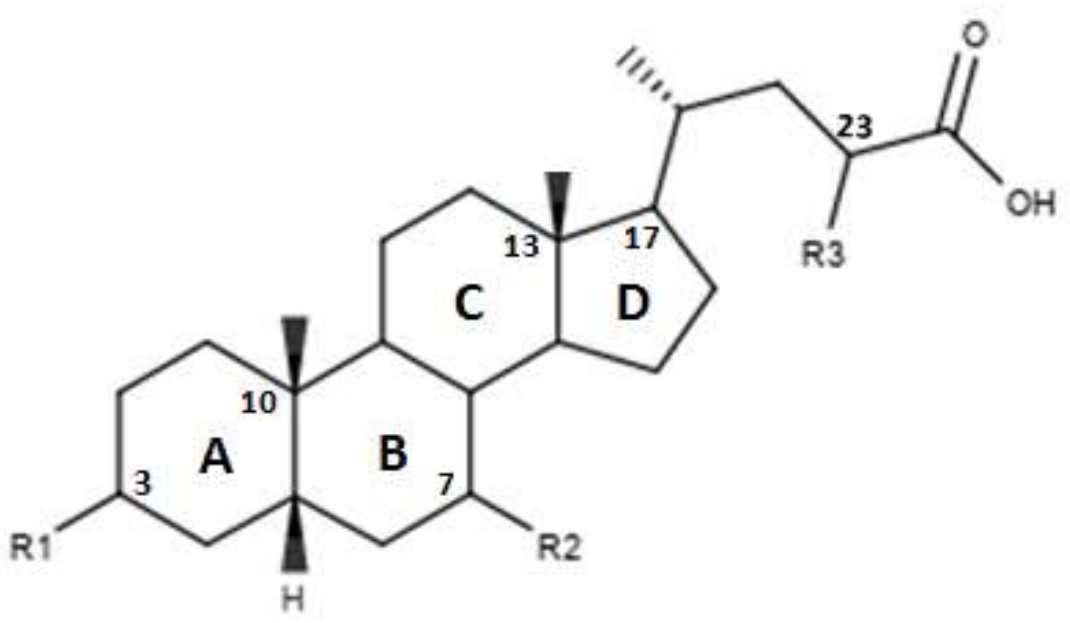
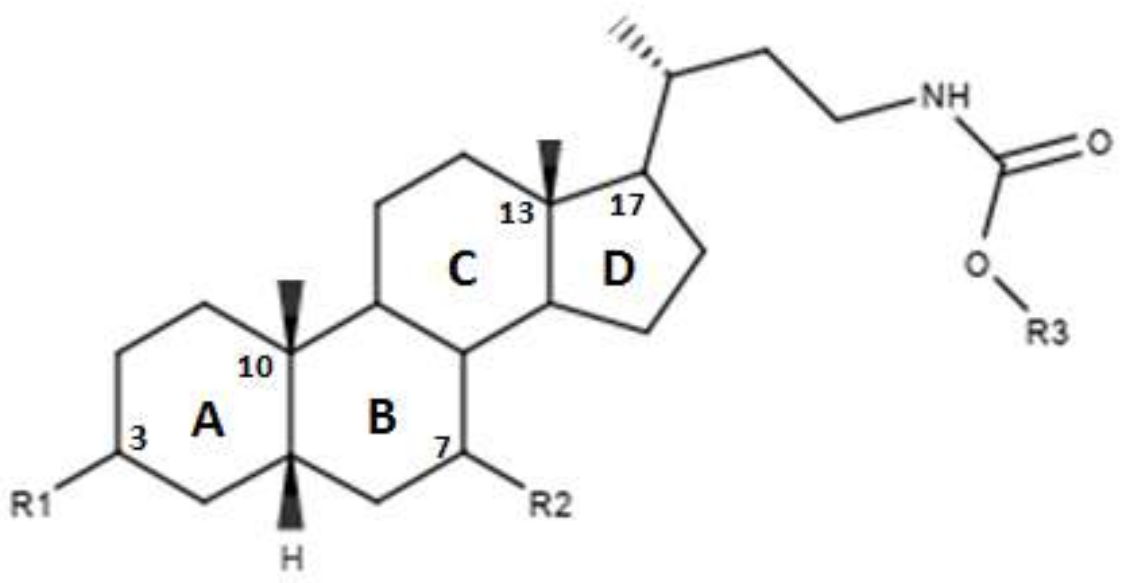
4. Structure–Activity Relationship of Bile Acid FXR Agonists
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Etherington, R.E.; Millar, B.J.M.; Innes, B.A.; Jones, D.E.J.; Kirby, J.A.; Brain, J.G. Bile acid receptor agonists in primary biliary cholangitis: Regulation of the cholangiocytesecretome and downstream T cell differentiation. FASEB Bioadv. 2019, 1, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Wang, L.; Woo, E.; He, X.; Yang, G.; Bowlus, C.; Leung, P.S.C.; Gershwin, M.E. Clinical Management of Primary Biliary Cholangitis-Strategies and Evolving Trends. Clin. Rev. Allergy Immunol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Hirschfield, G.M.; Adams, D.H.; Beuers, U.; Mann, D.A.; Lindor, K.D.; Jones, D.E. Novel Therapeutic Targets in Primary Biliary Cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Podda, M.; Selmi, C.; Lleo, A.; Moroni, L.; Invernizzi, P. The limitations and hidden gems of the epidemiology of primary biliary cirrhosis. J. Autoimmun. 2013, 46, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Jepsen, P.; Morenghi, E.; Carbone, M.; Moroni, L.; Battezzati, P.M.; Podda, M.; Mackay, I.R.; Gershwin, M.E.; Invernizzi, P. Evolving Trends in Female to Male Incidence and Male Mortality of Primary Biliary Cholangitis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Manno, V.; Gerussi, A.; Carbone, M.; Minelli, G.; Taruscio, D.; Conti, S.; Invernizzi, P. A National Hospital-Based Study of Hospitalized Patients with Primary Biliary Cholangitis. Hepatol. Commun. 2019, 3, 1250–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, E.J.; Ali, A.H.; Lindor, K.D. Primary biliary cirrhosis. Lancet 2015, 386, 1565–1575. [Google Scholar] [CrossRef]
