From Homogeneous to Heterogenized Molecular Catalysts for H2 Production by Formic Acid Dehydrogenation: Mechanistic Aspects, Role of Additives, and Co-Catalysts



Abstract
:1. Introduction
2. Type of Metal Complex-Catalysts
2.1. Noble Metal Catalysts
- (i)
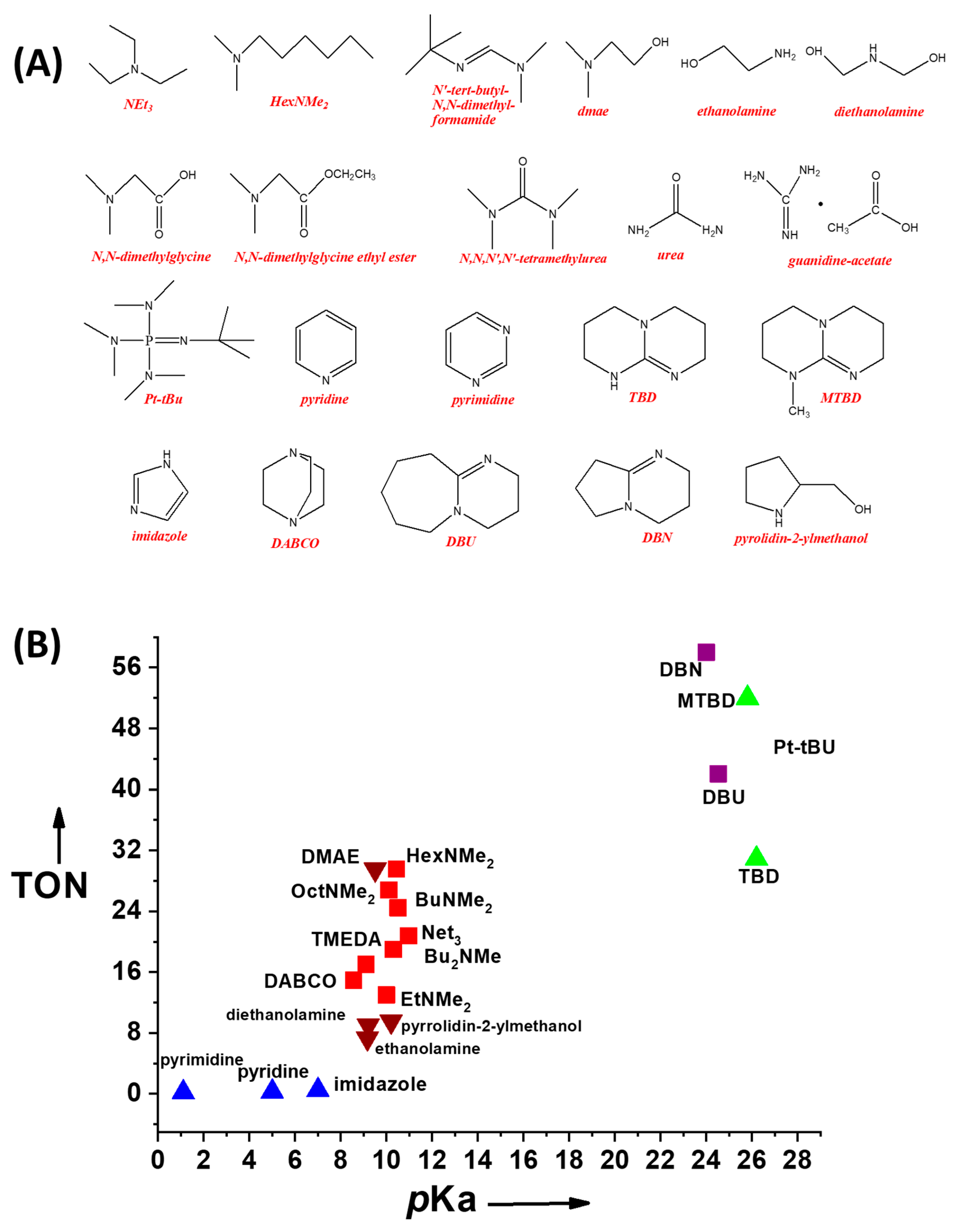
- in non-aqueous solvents, amines can be used to boost the FA deprotonation HCOOH→HCOO–,
- (ii)
- in aqueous solutions, the amines can have an effect on the HCOOH→HCOO– equilibrium, but also on the ligand itself. Thus, in aqueous solutions, the experimental observation is that pH can control gate the H2 production rate via gating of rate-limiting protonation/deprotonation events.
