
Characterisation of Moisture in Scots Pine (Pinus sylvestris L.) Sapwood Modified with Maleic Anhydride and Sodium Hypophosphite

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Modification
2.3. Leaching Test (Modified EN84)
2.4. Low-Field Nuclear Magnetic Resonance (LFNMR)
2.5. Determination of Hydroxyl Accessibility
2.6. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tiemann, H.D. Effect of Moisture upon the Strength and Stiffness of Wood; US Department of Agriculture, Forest Service: Washington, DC, USA, 1906. [Google Scholar]
- Rowell, R.M. Handbook of Wood Chemistry and Wood Composite; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Brischke, C.; Alfredsen, G. Wood-water relationships and their role for wood susceptibility to fungal decay. Appl. Microbiol. Biotechnol. 2020, 104, 3781–3795. [Google Scholar] [CrossRef]
- Rowell, R.M.; Ibach, R.E.; McSweeny, J.; Nilsson, T. Understanding decay resistance, dimensional stability and strength changes in heat-treated and acetylated wood. Wood Mater. Sci. Eng. 2009, 4, 14–22. [Google Scholar] [CrossRef]
- Thybring, E.E. The decay resistance of modified wood influenced by moisture exclusion and swelling reduction. Int. Biodeterior. Biodegrad. 2013, 82, 87–95. [Google Scholar] [CrossRef]
- Zelinka, S.L.; Ringman, R.; Pilgård, A.; Thybring, E.E.; Jakes, J.E.; Richter, K. The role of chemical transport in the brown-rot decay resistance of modified wood. Int. Wood Prod. J. 2016, 7, 66–70. [Google Scholar] [CrossRef]
- Ringman, R.; Beck, G.; Pilgård, A. The Importance of Moisture for Brown Rot Degradation of Modified Wood: A Critical Discussion. Forests 2019, 10, 522. [Google Scholar] [CrossRef] [Green Version]
- Thybring, E.E.; Fredriksson, M. Wood Modification as a Tool to Understand Moisture in Wood. Forests 2021, 12, 372. [Google Scholar] [CrossRef]
- Thygesen, L.G.; Elder, T. Moisture in untreated, acetylated, and furfurylated Norway spruce studied during drying using time domain NMR. Wood Fiber Sci. 2008, 40, 309–320. [Google Scholar]
- Thygesen, L.G.; Elder, T. Moisture in untreated, acetylated, and furfurylated Norway spruce monitored during drying below fiber saturation using time domain NMR. Wood Fiber Sci. 2009, 41, 194–200. [Google Scholar]
- Menon, R.S.; Mackay, A.L.; Hailey, J.R.T.; Bloom, M.; Burgess, A.E.; Swanson, J.S. A NMR determination of the physiological water distribution in wood during drying. J. Appl. Polym. Sci. 1987, 33, 1141–1155. [Google Scholar] [CrossRef]
- Labbé, N.; De Jéso, B.; Lartigue, J.C.; Daudé, G.; Pétraud, M.; Ratier, M. Moisture content and extractive materials in maritime pine wood by low field 1 H NMR. Holzforschung 2002, 56, 25–31. [Google Scholar] [CrossRef]