- Lindor, K.D.; Gershwin, M.E.; Poupon, R.; Kaplan, M.; Bergasa, N.V.; Heathcote, E.J.; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009, 50, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Carbone, M.; Lleo, A.; Invernizzi, P. Geoepidemiology, Genetic and Environmental Risk Factors for PBC. Dig. Dis. 2015, 2, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Lu, Y.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 658–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Invernizzi, P.; Ransom, M.; Raychaudhuri, S.; Kosoy, R.; Lleo, A.; Shigeta, R.; Franke, A.; Bossa, F.; Amos, C.I.; Gregersen, P.K.; et al. Classical HLA-DRB1 and DPB1 alleles account for HLA associations with primary biliary cirrhosis. Genes Immun. 2012, 13, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Invernizzi, P. Human leukocyte antigen in primary biliary cirrhosis: An old story now reviving. Hepatology 2011, 54, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Zhang, W.; McDonald, W.H.; Seeley, E.H.; Leung, P.S.; Coppel, R.L.; Ansari, A.A.; Adams, D.H.; Afford, S.; Invernizzi, P.; et al. Shotgun proteomics: Identification of unique protein profiles of apoptotic bodies from biliary epithelial cells. Hepatology 2014, 60, 1314–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New Paradigms in the Treatment of Hepatic Cholestasis: From UDCA to FXR, PXR and Beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abenavoli, L.; Falalyeyeva, T.; Boccuto, L.; Tsyryuk, O.; Kobyliak, N. Obeticholic Acid: A New Era in the Treatment of Nonalcoholic Fatty Liver Disease. Pharmaceuticals 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpechot, C.; Abenavoli, L.; Rabahi, N.; Chrétien, Y.; Andréani, T.; Johanet, C.; Chazouillères, O.; Poupon, R. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 2008, 48, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Matsuzaki, Y. Ursodeoxycholic acid: Mechanism of action and novel clinical applications. Hepatol. Res. 2008, 38, 123–131. [Google Scholar] [CrossRef]