2.2. Non-Noble Metal Catalysts
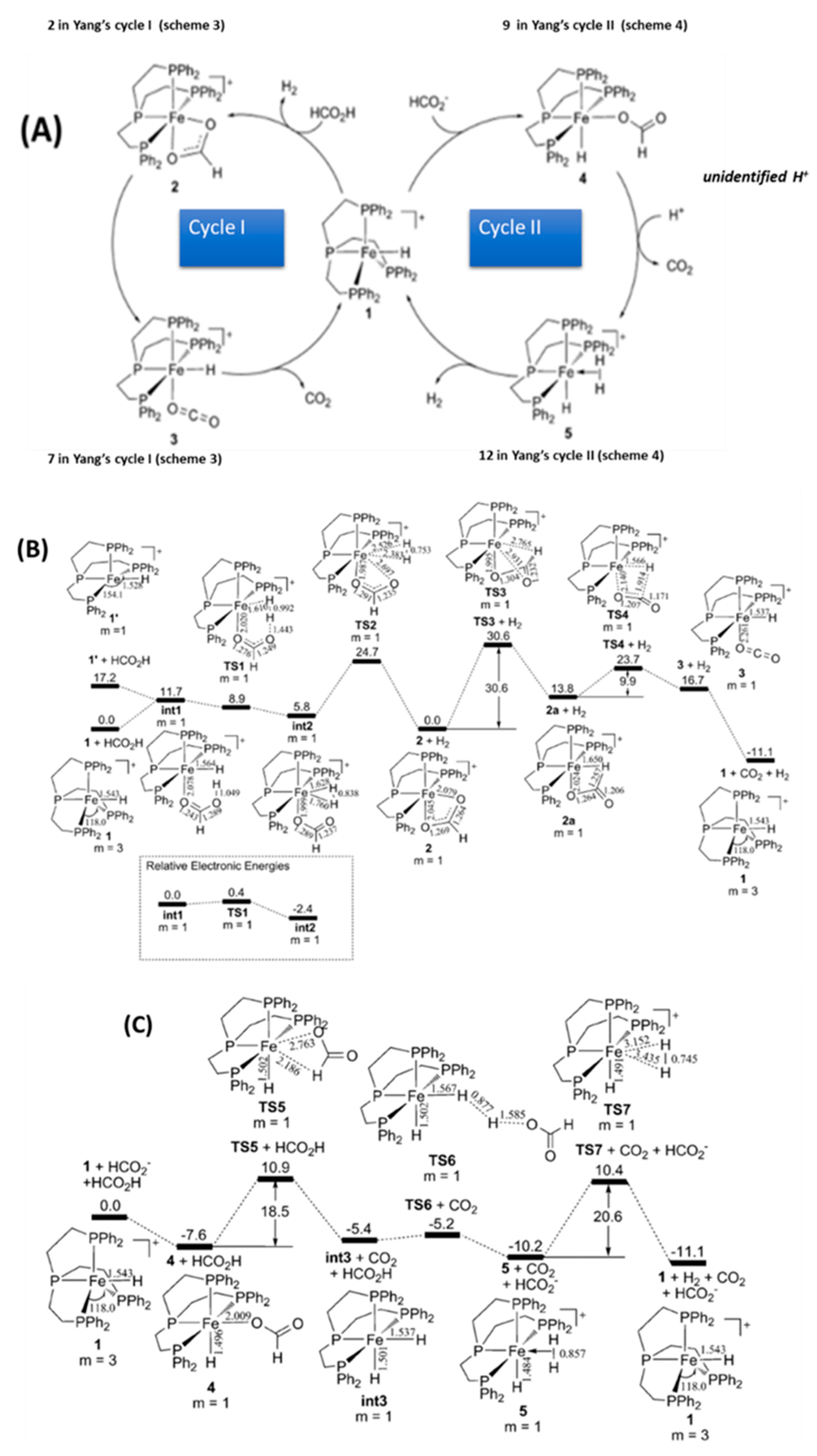
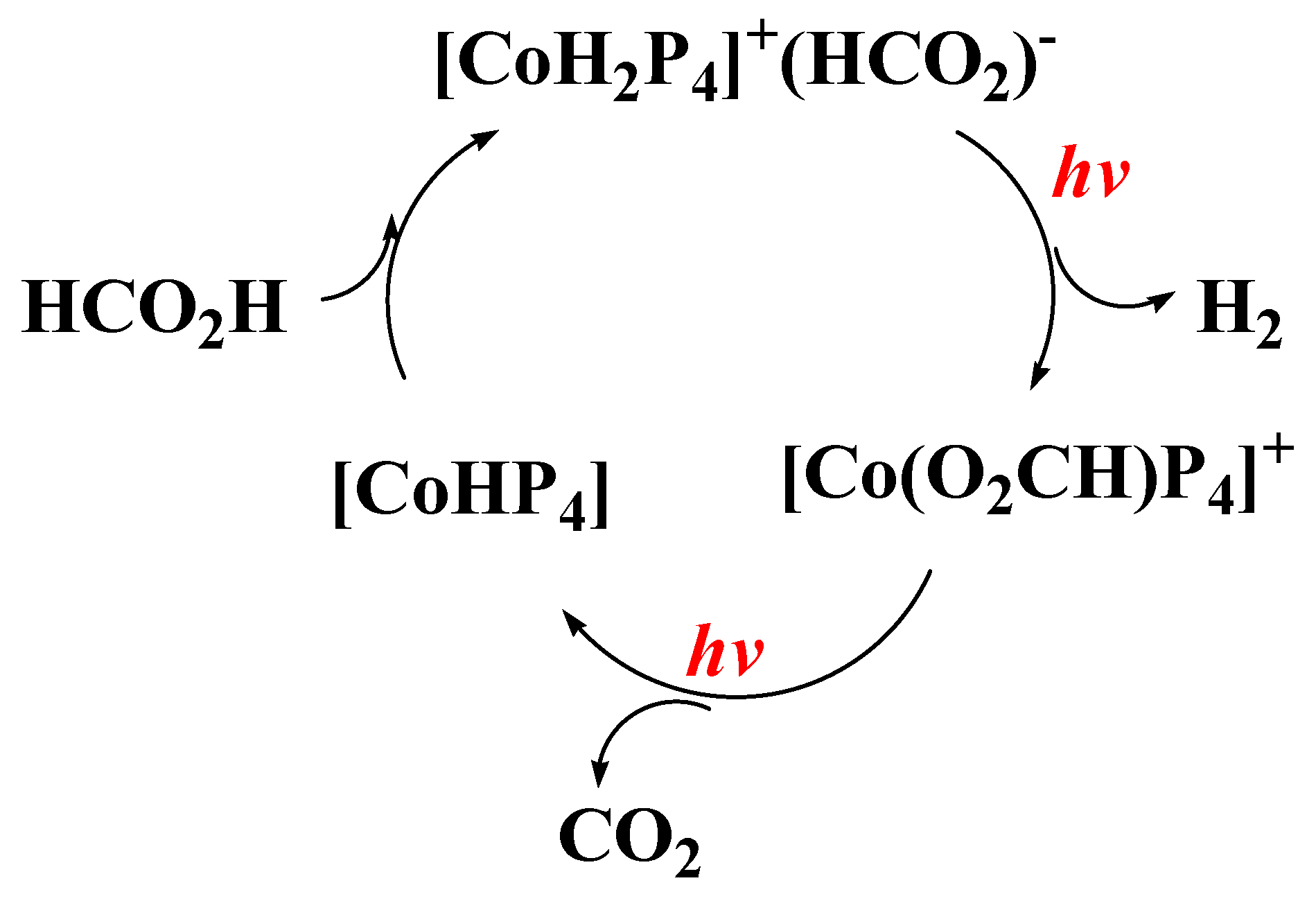
2.3. Outline of the Catalytic Mechanisms
- (i)
- FA activation i.e., formation of a HCOO– anion. This can be achieved via ionization of HCOONa in aqueous solvent or by deprotonation of formic acid HCOOH in aprotic solvents, see Figure 2.
- (ii)
- Catalyst activation: This might involve coordination of one HCOO– on the (LM) complex, or formation of the (LM-H) hydride via LnM/H2 interaction at high H2 pressure.
- (iii)
- Catalytic Η2 production. This critical step can be accomplished with two alternative routes, exemplified as Cycle-I and Cycle-II in Figure 2.
2.4. Catalyst Activation
2.4.1. Based on the Applied Preparation Method
2.4.2. Light-Assisted Activation (LAA) of Catalyst
2.5. Formic Acid Deprotonation
2.5.1. Deprotonation by Homogeneous Molecular Co-catalysts
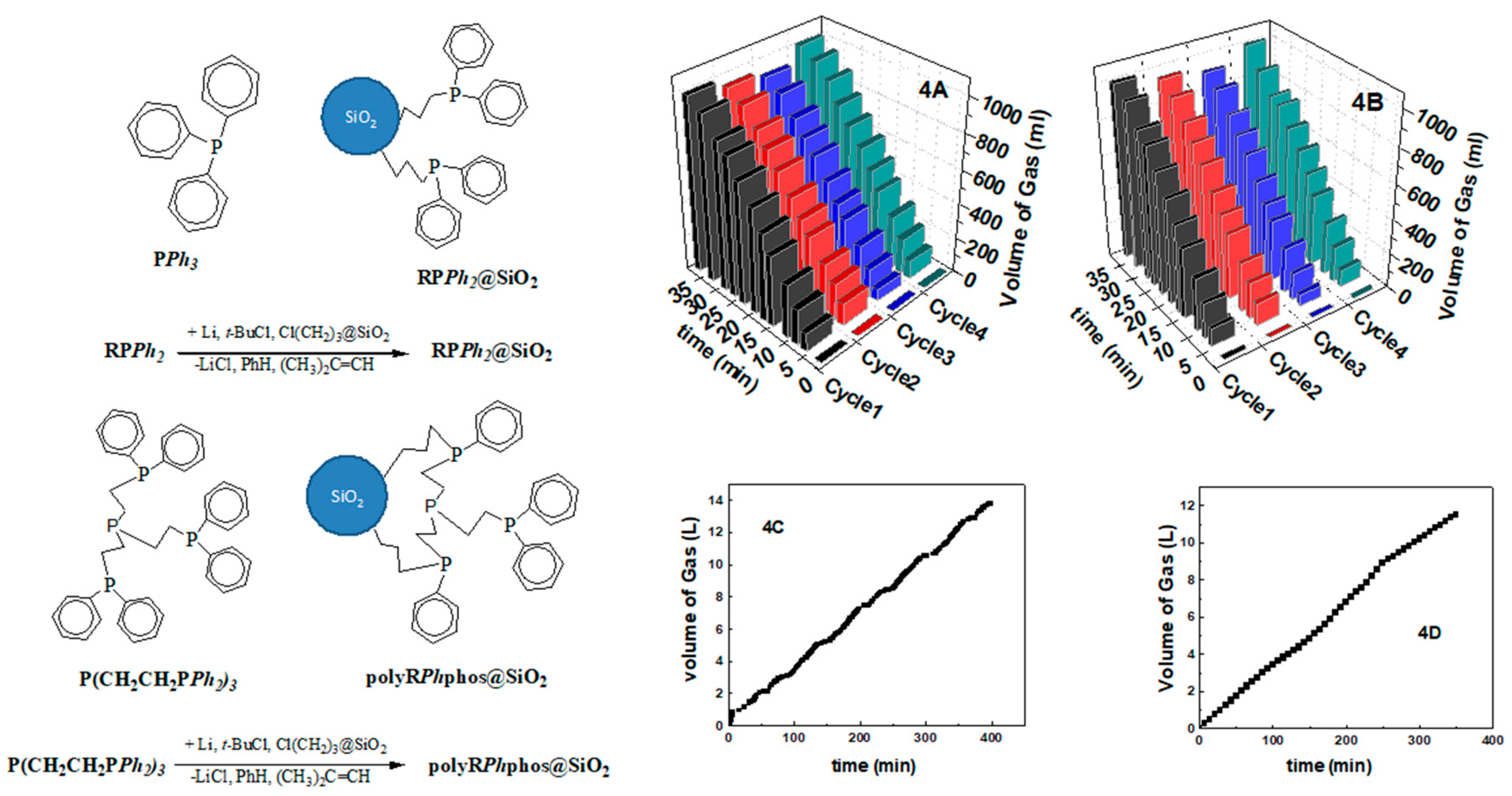
2.5.2. Deprotonation of FA by Heterogeneous Co-catalytic-particles
3. Experimental Issues—Limitations
3.1. Solution-Chemistry Issues
3.2. Hydrogen Production Under Continuous Operation
3.3. Heterogeneous Catalysts
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DPPM | Ph2PCH2PPh2 |
| tos | toluene-4-sulfonate |
| DPPE | 1,1-bis(diphenylphosphino)ethane |
| PNP | 2,6-bis(di-tert-butylphosphinomethyl)pyridine |
| CP* | 1,2,3,4,5-pentamethylcyclopentadienyl |
| TMBI | tetramethylbiimidazole |
| PHEN | 1,10-phenanthroline |
| COD | cyclo-1,5-octadiene |
| NP | 2-(di-tert-butylphosphinomethyl)pyridine |
| NHC | N-heterocyclic carbene |
| TPY | 2,2′:6′,2″-terpyridine |
| PNP1 | HN(CH2CH2(PiPr2))2 |
| PPh3 | tris[2-(diphenylphosphino)ethyl]phosphine |
| TON | Turn over number (TON = mol of produced gasses/mol of catalyst) |
| TOF | Turn over frequency (TOF = TON/t) |
References
- U.S. Energy Information Administration (EIA). Available online: https://www.eia.gov/ (accessed on 4 December 2019).
- Mason, J.E. World energy analysis: H2 now or later? Energy Policy 2007, 35, 1315–1329. [Google Scholar] [CrossRef]
- Markiewicz, M.; Zhang, Y.Q.; Bösmann, A.; Brückner, N.; Thöming, J.; Wasserscheid, P.; Stolte, S. Environmental and health impact assessment of Liquid Organic Hydrogen Carrier (LOHC) systems-challenges and preliminary results. Energy Environ. Sci. 2015, 8, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Ogden, J.M. Prospects for building a hydrogen energy infrastructure. Annu. Rev. Energy Environ. 1999, 24, 227–279. [Google Scholar] [CrossRef]
- Holdren, J.P. Energy and sustainability. Science 2007, 315, 737–738. [Google Scholar] [CrossRef] [Green Version]
- Eppinger, J.; Huang, K.W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, M. Hydrogenases for biological hydrogen production. Bioresource Techn. 2011, 102, 8423–8431. [Google Scholar] [CrossRef]
