CO2 Electrochemical Reduction by Exohedral N-Pyridine Decorated Metal-Free Carbon Nanotubes


 , , and
, , and
Abstract
:
1. Introduction
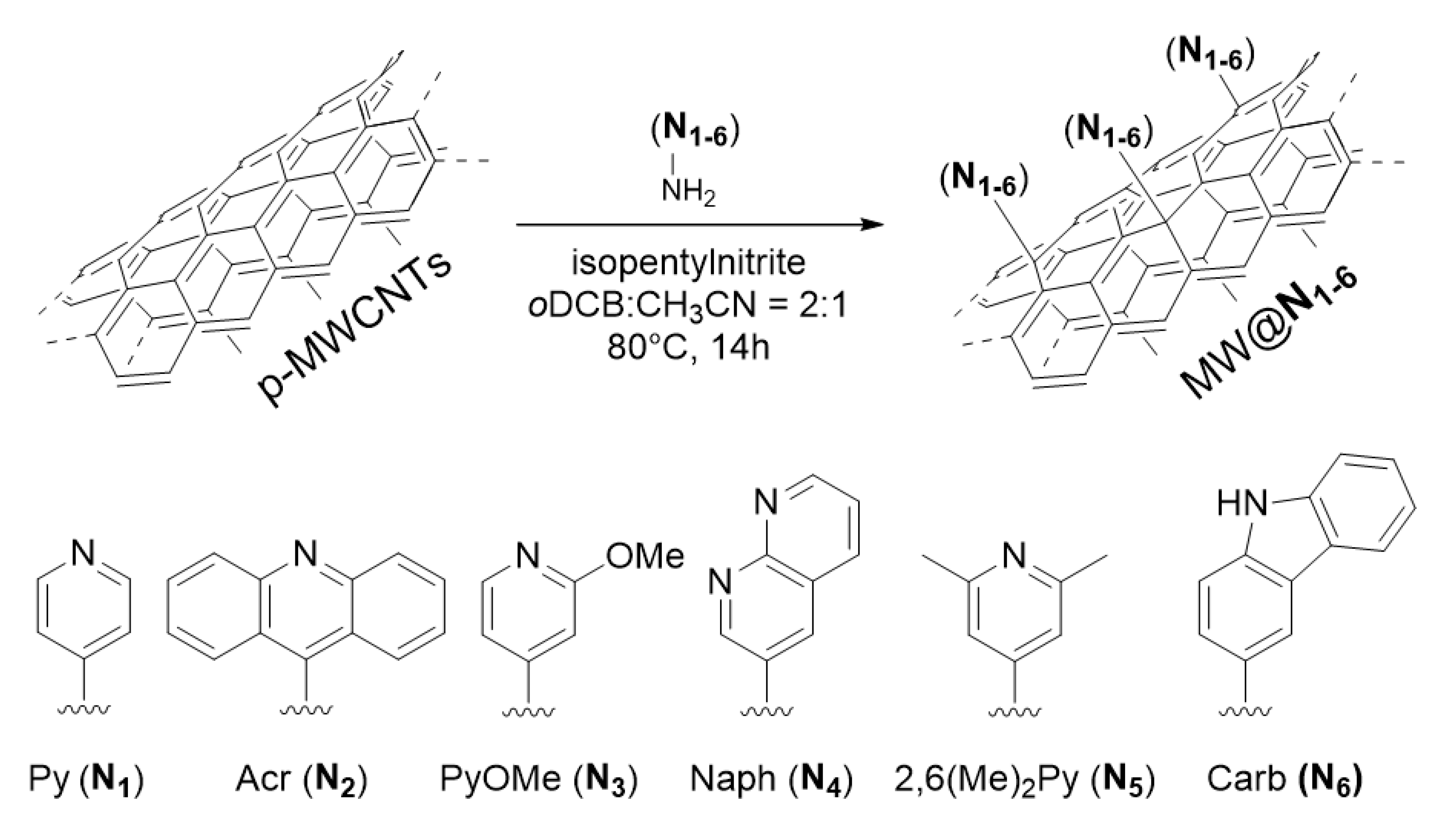
2. Materials and Methods
2.1. Electrochemical Measurements
2.1.1. Working Electrode Preparation
2.1.2. Cell Assembly
2.1.3. Electrolysis Experiments and Products Analysis
2.2. Computational Methods
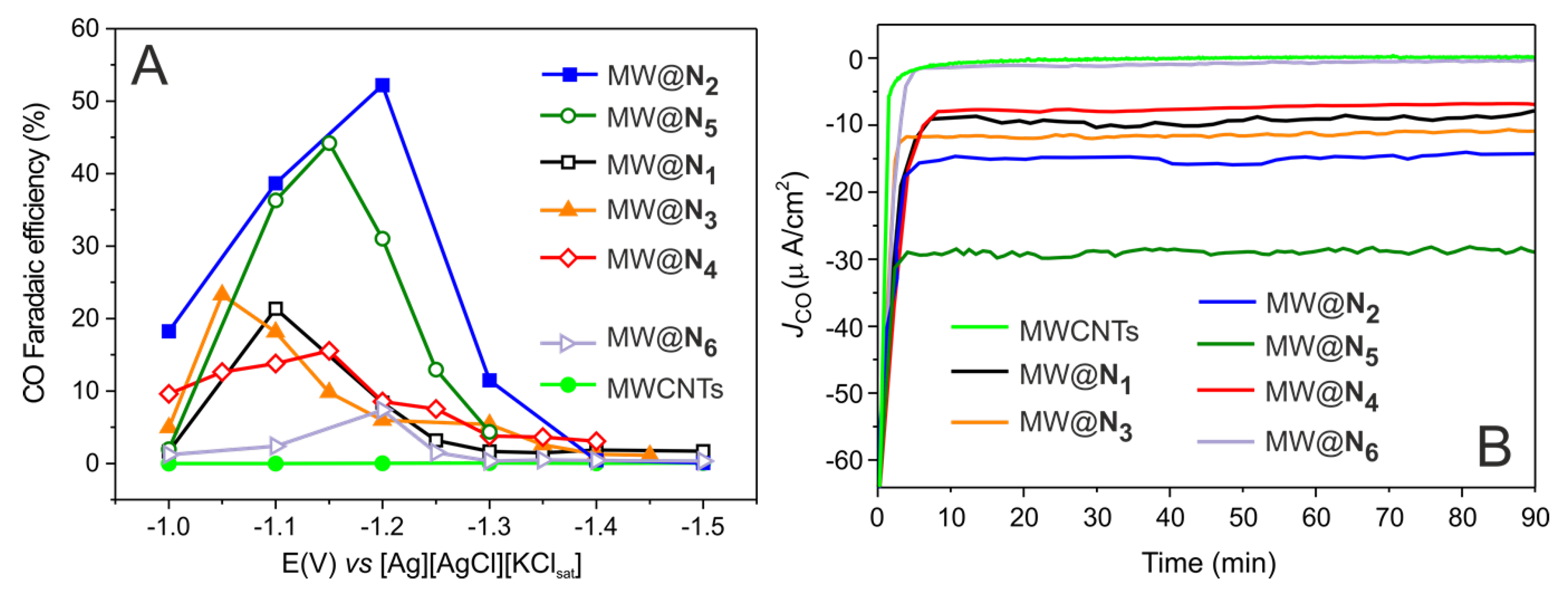
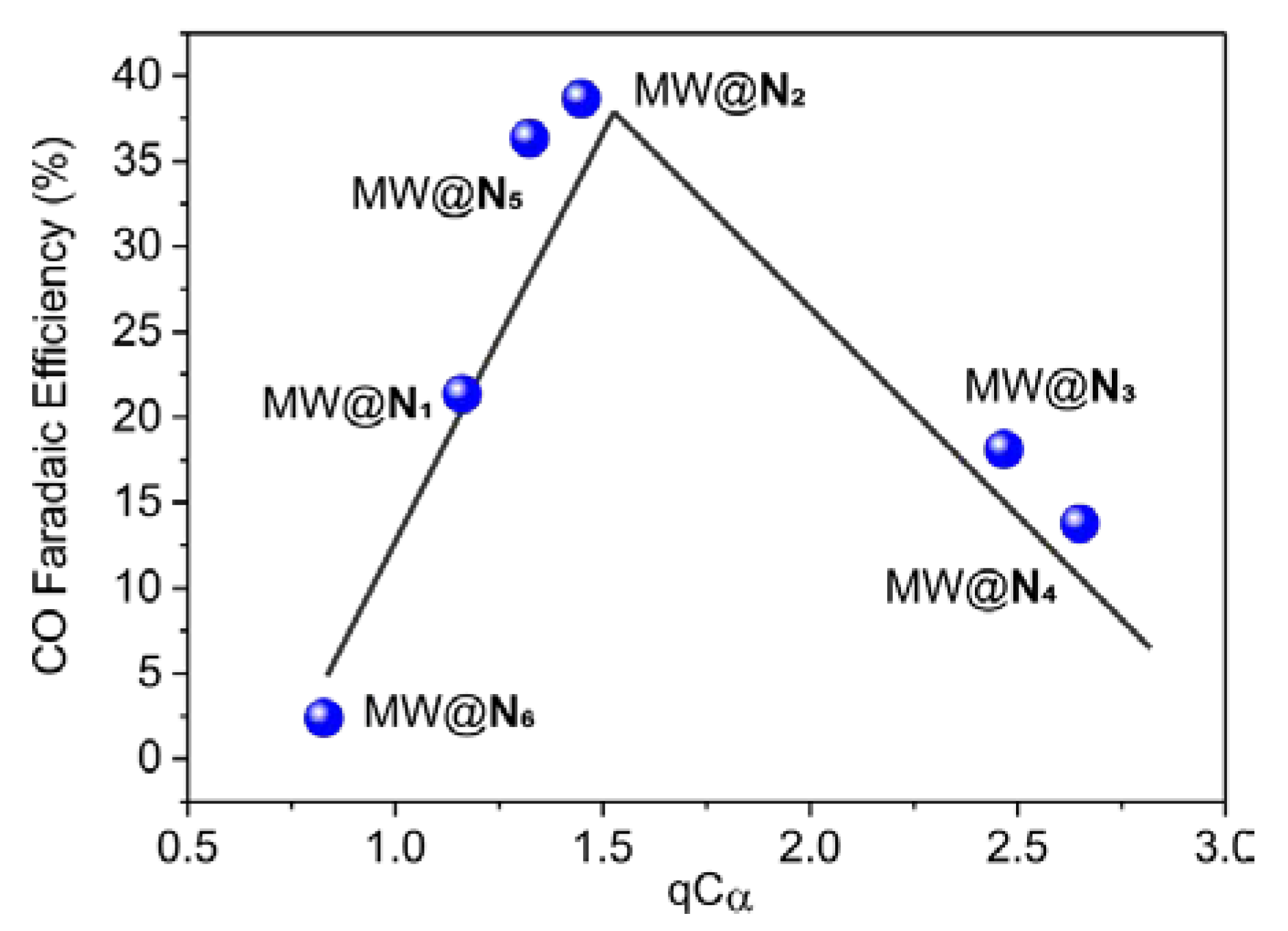
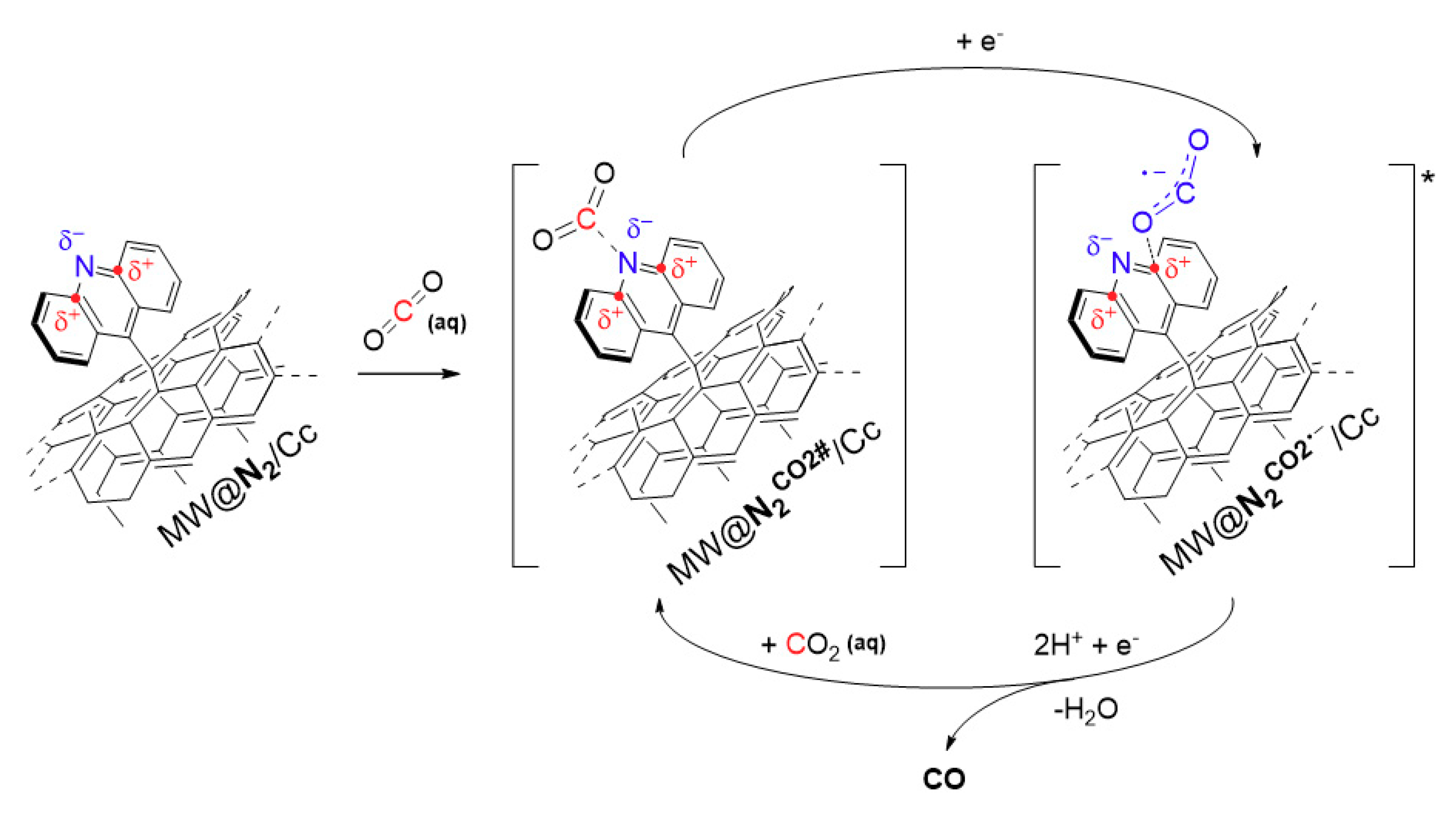
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martens, J.A.; Bogaerts, A.; De Kimpe, N.; Jacobs, P.A.; Marin, G.B.; Rabaey, K.; Saeys, M.; Verhelst, S. The Chemical Route to a Carbon Dioxide Neutral World. ChemSusChem 2010, 10, 1039–1055. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Cherevotan, A.; Peter, S.C. Thermochemical CO2 Hydrogenation to Single Carbon Products: Scientific and Technological Challenges. ACS Energy Lett. 2018, 3, 1938–1966. [Google Scholar] [CrossRef] [Green Version]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Jiao, F. Electrochemical CO2 reduction: Electrocatalyst, reaction mechanism, and process engineering. Nano Energy 2016, 29, 439–456. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, A.; Mehrpooya, M. A comprehensive review on coupling different types of electrolyzer to renewable energy sources. Energy 2018, 158, 632–655. [Google Scholar] [CrossRef]
- Hori, Y. Modern Aspects of Electrochemistry; Vayenas, G.C., White, E.R., Eds.; Springer: Patras, Greece, 2008; p. 89. [Google Scholar]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Zheng, T.; Jiang, K.; Wang, H. Recent advances in electrochemical CO2-to-CO conversion on heterogeneous catalysts. Adv. Mater. 2018, 30, 1802066. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and Prospects in the Chemical Recycling of Carbon Dioxide to Fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Jiang, K.; Siahrostami, S.; Akey, A.J.; Li, Y.; Lu, Z.; Lattimer, J.; Hu, Y.; Stokes, C.; Gangishetty, M.; Chen, G.; et al. Transition-Metal Single Atoms in a Graphene Shell as Active Centers for Highly Efficient Artificial Photosynthesis. Chem 2017, 3, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.