- Beuers, U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Marschall, H.U.; Wagner, M.; Zollner, G.; Fickert, P.; Diczfalusy, U.; Gumhold, J.; Silbert, D.; Fuchsbichler, A.; Benthin, L.; Grundström, R.; et al. Complementary Stimulation of Hepatobiliary Transport and Detoxification Systems by Rifampicin and Ursodeoxycholic Acid in Humans. Gastroenterology 2005, 129, 476–485. [Google Scholar] [CrossRef]
- Pellicciari, R.; Costantino, G.; Fiorucci, S. Farnesoid X receptor: From structure to potential clinical applications. J. Med. Chem. 2005, 48, 5383–5403. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Marchianò, S.; Finamore, C.; Baronissi, G.; Di Leva, F.S.; Carino, A.; Biagioli, M.; Fiorucci, C.; Cassiano, C.; Monti, M.C.; et al. Novel Isoxazole Derivatives with Potent FXR Agonistic Activity Prevent Acetaminophen-Induced Liver Injury. ACS Med. Chem. Lett. 2018, 10, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Leva, F.S.; Di Marino, D.; Limongelli, V. Structural Insight Into the Binding Mode of FXR and GPBAR1 Modulators. Handb. Exp. Pharmacol. 2019, 256, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 18, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Parés, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholicacid. Gastroenterology 2015, 148, 751–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. Fibrates and cholestasis. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 7, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M.; Namisaki, T.; Hashimoto, N.; Kawata, K.; Takahashi, A.; et al. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients with Primary Biliary Cholangitis. Hepatology 2019, 70, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.C.; Lapointe-Shaw, L.; Kowgier, M.; Meza-Cardona, J.; Hirschfield, G.M.; Janssen, H.L.; Feld, J.J. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment. Pharmacol. Ther. 2016, 43, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hegade, V.S.; Khanna, A.; Walker, L.J.; Wong, L.L.; Dyson, J.K.; Jones, D.E.J. Long-Term Fenofibrate Treatment in Primary Biliary Cholangitis Improves Biochemistry but Not the UK-PBC Risk Score. Dig. Dis. Sci. 2016, 61, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: a double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J.; Weltman, M.; Carey, E.J.; Muir, A.J.; Ling, L.; Rossi, S.J.; et al. NGM282 for Treatment of Patients With Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol. Commun. 2018, 2, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X.; et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 60, 9960–9973. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non-Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug. Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; Mayo, M.J.; Levy, C.; Zou, B.; Johanns, J.; Nnane, I.P.; Dasgupta, B.; Li, K.; et al. PURIFI Study Group. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: A proof-of-concept study. Hepatology 2016, 64, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Bowlus, C.L.; Yang, G.X.; Liu, C.H.; Johnson, C.R.; Dhaliwal, S.S.; Frank, D.; Levy, C.; Peters, M.G.; Vierling, J.M.; Gershwin, M.E. Therapeutic Trials of Biologics in Primary Biliary Cholangitis: An Open Label Study of Abatacept and Review of the Literature. J. Autoimmun. 2019, 101, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; Renga, B.; D’Amore, C.; Sepe, V.; Finamore, C.; De Marino, S.; Carino, A.; Cipriani, S.; Monti, M.C.; Zampella, A.; et al. Exploitation of cholane scaffold for the discovery of potent and selective farnesoid X receptor (FXR) and G-protein coupled bile acid receptor 1 (GP-BAR1) ligands. J. Med. Chem. 2014, 57, 8477–8495. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholicacid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Di Leva, F.S.; Sepe, V.; Renga, B.; Del Gaudio, C.; D’Auria, M.V.; Zampella, A.; Fiorucci, S.; Limongelli, V. Design, synthesis, and biological evaluation of potent dual agonists of nuclear and membrane bile acid