- Deligiannakis, Y. Nanomaterials for environmental solar energy technologies: Applications & limitations. KONA Powder Part. J. 2018, 35, 14–31. [Google Scholar]
- Klerke, A.; Christensen, C.H.; Nørskov, J.K.; Vegge, T. Ammonia for hydrogen storage: Challenges and opportunities. J. Mater. Chem. 2008, 18, 2304–2310. [Google Scholar] [CrossRef]
- Jonston, T.C.; Morris, D.J.; Wills, M. Hydrogen generation from formic acid and alcohols using homogeneous catalysts. Chem. Soc. Rev. 2010, 39, 81–88. [Google Scholar] [CrossRef]
- McEvoy, J.P.; Brudvig, G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef]
- Navarro, R.M.; Peña, M.A.; Fierro, J.L.G. Hydrogen production reactions from carbon feedstocks: Fossil fuels and biomass. Chem. Rev. 2007, 107, 3952–3991. [Google Scholar] [CrossRef] [PubMed]
- Kelly, N.A. Advances in Hydrogen Production, Storage, Distribution; Wood Head Publishing: Cambrige, UK, 2014. [Google Scholar]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid organic hydrogen carriers (LOHCs): Toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material-development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef] [PubMed]
- Sang, R.; Kucmierczyk, P.; Dong, K.; Franke, R.; Neumann, H.; Jackstell, R.; Beller, M. Palladium-Catalyzed Selective Generation of CO from Formic Acid for Carbonylation of Alkenes. J. Am. Chem. Soc. 2018, 140, 5217–5223. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, Q.; Liu, W.; Sun, N.; Jiang, K.; Ma, L.; Peng, Z.; Cai, W.; Liu, C.; Ge, J.; et al. Pd–PdO Interface as Active Site for HCOOH Selective Dehydrogenation at Ambient Condition. J. Phys. Chem. C 2018, 122, 2081–2088. [Google Scholar] [CrossRef]
- Marcinkowski, M.D.; Liu, J.; Murphy, C.J.; Liliano, M.L.; Wasio, N.A.; Lucci, F.R.; Flytzani-Stephanopoulos, M.; Sykes, C.H. Selective Formic Acid Dehydrogenation on Pt-Cu Single-Atom Alloys. ACS Catal. 2017, 7, 413–420. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Wang, Z.L.; Yan, J.M.; Wang, H.L.; Ping, Y.; Jiang, Q. Pd/C synthesized with citric acid: An efficient catalyst for hydrogen generation from formic acid/sodium formate. Sci. Rep. 2012, 2, 598. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Onishi, N.; Laurenczy, G.; Beller, M.; Himeda, Y. Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol. Coord. Chem. Rev. 2018, 373, 317–332. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Van Putten, R.; Schulpen, E.N.; Hensen, E.J.M.; Pidko, E.A. Highly Efficient Reversible Hydrogenation of Carbon Dioxide to Formates Using a Ruthenium PNP-Pincer Catalyst. Chem. Cat Chem. 2014, 6, 1526–1530. [Google Scholar]
- Iglesias, M.; Oro, L.A. Mechanistic Considerations on Homogeneously Catalyzed Formic Acid Dehydrogenation. Eur. J. Inorg. Chem. 2018, 2018, 2125–2138. [Google Scholar] [CrossRef]
- Stathi, P.; Deligiannakis, Y.; Avgouropoulos, G.; Louloudi, M. Efficient H2 production from formic acid by a supported iron catalyst on silica. Appl. Catal. A Gen. 2015, 498, 176–184. [Google Scholar] [CrossRef]