-M.; Zhu, Y.-P.; Chen, P.; Ma, T.-Y. Recent advances in transition-metal-mediated electrocatalytic CO2 reduction: From homogeneous to heterogeneous systems. Catalysts 2017, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide-Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ajmal, S.; Zheng, X.; Zhang, L. Efficient nanomaterials for harvesting clean fuels from electrochemical and photoelectrochemical CO2 reduction. Sustain. Energy Fuels 2018, 2, 510–537. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Z.-J.; Gong, J. Nanostructured materials for heterogeneous electrocatalytic CO2 reduction and their related reaction mechanisms. Angew. Chem. Int. Ed. 2017, 56, 11326–11353. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef]
- Bevilacqua, M.; Filippi, J.; Miller, H.A.; Vizza, F. Recent technological progress in CO2 electroreduction to fuels and energy carriers in aqueous environments. Energy Technol. 2015, 3, 197–210. [Google Scholar] [CrossRef]
- Sharma, P.P.; Wu, J.; Yadav, R.M.; Liu, M.; Wright, C.J.; Tiwary, C.S.; Yakobson, B.I.; Lou, J.; Ajayan, P.M.; Zhou, X.-D. Nitrogen-doped carbon nanotube arrays for high-efficiency electrochemical reduction of CO2: On the understanding of defects, defect density, and selectivity. Angew. Chem. Int. Ed. 2015, 54, 13701–13705. [Google Scholar] [CrossRef]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-Free Carbon Materials for CO2 Electrochemical Reduction. Adv. Mater. 2017, 29, 1701784. [Google Scholar] [CrossRef]
- Chai, G.-L.; Guo, Z.-X. Highly effective sites and selectivity of nitrogendoped graphene/CNT catalysts for CO2 electrochemical reduction. Chem. Sci. 2016, 7, 1268–1275. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Asadi, M.; Pisasale, D.; Sinha-Ray, S.; Rosen, B.A.; Haasch, R.; Abiade, J.; Yarin, A.L.; Salehi-Khojin, A. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nat. Commun. 2013, 4, 2819. [Google Scholar] [CrossRef]