- Mitchell, J.; Stark, S.C.; Strange, J.H. Probing surface interactions by combining NMR cryoporometry and NMR relaxometry. J. Phys. D Appl. Phys. 2005, 38, 1950. [Google Scholar] [CrossRef]
- Fredriksson, M.; Thygesen, L.G. The states of water in Norway spruce (Picea abies (L.) Karst.) studied by low-field nuclear magnetic resonance (LFNMR) relaxometry: Assignment of free-water populations based on quantitative wood anatomy. Holzforschung 2017, 71, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Flibotte, S.; Menon, R.S.; MacKay, A.L.; Hailey, J.R.T. Proton magnetic resonance of Western red cedar. Wood Fiber Sci. 1990, 22, 362–376. [Google Scholar]
- Araujo, C.D.; Mackay, A.L.; Hailey, J.R.T.; Whittall, K.P.; Le, H. Proton magnetic resonance techniques for characterization of water in wood–application to white spruce. Wood Sci. Technol. 1992, 26, 101–113. [Google Scholar] [CrossRef]
- Telkki, V.V.; Yliniemi, M.; Jokisaari, J. Moisture in softwoods: Fiber saturation point, hydroxyl site content, and the amount of micropores as determined from NMR relaxation time distributions. Holzforschung 2013, 67, 291–300. [Google Scholar] [CrossRef]
- Kekkonen, P.M.; Ylisassi, A.; Telkki, V.V. Absorption of water in thermally modified pine wood as studied by nuclear magnetic resonance. J. Phys. Chem. C 2014, 118, 2146–2153. [Google Scholar] [CrossRef]
- Menon, R.S.; Mackay, A.L.; Flibotte, S.; Hailey, J.R.T. Quantitative separation of NMR images of water in wood on the basis of T2. J. Magn. Reason. 1989, 82, 205–210. [Google Scholar] [CrossRef]
- Beck, G.; Thybring, E.E.; Thygesen, L.G.; Hill, C. Characterization of moisture in acetylated and propionylated radiata pine using low-field nuclear magnetic resonance (LFNMR) relaxometry. Holzforschung 2018, 72, 225–233. [Google Scholar] [CrossRef]
- Atalla, R.H.; Brady, J.W.; Matthews, J.F.; Ding, S.; Himmel, M.E. Structures of plant cell wall celluloses. In Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy; Himmel, M.E., Ed.; Blackwell Publishing: Chichester, UK, 2008; pp. 188–212. [Google Scholar]
- Englander, S.W.; Downer, N.W.; Teitelbaum, H. Hydrogen-exchange. Annu. Rev. Biochem. 1972, 41, 903–924. [Google Scholar] [CrossRef]
- Gold, V.; Satchell, D.P.N. The principles of hydrogen isotope exchange reactions in solution. Q. Rev. Chem. Soc. 1955, 9, 51–72. [Google Scholar] [CrossRef]
- Mann, J.; Marrinan, H.J. The reaction between cellulose and heavy water 1. A qualitative study by infra-red spectroscopy. Trans. Faraday Soc. 1956, 52, 481–487. [Google Scholar] [CrossRef]