- Solakidou, M.; Deligiannakis, Y.; Louloudi, M. Heterogeneous amino-functionalized particles boost hydrogen production from Formic Acid by a ruthenium complex. Int. J. Hydrogen Energy 2018, 21386–21397. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Q. Metal-Nanoparticle-Catalyzed Hydrogen Generation from Formic Acid. Acc. Chem. Res. 2017, 50, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Getoff, N. Photoelectrochemical and photocatalytic methods of hydrogen production: A short review. Int. J. Hydrogen Energy 1990, 15, 407–417. [Google Scholar] [CrossRef]
- Coffey, R.S. The decomposition of formic acid catalysed by soluble metal complexes. Chem. Commun. 1967, 923–924. [Google Scholar] [CrossRef]
- Gao, Y.; Kuncheria, J.; Yap, G.P.A.; Puddephatt, R.J. An efficient binuclear catalyst for decomposition of formic acid. Chem. Commun. 1998, 2365–2366. [Google Scholar] [CrossRef]
- Fellay, C.; Dyson, P.J.; Laurenczy, G. A viable hydrogen-storage system based on selective formic acid decomposition with a ruthenium catalyst. Angew. Chem. Int. Ed. 2008, 47, 3966–3968. [Google Scholar] [CrossRef]
- Durbin, D.J.; Malardier-Jugroot, C. Review of hydrogen storage techniques for on board vehicle applications. Int. J. Hydrogen Energy 2013, 38, 14595–14617. [Google Scholar] [CrossRef]
- Celaje, J.J.A.; Lu, Z.; Kedzie, E.A.; Terrile, N.J.; Lo, J.N.; Williams, T.J. A prolific catalyst for dehydrogenation of neat formic acid. Nat. Commun. 2016, 7, 11308. [Google Scholar] [CrossRef] [PubMed]
- Jantke, D.; Pardatscher, L.; Drees, M.; Cokoja, M.; Herrmann, W.A.; Kuhn, F.E. Hydrogen Production and Storage on a Formic Acid/Bicarbonate Platform Using Water-Soluble N-Heterocyclic Carbene Complexes of Late Transition Metals. ChemSusChem 2016, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, M.; Onishi, N.; Himeda, Y.; Kawanami, H. Ligand Effect on the Stability of Water-Soluble Iridium Catalysts for High-Pressure Hydrogen Gas Production by Dehydrogenation of Formic Acid. ChemPhysChem 2019, 20, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kuncheria, J.K.; Jcnkins, H.A.; Puddephatt, R.J.; Payap, G. The interconversion of formic acid and hydrogen/carbon dioxide using a binuclear ruthenium complex catalyst. J. Chem. Soc. Dalt. Trans. 2000, 3212–3217. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Junge, H.; Beller, M. Controlled generation of hydrogen from formic acid amine adducts at room temperature and application in H2/O2 fuel cells. Angew. Chem. Int. Ed. 2008, 47, 3962–3965. [Google Scholar] [CrossRef] [PubMed]