- Li, W.; Seredych, M.; Rodriguez-Castellon, E.; Bandosz, T.J. Metal-free nanoporous carbon as a catalyst for electrochemical reduction of CO2 to CO and CH4. ChemSusChem 2016, 9, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Tan, T.H.; Ng, Y.H.; Amal, R. Highly selective and stable reduction of CO2 to CO by a graphitic carbon nitride/carbon nanotube composite electrocatalyst. Chem. Eur. J. 2016, 22, 11991–11996. [Google Scholar] [CrossRef]
- Molly Jhong, H.-R.; Tornow, C.E.; Smid, B.; Gewirth, A.A.; Lyth, S.M.; Kenis, P.J.A. A nitrogen-doped carbon catalyst for electrochemical CO2 conversion to CO with high selectivity and current density. ChemSusChem 2017, 10, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.-D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of nitrogen defects for efficient reduction of CO2 via two-electron pathway on three-dimensional graphene foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef]
- Wu, J.; Yadav, R.M.; Liu, M.; Sharma, P.P.; Sekhar Tiwary, C.; Ma, L.; Zou, X.; Zhou, X.-D.; Yakobson, B.I.; Lou, J.; et al. Achieving highly efficient, selective, and stable CO2 reduction on nitrogen-doped carbon nanotubes. ACS Nano 2015, 9, 5364–5371. [Google Scholar] [CrossRef]
- Xu, J.; Kan, Y.; Huang, R.; Zhang, B.; Wang, B.; Wu, K.-H.; Lin, Y.; Sun, X.; Li, Q.; Centi, G.; et al. Revealing the Origin of Activity in Nitrogen-Doped Nanocarbons towards Electrocatalytic Reduction of Carbon Dioxide. ChemSusChem 2016, 9, 1085–1089. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhao, T.-J.; Feng, W.-J.; Liu, Y.-X.; Wang, H.-H.; Su, H.; Lv, L.-B.; Li, X.-H.; Chen, J.-S. Polarized few-layer g-C3N4 as metal-free electrocatalyst for highly efficient reduction of CO2. Nano Res. 2018, 11, 2450–2459. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Cai, Q. Pyrrolic-nitrogen doped graphene: A metal-free electrocatalyst with high efficiency and selectivity for the reduction of carbon dioxide to formic acid: A computational study. Phys. Chem. Chem. Phys. 2016, 18, 5491–5498. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Hou, X.; Ma, C.; Tan, T. Nitrogen-doped graphenes as efficient electrocatalysts for the selective reduction of carbon dioxide to formate in aqueous solution. Green Chem. 2016, 18, 3250–3256. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Ubnoske, S.; Brennaman, M.K.; Song, N.; House, R.L.; Glass, J.T.; Meyer, T.J. Polyethylenimine-enhanced electrocatalytic reduction of CO2 to formate at nitrogen-doped carbon nanomaterials. J. Am. Chem. Soc. 2014, 136, 7845–7848. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Quan, X.; Yu, H. Efficient Electrochemical Reduction of Carbon Dioxide to Acetate on Nitrogen-Doped Nanodiamond. J. Am. Chem. Soc. 2015, 137, 11631–11636. [Google Scholar] [CrossRef] [PubMed]
- Lum, Y.; Kwon, Y.; Lobaccaro, P.; Chen, L.; Clark, E.L.; Bell, A.T.; Ager, J.W. Trace levels of copper in carbon materials show significant electrochemical CO2 reduction activity. ACS Catal. 2016, 6, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Siahrostami, S.; Jiang, K.; Karamad, M.; Chan, K.; Wang, H.; Nørskov, J. Theoretical investigations into defected graphene for electrochemical reduction of CO2. ACS Sustain. Chem. Eng. 2017, 5, 11080–11085. [Google Scholar] [CrossRef]
- Wu, J.; Ma, S.; Sun, J.; Gold, J.I.; Tiwary, C.S.; Kim, B.; Zhu, L.; Chopra, N.; Odeh, I.N.; Vajtai, R.; et al. A metal-free electrocatalyst for carbon dioxide reduction to multi-carbon hydrocarbons and oxygenates. Nat. Commun. 2016, 7, 13869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuci, G.; Filippi, J.; Ba, H.; Rossin, A.; Luconi, L.; Pham-Huu, C.; Vizza, F.; Giambastiani, G. How to Teach an Old Dog New (Electrochemical) Tricks: Aziridine-Functionalized CNTs as Efficient Electrocatalysts for the Selective CO2 Reduction to CO. J. Mater. Chem. A 2018, 6, 16383–16389. [Google Scholar] [CrossRef]