- Taniguchi, T.; Harada, H.; Nakato, K. Determination of water-adsorption sites in wood by a hydrogen deuterium exchange. Nature 1978, 272, 230–231. [Google Scholar] [CrossRef]
- Hofstetter, K.; Hinterstoisser, B.; Salmén, L. Moisture uptake in native cellulose–the roles of different hydrogen bonds: A dynamic FT-IR study using deuterium exchange. Cellulose 2006, 13, 131–145. [Google Scholar] [CrossRef]
- Schmidt, M.; Gierlinger, N.; Schade, U.; Rogge, T.; Grunze, M. Polarized infrared microspectroscopy of single spruce fibers: Hydrogen bonding in wood polymers. Biopolymers 2006, 83, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Morita, S.; Kokot, S.; Matsubara, M.; Fukai, K.; Ozaki, Y. Drying process of microcrystalline cellulose studied by attenuated total reflection IR spectroscopy with two dimensional correlation spectroscopy and principal component analysis. J. Mol. Struct. 2006, 799, 102–110. [Google Scholar] [CrossRef]
- Suchy, M.; Virtanen, J.; Kontturi, E.; Vuorinen, T. Impact of drying on wood ultrastructure observed by deuterium exchange and photoacoustic FT-IR spectroscopy. Biomacromol. 2010, 11, 515–520. [Google Scholar] [CrossRef]
- Tarmian, A.; Burgert, I.; Thybring, E. Hydroxyl accessibility in wood by deuterium exchange and ATR-FTIR spectroscopy: Methodological uncertainties. Wood Sci. Technol. 2017, 51, 845–853. [Google Scholar] [CrossRef]
- Javed, M.A.; Kekkonen, P.M.; Ahola, S.; Telkki, V.V. Magnetic resonance imaging study of water absorption in thermally modified pine wood. Holzforschung 2015, 69, 899–907. [Google Scholar] [CrossRef]
- Kim, I.; Karlsson, O.; Jones, D.; Mantanis, G.; Sandberg, D. Dimensional stabilisation of Scots pine (Pinus sylvestris L.) sapwood by reaction with maleic anhydride and sodium hypophosphite. Eur. J. Wood Wood Prod. 2021, 79, 589–596. [Google Scholar] [CrossRef]
- Lande, S.; Westin, M.; Schneider, M. Properties of furfurylated wood. Scand. J. For. Res. 2004, 19 (Suppl. 5), 22–30. [Google Scholar] [CrossRef]
- European Committee for Standardization. EN 84 Durability of wood and wood-based products. In Accelerated Ageing of Treated Wood Prior to Biological Testing; Leaching procedure; European Committee for Standardisation: Brussels, Belgium, 2020. [Google Scholar]
- Thybring, E.E.; Digaitis, R.; Nord-Larsen, T.; Beck, G.; Fredriksson, M. How much water can wood cell walls hold? A triangulation approach to determine the maximum cell wall moisture content. PLoS ONE 2020, 15, e0238319. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Akitsu, H.; Norimoto, M.; Morooka, T.; Rowell, R.M. Effect of humidity on vibrational properties of chemically modified wood. Wood Fiber Sci. 1993, 25, 250–260. [Google Scholar]