- Himeda, Y. Highly efficient hydrogen evolution by decomposition of formic acid using an iridium catalyst with 4,4′-dihydroxy-2,2′-bipyridine. Green Chem. 2009, 11, 2018–2022. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Unusually large tunneling effect on highly efficient generation of hydrogen and hydrogen isotopes in pH-selective decomposition of formic acid catalyzed by a heterodinuclear iridium-ruthenium complex in water. J. Am. Chem. Soc. 2010, 132, 1496–1497. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Catalytic (de)hydrogenation promoted by non-precious metals-Co, Fe and Mn: Recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. [Google Scholar] [CrossRef] [Green Version]
- Boddien, A.; Loges, B.; Garthner, F.; Torborg, C.; Fumino, K.; Junge, H.; Ludwig, R.; Beller, M. Iron-catalyzed Hydrogen production from formic acid. J. Am. Chem. Soc. 2010, 132, 8924–8934. [Google Scholar] [CrossRef]
- Scotti, N.; Psaro, R.; Ravasio, N.; Zaccheria, F. A new Cu-based system for formic acid dehydrogenation. RSC Adv. 2014, 4, 61514–61517. [Google Scholar] [CrossRef]
- Boddien, A.; Mellmann, D.; Gärtner, F.; Jackstell, R.; Junge, H.; Dyson, P.J.; Laurenczy, G.; Ludwig, R.; Beller, M. Efficient dehydrogenation of formic acid using an iron catalyst. Science 2011, 333, 1733–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zell, T.; Butschke, B.; Ben-David, Y.; Milstein, D. Efficient hydrogen liberation from formic acid catalyzed by a well-defined iron pincer complex under mild conditions. Chem. Eur. J. 2013, 19, 8068–8072. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Nerush, A.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Reversible CO2 binding triggered by metal-ligand cooperation in a rhenium(i) PNP pincer-type complex and the reaction with dihydrogen. Chem. Sci. 2014, 5, 2043–2051. [Google Scholar] [CrossRef]
- Zhang, Y.; Würtele, C.; Lagaditis, P.O.; Bielinski, E.A.; Hazari, N.; Schneider, S.; Bernskoetter, W.H.; Mercado, B.Q. Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst. J. Am. Chem. Soc. 2014, 136, 10234–10237. [Google Scholar]
- Mellone, I.; Gorgas, N.; Bertini, F.; Peruzzini, M.; Kirchner, K.; Gonsalvi, L. Selective Formic Acid Dehydrogenation Catalyzed by Fe-PNP Pincer Complexes Based on the 2,6-Diaminopyridine Scaffold. Organometallics 2016, 35, 3344–3349. [Google Scholar] [CrossRef]
- Curley, J.B.; Smith, N.E.; Bernskoetter, W.H.; Hazari, N.; Mercado, B.Q. Catalytic Formic Acid Dehydrogenation and CO2 Hydrogenation Using Iron PNRP Pincer Complexes with Isonitrile Ligands. Organometallics 2018, 37, 3846–3853. [Google Scholar] [CrossRef]
- Fellay, C.; Yan, N.; Dyson, P.J.; Laurenczy, G. Selective formic acid decomposition for high-pressure hydrogen generation: A mechanistic study. Chem. Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef]
- Stathi, P.; Mitrikas, G.; Sanakis, Y.; Louloudi, M.; Deligiannakis, Y. Back-clocking of Fe2 +/Fe1 + spin states in a H2-producing catalyst by advanced EPR. Mol. Phys. 2013, 111, 18–19. [Google Scholar] [CrossRef]
- Mellmann, D.; Barsch, E.; Bauer, M.; Grabow, K.; Boddien, A.; Kammer, A.; Sponholz, P.; Bentrup, U.; Jackstell, R.; Junge, H.; et al. Base-free non-noble-metal-catalyzed hydrogen generation from formic acid: Scope and mechanistic insights. Chem. Eur. J. 2014, 20, 13589–13602. [Google Scholar] [CrossRef]