- Tuci, G.; Luconi, L.; Rossin, A.; Berretti, E.; Ba, H.; Innocenti, M.; Yakhvarov, D.; Caporali, S.; Pham-Huu, C.; Giambastiani, G. Aziridine-functionalized multiwalled carbon nanotubes: Robust and versatile catalysts for the oxygen reduction reaction and Knoevenagel condensation. ACS Appl. Mater. Interfaces 2016, 8, 30099–30106. [Google Scholar] [CrossRef]
- Tuci, G.; Luconi, L.; Rossin, A.; Giambastiani, G. Chemical Functionalization of Carbon Nanomaterials: Bridging the Gap between Simple Carriers and Smart (Metalfree) Catalysts. Chimia 2017, 71, 568–572. [Google Scholar] [CrossRef]
- Tuci, G.; Rossin, A.; Luconi, L.; Pham-Huu, C.; Cicchi, S.; Ba, H.; Giambastiani, G. Pyridine-decorated carbon nanotubes as a metal-free heterogeneous catalyst for mild CO2 reduction to methanol with hydroboranes. Catal. Sci. Technol. 2017, 7, 5833–5837. [Google Scholar] [CrossRef] [Green Version]
- Tuci, G.; Zafferoni, C.; D’Ambrosio, P.; Caporali, S.; Ceppatelli, M.; Rossin, A.; Tsoufis, T.; Innocenti, M.; Giambastiani, G. Tailoring Carbon Nanotube N-Dopants while Designing Metal-Free Electrocatalysts for the Oxygen Reduction Reaction in Alkaline Medium. ACS Catal. 2013, 3, 2108–2111. [Google Scholar] [CrossRef]
- Tuci, G.; Zafferoni, C.; Rossin, A.; Luconi, L.; Milella, A.; Ceppatelli, M.; Innocenti, M.; Liu, Y.; Pham-Huu, C.; Giambastiani, G. Chemical functionalization of N-doped carbon nanotubes: A powerful approach to cast light on the electrochemical role of specific N-functionalities in the Oxygen Reduction Reaction. Catal. Sci. Technol. 2016, 6, 6226–6236. [Google Scholar] [CrossRef]
- Tuci, G.; Zafferoni, C.; Rossin, A.; Milella, A.; Luconi, L.; Innocenti, M.; Truong Phuoc, L.; Duong-Viet, C.; Pham-Huu, C.; Giambastiani, G. Chemically functionalized carbon nanotubes with pyridine groups as easily tunable N-decorated nanomaterials for the oxygen reduction reaction in alkaline medium. Chem. Mater. 2014, 26, 3460–3470. [Google Scholar] [CrossRef]
- Wang, H.; Jia, J.; Song, P.; Wang, Q.; Li, D.; Min, S.; Qian, C.; Wang, L.; Li, Y.F.; Ma, C.; et al. Efficient electrocatalytic reduction of CO2 by nitrogen-doped nanoporous carbon/carbon nanotube membranes: A step towards the electrochemical CO2 refinery. Angew. Chem. Int. Ed. 2017, 56, 7847–7852. [Google Scholar] [CrossRef] [Green Version]