receptors. J. Med. Chem. 2014, 57, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Passeri, D.; De Franco, F.; Mostarda, S.; Filipponi, P.; Colliva, C.; Gadaleta, R.M.; Franco, P.; Carotti, A.; Macchiarulo, A.; et al. Discovery of 3α,7α,11β-Trihydroxy-6α-ethyl-5β-cholan-24-oic Acid (TC-100), a Novel Bile Acid as Potent and Highly Selective FXR Agonist for Enterohepatic Disorders. J. Med. Chem. 2016, 59, 9201–9214. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Gioiello, A.; Costantino, G.; Sadeghpour, B.M.; Rizzo, G.; Meyer, U.; Parks, D.J.; Entrena-Guadix, A.; Fiorucci, S. Back door modulation of the farnesoid X receptor: design, synthesis, and biological evaluation of a series of side chain modified chenodeoxycholic acid derivatives. J. Med. Chem. 2006, 49, 4208–4215. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
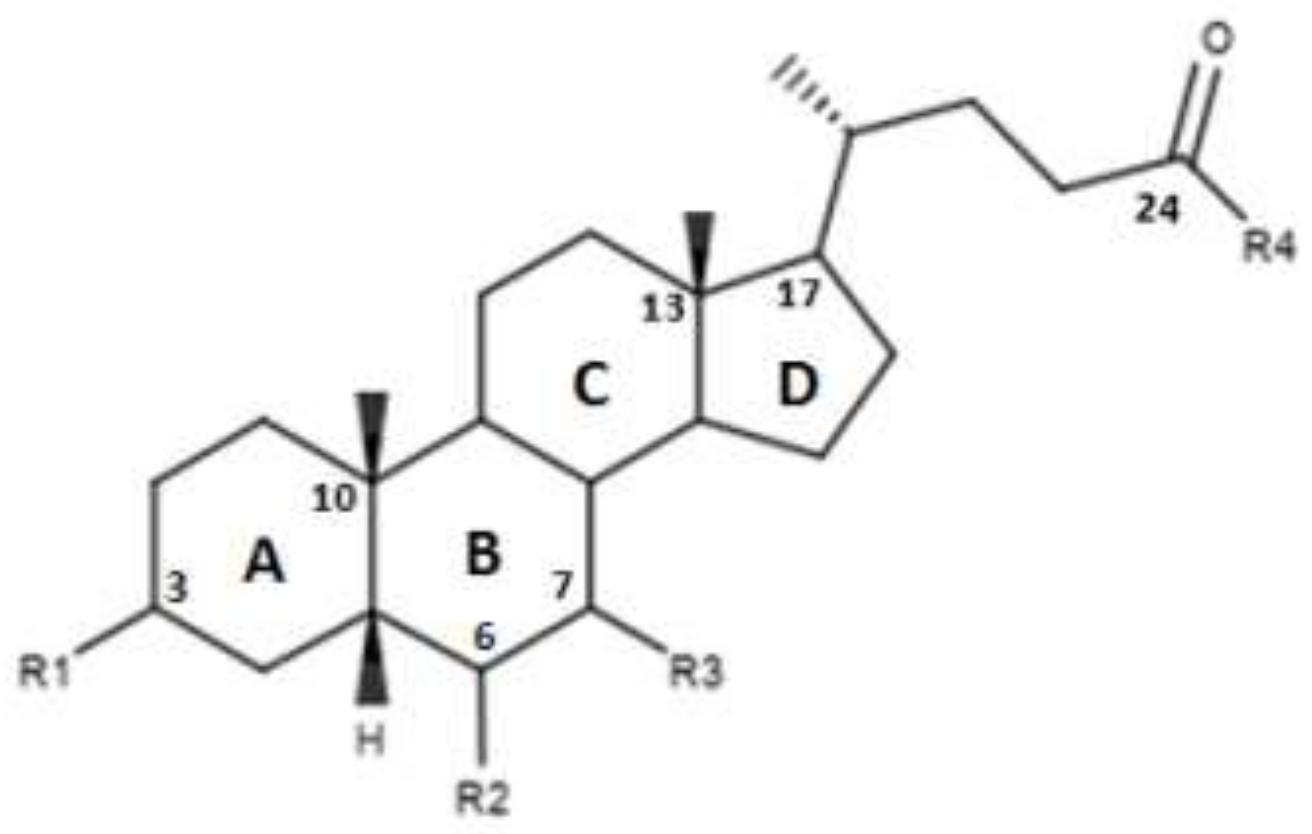
| Compounds | R1 | R2 | R3 | R4 | EC50 (µM) |
|---|---|---|---|---|---|
| OCA | α-OH | α-ethyl | α-OH | OH | 0.099 ± 0.01 |
| 1 | α-OH | α-methyl | α-OH | OH | 0.75 ± 0.08 |
| 2 | α-OH | α-propyl | α-OH | OH | 1.11 ± 0.13 |
| 3 | α-OH | α-butane | α-OH | OH | >30 |
| 4 | H | H | α-OH | OH | - |
| 5 | H | H | α-OH | NHCH2CH2SO3Na | - |
| 6 | H | H | β-OH | OH | - |
| 7 | H | H | β-OH | NHCH2CH2SO3Na | - |
| 8 | β-OH | H | α-OH | OH | - |
| 9 | α-OH |  | α-OH | OH | 0.48 |
| 10 | α-OH |  | α-OH | OH | 61.15 |
| 11 | α-OH |  | α-OH | OH | 0.54 |
| 12 | α-OH | α-OH | α-OH | OH | >30 |
| 13 | α-OH | α-OCH3 | α-OH | OH | 14.73 |
| 14 | α-OH | α-F | α-OH | OH | 15.11 |
| 15 | α-OH | β-F | α-OH | OH | 1.21 |
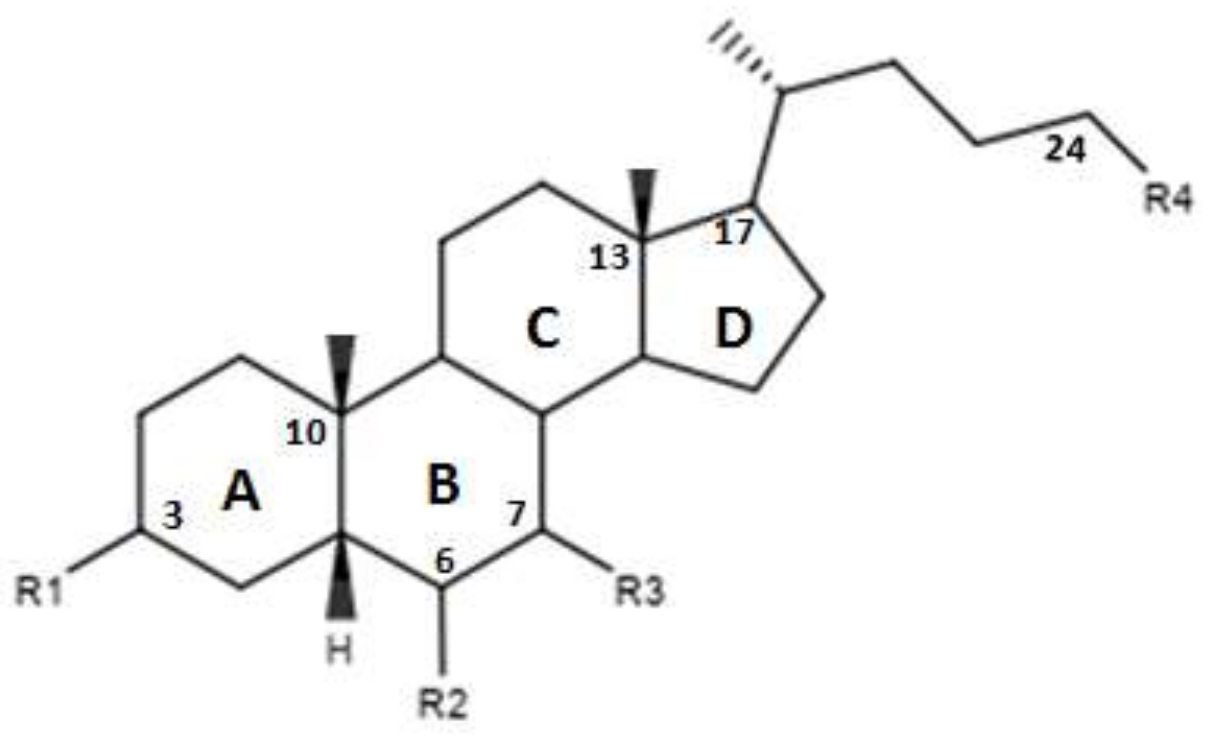
| Compounds | R1 | R2 | R3 | R4 | Ec50 (µM) |
|---|---|---|---|---|---|
| 16 | H | H | α-OH | OH | - |
| 17 | H | H | α-OH | OSO3Na | ~9 |
| 18 | H | H | α OSO3Na | OSO3Na | - |
| 19 | H | H | β-OH | OH | - |
| 20 | H | H | β-OH | OSO3Na | - |
| 21 | H | H | β-OSO3Na | OSO3Na | - |
| 22 | H | H | H | OH | - |
| 23 | H | H | H | OSO3Na | - |