- Yang, X. Mechanistic insights into iron catalyzed dehydrogenation of formic acid: β-hydride elimination vs. direct hydride transfer. Dalt. Trans. 2013, 42, 11987–11991. [Google Scholar] [CrossRef]
- Sánchez-De-Armas, R.; Xue, L.; Ahlquist, M.S.G. One site is enough: A theoretical investigation of iron-catalyzed dehydrogenation of formic acid. Chem. Eur. J. 2013, 19, 11869–11873. [Google Scholar] [CrossRef] [PubMed]
- Stathi, P.; Deligiannakis, Y.; Louloudi, M. Co-catalytic enhancement of H2 production by SiO2 nanoparticles. Catal. Today 2015, 242, 146–152. [Google Scholar] [CrossRef]
- Stathi, P.; Louloudi, M.; Deligiannakis, Y. Efficient Low-Temperature H2 Production from HCOOH/HCOO– by [Pd0@SiO2-Gallic Acid] Nanohybrids: Catalysis and the Underlying Thermodynamics and Mechanism. Energy Fuels 2016, 30, 8613–8622. [Google Scholar] [CrossRef]
- Wang, W.-H.; Xu, S.; Manaka, Y.; Suna, Y.; Kambayashi, H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Formic Acid Dehydrogenation with Bioinspired Iridium Complexes: A Kinetic Isotope Effect Study and Mechanistic Insight. ChemSusChem 2014, 7, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Manca, G.; Mellone, I.; Bertini, F.; Peruzzini, M.; Rosi, L.; Mellmann, D.; Junge, H.; Beller, M.; Ienco, A.; Gonsalvi, L. Inner-versus outer-sphere ru-catalyzed formic acid dehydrogenation: A computational study. Organometallics 2013, 32, 7053–7064. [Google Scholar] [CrossRef]
- Linn, D.E.; King, R.B.; King, A.D. Catalytic reactions of formate: Part 1. Photocatalytic hydrogen production from formate with chromium hexacarbonyl. J. Mol. Catal. 1993, 80, 151–163. [Google Scholar] [CrossRef]
- Onishi, M. Decomposition of formic acid catalyzed by hydrido (phosphonite) cobalt (I) under photoirradiation. J. Mol. Catal. 1993, 80, 145–149. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Junge, H.; Noyes, J.R.; Baumann, W.; Beller, M. Hydrogen generation: Catalytic acceleration and control by light. Chem. Commun. 2009, 4185–4187. [Google Scholar] [CrossRef]
- Junge, H.; Boddien, A.; Capitta, F.; Loges, B.; Noyes, J.R.; Gladiali, S.; Beller, M. Improved hydrogen generation from formic acid. Tetrahedron Lett. 2009, 50, 1603–1606. [Google Scholar] [CrossRef] [Green Version]
- Stathi, P.; Deligiannakis, Y.; Louloudi, M. Co-catalytic Effect of Functionalized SiO2 Material on H2 Production from Formic Acid by an Iron Catalyst. Mater. Res. Soc. Symp. Proc. 2014, 1641. [Google Scholar] [CrossRef]
- Papp, G.; Ölveti, G.; Horváth, H.; Kathó, A.; Joó, F. Highly efficient dehydrogenation of formic acid in aqueous solution catalysed by an easily available water-soluble iridium(III) dihydride. Dalt. Trans. 2016, 45, 14516–14519. [Google Scholar] [CrossRef] [PubMed]