- ABS (Acrylonitrile-Butadiene-Styrene) is a common base-resistant thermoplastic polymer for 3D-printing.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.A.J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Singh, S.K.; Takeyasu, K.; Nakamura, J. Active Sites and Mechanism of Oxygen Reduction Reaction Electrocatalysis on Nitrogen-Doped Carbon Materials. Adv. Mater. 2019, 31, 1804297. [Google Scholar] [CrossRef]
- Dai, L.; Xue, Y.; Qu, L.; Choi, H.-J.; Baek, J.-B. Metal-Free Catalysts for Oxygen Reduction Reaction. Chem. Rev. 2015, 115, 4823–4892. [Google Scholar] [CrossRef]
- Chizari, K.; Vena, A.; Laurentius, L.; Sundararaj, U. The effect of temperature on the morphology and chemical surface properties of nitrogen-doped carbon nanotubes. Carbon 2014, 68, 369–379. [Google Scholar] [CrossRef]
- Louette, P.; Bodino, F.; Pireaux, J.-J. Poly(pyrrole) (PPY) XPS Reference Core Level and Energy Loss Spectra. Surf. Sci. Spectra 2005, 12, 84–89. [Google Scholar] [CrossRef]
- Dyke, C.A.; Stewart, M.P.; Maya, F.; Tour, J.M. Diazonium-based functionalization of carbon nanotubes: XPS and GC-MS analysis and mechanistic implications. Synlett 2004, 2004, 155–160. [Google Scholar] [CrossRef]
- Price, B.K.; Tour, J.M. Functionalization of Single-Walled Carbon Nanotubes “On Water”. J. Am. Chem. Soc. 2006, 128, 12899–12904. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.A.; Chua, C.K.; Khezri, B.; Webster, R.D.; Pumera, M. Graphene Oxide Nanoribbons from the Oxidative Opening of Carbon Nanotubes Retain Electrochemically Active Metallic Impurities. Angew. Chem. Int. Ed. 2013, 52, 8685–8688. [Google Scholar] [CrossRef]
- The higher N-loading obtained for MW@N6 is in line with the electron rich character of carbazole heterocycles and the consequent higher reactivity of their postulated aryl radical intermediates obtained during the Tour functionalization protocol.