| 24 | α-OH | H | α-OH | OSO3Na | - |
| 25 | α-OH | α-ethyl | α-OH | OSO3Na | ~1 |
| 26 | α-OSO3Na | H | α-OSO3Na | OSO3Na | - |
| 27 | α-OSO3Na | α-ethyl | α-OSO3Na | OSO3Na | - |
| 28 | α-OH | H | β-OH | OSO3Na | - |
| 29 | α-OSO3Na | H | β-OSO3Na | OSO3Na | - |
| 30 | α-OSO3Na | H | β-OH | OSO3Na | - |
| 31 | α-OSO3Na | H | β-OH | OH | - |
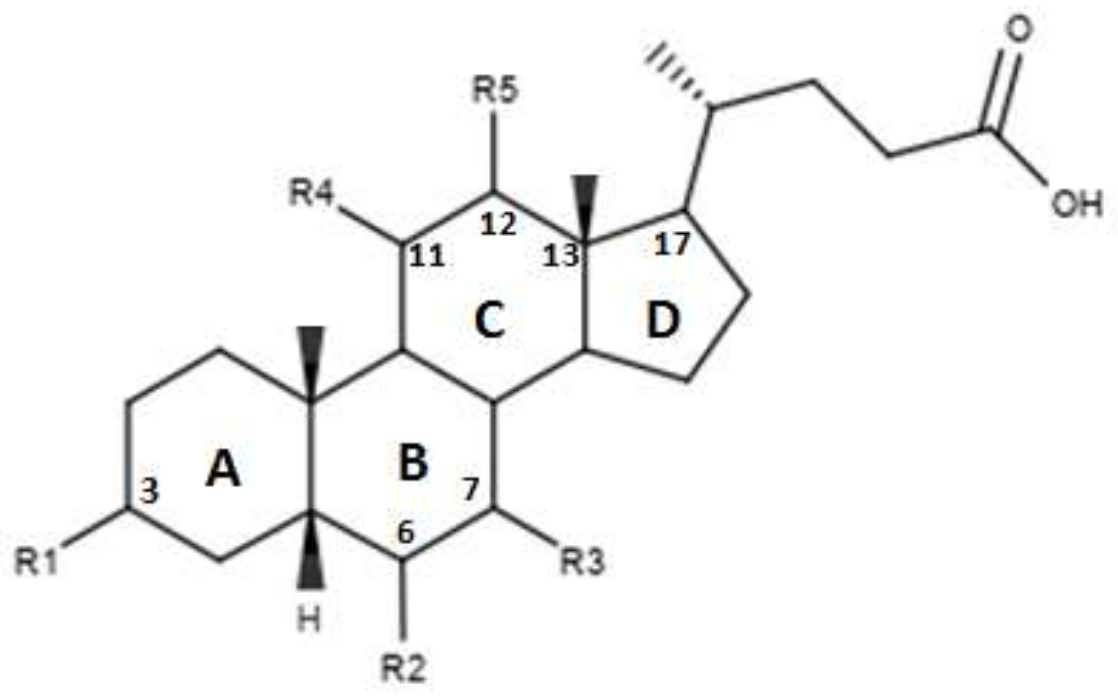
| Compounds | R1 | R2 | R3 | R4 | R5 | EC50 (µM) |
|---|---|---|---|---|---|---|
| TC-100 | α-OH | α ethyl | α-OH | β-OH | H | 0.14 ± 0.05 |
| 32 | α-OH | α ethyl | α-OH | α-OH | H | 3 ± 2 |
| 33 | α-OH | α ethyl | α-OH | H | β-OH | 4.7 ± 3.0 |
| 34 | α-OH | α ethyl | α-OH | H | α-OH | 7.0 ± 2.3 |
| Compounds | R1 | R2 | R3 | EC50 (µM) |
|---|---|---|---|---|
| 35 | α-OH | α-OH | β-OH | 13.20 |
| 36 | α-OH | α-OH | α-OH | 10.23 |
| Compounds | R1 | R2 | R3 | EC50 (µM) |
|---|---|---|---|---|
| 37 | α OH | α OH |  | 2.50 |
| 38 | α OH | α OH |  | 1.14 |
| 39 | α OH | α OH |  | 0.79 |
| 40 | α OH | α OH |  | 1.48 |
| 41 | α OH | α OH |  | 7.11 |
| 42 | α OH | α OH |  | 0.41 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abenavoli, L.; Procopio, A.C.; Fagoonee, S.; Pellicano, R.; Carbone, M.; Luzza, F.; Invernizzi, P. Primary Biliary Cholangitis and Bile Acid Farnesoid X Receptor Agonists. Diseases 2020, 8, 20. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases8020020
Abenavoli L, Procopio AC, Fagoonee S, Pellicano R, Carbone M, Luzza F, Invernizzi P. Primary Biliary Cholangitis and Bile Acid Farnesoid X Receptor Agonists. Diseases. 2020; 8(2):20. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases8020020
Chicago/Turabian StyleAbenavoli, Ludovico, Anna Caterina Procopio, Sharmila Fagoonee, Rinaldo Pellicano, Marco Carbone, Francesco Luzza, and Pietro Invernizzi. 2020. "Primary Biliary Cholangitis and Bile Acid Farnesoid X Receptor Agonists" Diseases 8, no. 2: 20. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases8020020