- Boddien, A.; Loges, B.; Junge, H.; Gärtner, F.; Noyes, J.R.; Beller, M. Continuous hydrogen generation from formic acid: Highly active and stable ruthenium catalysts. Adv. Synth. Catal. 2009, 351, 2517–2520. [Google Scholar] [CrossRef]
- Morris, D.J.; Clarkson, G.J.; Wills, M. Insights into hydrogen generation from formic acid using ruthenium complexes. Organometallics 2009, 28, 4133–4140. [Google Scholar] [CrossRef]
- Fujita, E.; Muckerman, J.T.; Himeda, Y. Interconversion of CO2 and formic acid by bio-inspired Ir complexes with pendent bases. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1031–1038. [Google Scholar] [CrossRef] [Green Version]
- Van Putten, R.; Wissink, T.; Swinkels, T.; Pidko, E.A. Fuelling the hydrogen economy: Scale-up of an integrated formic acid-to-power system. Int. J. Hydrogen Energy 2019, 44, 28533–28541. [Google Scholar] [CrossRef]
- Serafimidou, A.; Stamatis, A.; Louloudi, M. Manganese(II) complexes of imidazole based-acetamide as homogeneous and heterogenised catalysts for alkene epoxidation with H2O2. Catal. Commun. 2008, 9, 35–39. [Google Scholar] [CrossRef]
- Bilis, G.; Stathi, P.; Mavrogiorgou, A.; Deligiannakis, Y.; Louloudi, M. Improved robustness of heterogeneous Fe-non-heme oxidation catalysts: A catalytic and EPR study. Appl. Catal. A Gen. 2014, 470, 376–389. [Google Scholar] [CrossRef]
- Mavrogiorgou, A.; Papastergiou, M.; Deligiannakis, Y.; Louloudi, M. Activated carbon functionalized with Mn(II) Schiff base complexes as efficient alkene oxidation catalysts: Solid support matters. J. Mol. Catal. A Chem. 2014, 393, 8–17. [Google Scholar] [CrossRef]
- Gan, W.; Dyson, P.J.; Laurenczy, G. Heterogeneous silica-supported ruthenium phosphine catalysts for selective formic acid decomposition. ChemCatChem 2013, 5, 3124–3130. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, L.; Tang, S.Y.; Lai, D.M.; Liao, B.; Fu, Y.; Guo, Q.X. Selective decomposition of formic acid over immobilized catalysts. Energy Fuels 2011, 25, 3693–3697. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Kim, H.; Yoon, S. Dehydrogenation of formic acid using molecular Rh and Ir catalysts immobilized on bipyridine-based covalent triazine frameworks. Sustain. Energy Fuels 2019, 3, 1042–1047. [Google Scholar] [CrossRef]
- Kuo, C.; Lu, Y.; Kovarik, L.; Engelhard, M.H.; Karim, A.M. Structure Sensitivity of Acetylene Semi-Hydrogenetion on Pt Single atoms and Subnanometer clusters. ACS Catal. 2019, 9, 11030–11041. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalytic Complex | Temp. °C | Operation Time (h) | TONs | TOFs (h−1) | Substrate/Solvent | Additive | FA/Additive Ratio | Ref. |
|---|---|---|---|---|---|---|---|---|
| Noble Metals (Homogeneous Catalyst/Homogenous Base Additive) | ||||||||
| Ru2(μ-CO)(CO4)(μ-DPPM)2 | RT | - | 500 | FA/Acetone | - | - | [30] | |
| [Ru(H2O)6](tos)2 | 120 | 90 | 40,000 | 460 | FA, SF/H2O | - | - | [31] |
| [RuCl2(C6H6)]2/DPPE | 40 | 264 | 260,000 | 900 | FA/Dimethylamine | - | - | [65] |