- Gouse Peera, S.; Sahu, A.K.; Arunchander, A.; Bhat, S.D.; Karthikeyan, J.; Murugan, P. Nitrogen and fluorine co-doped graphite nanofibers as high durable oxygen reduction catalyst in acidic media for polymer electrolyte fuel cells. Carbon 2015, 93, 130–142. [Google Scholar] [CrossRef]
- Lin, I.-H.; Lu, Y.-H.; Chen, H.-T. Nitrogen-doped carbon nanotube as a potential metal-free catalyst for CO oxidation. Phys. Chem. Chem. Phys. 2016, 18, 12093–12100. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Ali, S.; Li, B.; Su, D.S. Revealing the role of sp2@sp3 structure of nanodiamond in direct dehydrogenation: Insight from DFT study. ACS Catal. 2017, 7, 3779–3785. [Google Scholar] [CrossRef]
- Zhu, Y.; Lv, K.; Wang, X.; Yang, H.; Xiao, G.; Zhu, Y. 1D/2D nitrogen-doped carbon nanorod arrays/ultrathin carbon nanosheets: Outstanding catalysts for the highly efficient electroreduction of CO2 to CO. J. Mater. Chem. A 2019, 7, 14895–14903. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
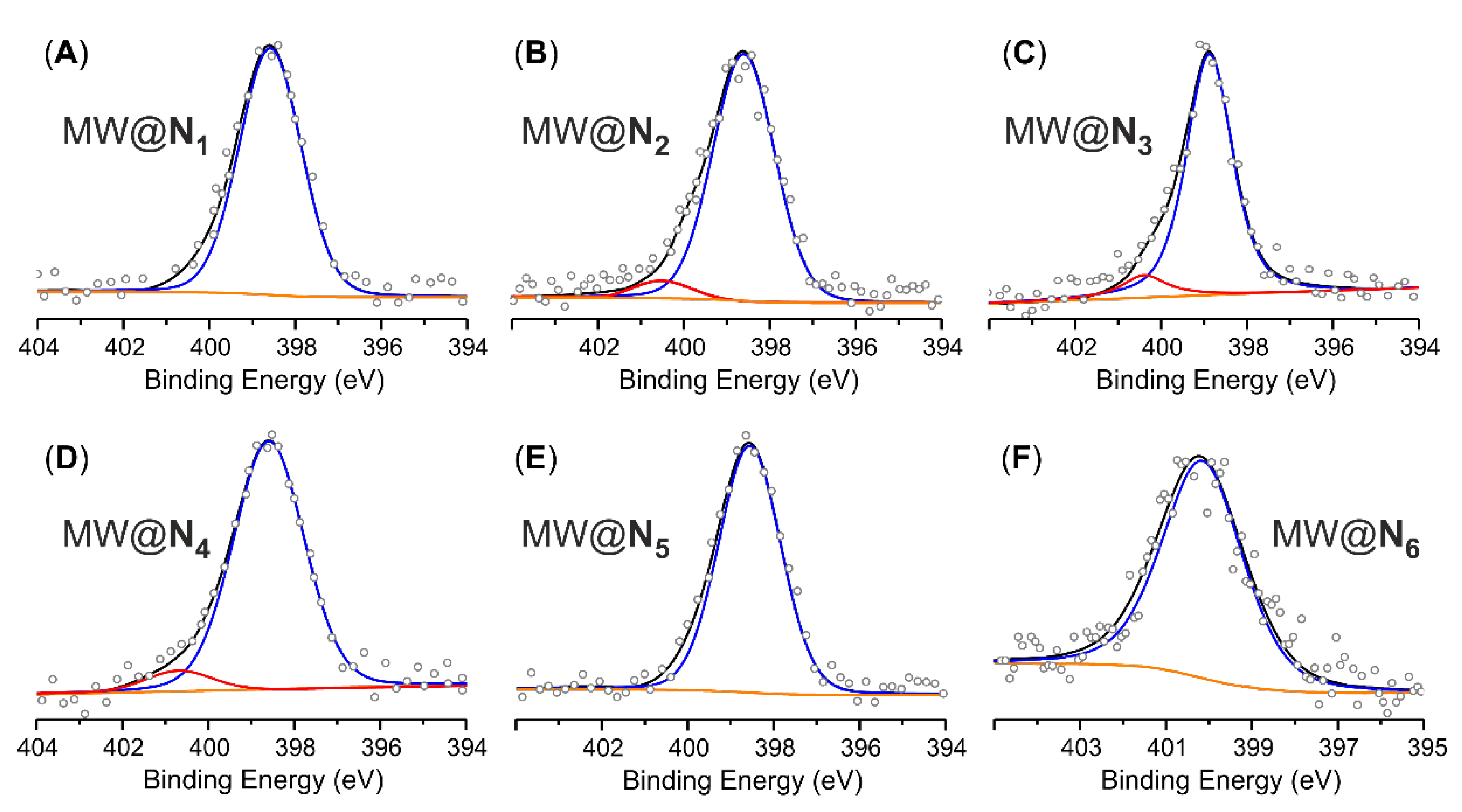
| Samples | Elemental Analysisa | Acid-base titrationb | XPS analysis | ||
|---|---|---|---|---|---|
| C (%) | N (%) | N (wt.%)c [mmol g−1]d | NPy (wt.%) [mmol g−1]d | N (at.%) [mmol g−1]f | |
| MWCNTse MW@N1 | 94.08 90.18 | 0.20 1.76 | 1.56; [1.11] | 1.42; [1.01] | 2.0; [1.01] |
| MWCNTse,g MW@N2 | 92.34 91.35 | 0.29 1.57 | 1.28; [0.91] | 1.27; [0.91] | 1.6; [1.33] |
| MWCNTse MW@N3 | 94.56 90.47 | 0.07 1.24 | 1.17; [0.84] | 1.00; [0.71] | 1.2; [1.00] |