- Hill, C.A.S. The reduction in the fibre saturation point of wood due to chemical modification using anhydride reagents: A reappraisal. Holzforschung 2008, 62, 423–428. [Google Scholar] [CrossRef]
- Kekkonen, P.M.; Telkki, V.V.; Jokisaari, J. Effect of thermal modification on wood cell structures observed by pulsed-field-gradient stimulated-echo NMR. J. Phys. Chem. C 2010, 114, 18693–18697. [Google Scholar] [CrossRef]
- Izumi, K. NMR spectra of some monosaccharides of galactopyranose series in deuterium oxide. Agric. Biol. Chem. 1971, 35, 1816–1818. [Google Scholar] [CrossRef]



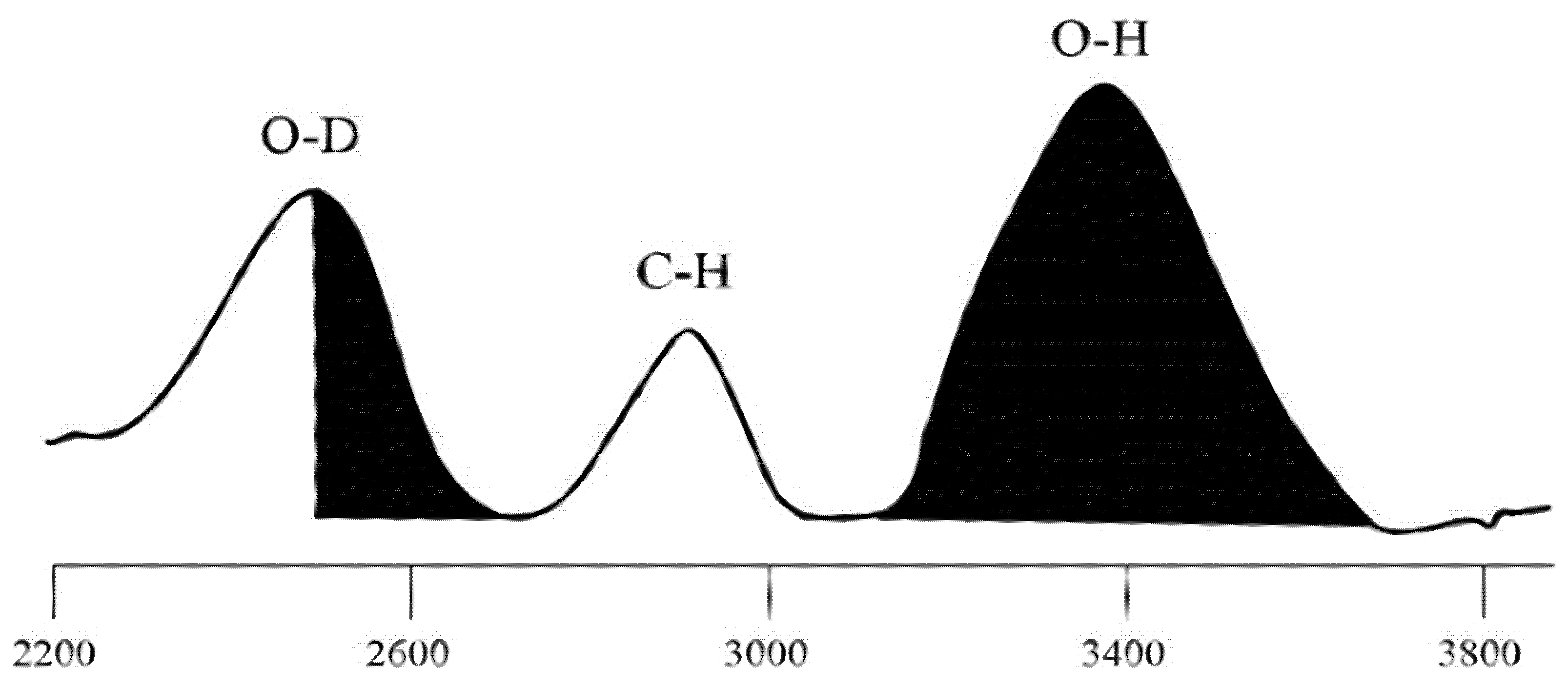{kind=link}
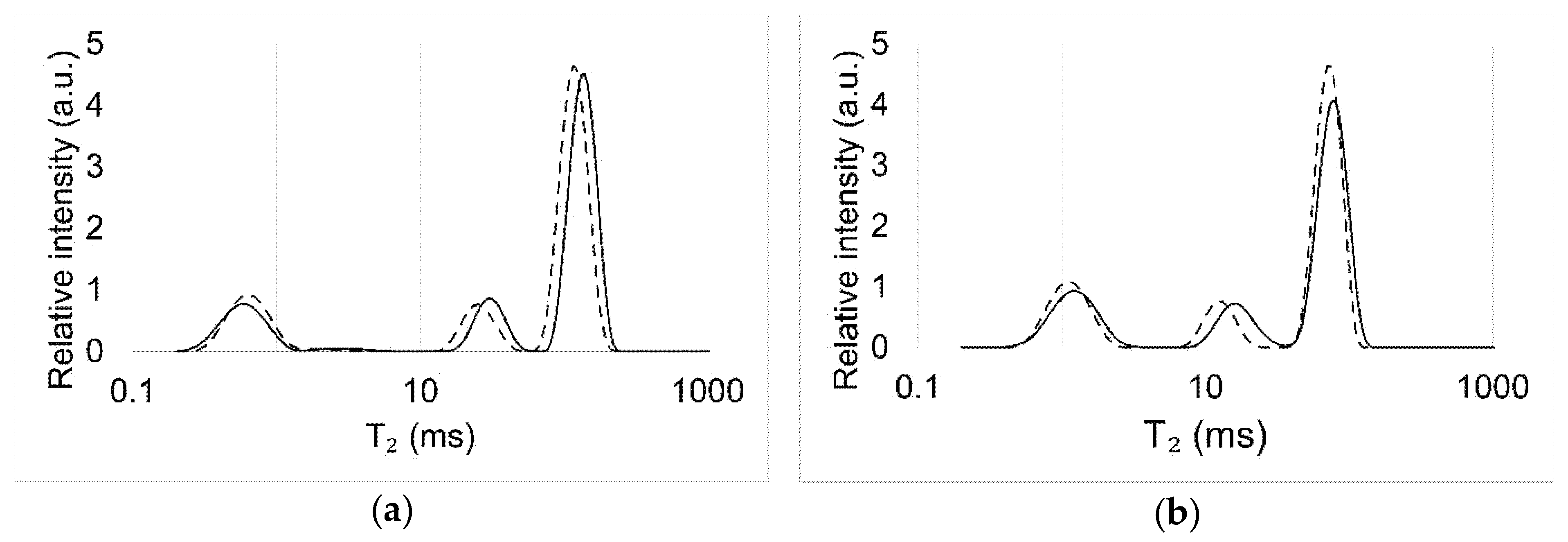{kind=link}
| Sample Group | A (%) | ωt (%) | ωcw (%) |
|---|---|---|---|
| M | 11.8 ± 1.0 | 125.9 (7.1) | 23.6 (1.8) |
| ML | 9.4 ± 1.3 | 126.5 (7.1) | 26.1 (2.1) |
| R | - | 150.5 (8.6) | 34.6 (1.7) |
| RL | −1.6 ± 0.4 | 150.8 (7.6) | 35.4 (2.2) |
| Sample Group | T2 (ms) | ||
|---|---|---|---|
| Peak 1 | Peak 2 | Peak 3 | |
| M | 0.5 (0.1) | 30.4 (3.4) | 137.7 (7.2) |
| ML | 0.6 (0.1) | 25.4 (4.1) | 118.8 (10.9) |
| R | 1.2 (0.1) | 16.4 (2.7) | 78.4 (4.7) |
| RL | 1.1 (0.1) | 12.7 (1.3) | 72.4 (2.9) |
| Sample Group | OH/Lignin | Accessible OH (%) | |
|---|---|---|---|
| Undeuterated | Deuterated | ||
| M | 2.14 (0.81) | 0.99 (0.51) | 47.3 (5.8) |
| ML | 1.77 (0.75) | 0.97 (0.24) | 47.8 (3.5) |
| R | 2.22 (1.30) | 2.02 (1.00) | 41.5 (4.6) |
| RL | 2.42 (0.40) | 1.44 (0.39) | 42.5 (8.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, I.; Thybring, E.E.; Karlsson, O.; Jones, D.; Mantanis, G.I.; Sandberg, D. Characterisation of Moisture in Scots Pine (Pinus sylvestris L.) Sapwood Modified with Maleic Anhydride and Sodium Hypophosphite. Forests 2021, 12, 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/f12101333
Kim I, Thybring EE, Karlsson O, Jones D, Mantanis GI, Sandberg D. Characterisation of Moisture in Scots Pine (Pinus sylvestris L.) Sapwood Modified with Maleic Anhydride and Sodium Hypophosphite. Forests. 2021; 12(10):1333. https://0-doi-org.brum.beds.ac.uk/10.3390/f12101333
Chicago/Turabian StyleKim, Injeong, Emil Engelund Thybring, Olov Karlsson, Dennis Jones, George I. Mantanis, and Dick Sandberg. 2021. "Characterisation of Moisture in Scots Pine (Pinus sylvestris L.) Sapwood Modified with Maleic Anhydride and Sodium Hypophosphite" Forests 12, no. 10: 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/f12101333