| [Cp*Ir(TMBI)H2O]SO4 | 80 | 0.17 | 10,000 | 34,000 | FA/H2O | - | - | [65] |
| [Ir(COD)(NP)](tfO) | 90 | 2880 | 2,160,000 | - | FA/SF | - | - | [33] |
| Cp*Rh(bis-NHC)Cl]Na | 100 | 50 | 449,000 | 9000 | FA/H2O | - | - | [34] |
| [RuCl2(p-cymene)]2 | 40 | 3 | 76 | FA/H2O | NEt3 | 2/5 | [61] | |
| [RuCl2(p-cymene)]2 | 40 | 3 | 21 | FA/H2O | NEt3 | 3/4 | [61] | |
| [RuCl2(p-cymene)]2 | 40 | 3 | 40 | FA/H2O | HexNMe2 | 4/5 | [61] | |
| Homogeneous Catalyst/Heterogeneous Base Co-catalyst | ||||||||
| Ru/P(CH2CH2PPh2) | 80 | 696 | 3924 | 823 | FA/PC | SiO2-NH2 | 500/1 | [26] |
| Non-Noble metals (Homogeneous catalyst/homogenous additive) | ||||||||
| [Fe(CO)12/benzylphospine/TPY | 60 | 51 | 1266 | 25 | FA/DMF | light | - | [41] |
| Fe(BF4)2/PPh3 | 80 | 18 | 92,417 | 5390 | FA/PC | - | - | [43] |
| [(PNP)Fe(H)2(CO) | 40 | 240 | 100,000 | 420 | FA/THF | Net3 | 1/2 | [44] |
| [(PNP)Re(CO)2] | 180 | 1 | 3300 | 3300 | FA/Dioxane | - | [45] | |
| Homogeneous Catalyst/Heterogeneous Base Co-catalyst | ||||||||
| Fe(BF4)2/PPh3 | 80 | 200 | 8483 | 6245 | FA/PC | SiO2 | 200/1 | [54] |
| Fe(BF4)2/PPh3 | 80 | 88 | 8564 | 5773 | FA/PC | SiO2-COOH | 577/1 | [62] |
| Fe(BF4)2/PPh3 | 80 | 34 | 8668 | 14,942 | FA/PC | SiO2-NH2 | 500/1 | [62] |
| Homogeneous catalyst/homogenous additive: Lewis-Acid Additives | ||||||||
| [PNP1 Fe (H)(CO)(OOCH] | 80 | 4 | 231 | 999 | FA/Dioxane | LiBF4 | 1/0.1 | [46] |
| [PNP1 Fe (H)(CO)(OOCH] | 80 | 4 | 263 | 999 | FA/Dioxane | NaCl | 1/0.1 | [46] |
| [PNP1 Fe (H)(CO)(OOCH] | 80 | 4 | 323 | 999 | FA/Dioxane | NaBF4 | 1/0.1 | [46] |
| Immobilized catalysts | ||||||||
| Fe(BF4)2-PPh3@SiO2 | 80 | 1.25 | 7869 | 6295 | FA/PC | - | [25] | |
| Pd-P@ SiO2 | 85 | - | 719 | - | H2O | - | - | [72] |
| Ru-S-SiO2 | 85 | - | 344 | H2O | - | - | [72] | |
| Ru-P-SiO2 | 85 | - | 102 | H2O | - | - | [72] | |
| Pd-N-SiO2 | 85 | - | 115 | H2O | - | - | [72] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stathi, P.; Solakidou, M.; Louloudi, M.; Deligiannakis, Y. From Homogeneous to Heterogenized Molecular Catalysts for H2 Production by Formic Acid Dehydrogenation: Mechanistic Aspects, Role of Additives, and Co-Catalysts. Energies 2020, 13, 733. https://0-doi-org.brum.beds.ac.uk/10.3390/en13030733
Stathi P, Solakidou M, Louloudi M, Deligiannakis Y. From Homogeneous to Heterogenized Molecular Catalysts for H2 Production by Formic Acid Dehydrogenation: Mechanistic Aspects, Role of Additives, and Co-Catalysts. Energies. 2020; 13(3):733. https://0-doi-org.brum.beds.ac.uk/10.3390/en13030733
Chicago/Turabian StyleStathi, Panagiota, Maria Solakidou, Maria Louloudi, and Yiannis Deligiannakis. 2020. "From Homogeneous to Heterogenized Molecular Catalysts for H2 Production by Formic Acid Dehydrogenation: Mechanistic Aspects, Role of Additives, and Co-Catalysts" Energies 13, no. 3: 733. https://0-doi-org.brum.beds.ac.uk/10.3390/en13030733