| MWCNTse MW@N4 | 94.54 91.35 | 0.27 2.14 | 1.87; [1.33] | 1.63; [1.16] | 2.0; [1.66] |
| MWCNTse MW@N5 | 93.67 91.87 | 0.09 1.39 | 1.30; [0.93] | 1.17; [0.84] | 1.5; [1.24] |
| MWCNTse MW@N6 | 94.30 90.89 | 0.14 2.83 | 2.69; [1.92] | - | 2.9; [2.40] |
| Entry | N-heter. | n | qN |  | |
|---|---|---|---|---|---|
| qCα | qCα′ | ||||
| 1 | N1 | 1 | −2.670 | 1.161 | 1.161 |
| 2 | N2 | 1 | −2.696 | 1.448 | 1.448 |
| 3 | N3 | 1 | −2.845 | 2.467 | 1.208 |
| 4 | N4 | 1 | −2.745 | 2.649 | 1.293 |
| 5 | N5 | 1 | −2.760 | 1.291 | 1.291 |
| 6 | N6 | 0 | −2.356 | 0.828 | 0.828 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuci, G.; Filippi, J.; Rossin, A.; Luconi, L.; Pham-Huu, C.; Yakhvarov, D.; Vizza, F.; Giambastiani, G. CO2 Electrochemical Reduction by Exohedral N-Pyridine Decorated Metal-Free Carbon Nanotubes. Energies 2020, 13, 2703. https://0-doi-org.brum.beds.ac.uk/10.3390/en13112703
Tuci G, Filippi J, Rossin A, Luconi L, Pham-Huu C, Yakhvarov D, Vizza F, Giambastiani G. CO2 Electrochemical Reduction by Exohedral N-Pyridine Decorated Metal-Free Carbon Nanotubes. Energies. 2020; 13(11):2703. https://0-doi-org.brum.beds.ac.uk/10.3390/en13112703
Chicago/Turabian StyleTuci, Giulia, Jonathan Filippi, Andrea Rossin, Lapo Luconi, Cuong Pham-Huu, Dmitry Yakhvarov, Francesco Vizza, and Giuliano Giambastiani. 2020. "CO2 Electrochemical Reduction by Exohedral N-Pyridine Decorated Metal-Free Carbon Nanotubes" Energies 13, no. 11: 2703. https://0-doi-org.brum.beds.ac.uk/10.3390/en13112703