

Human Serum Albumin Grafted by Monomeric and Polymeric β-Cyclodextrin as Drug Delivery System for Levofloxacin with Improved Pharmacological Properties
 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
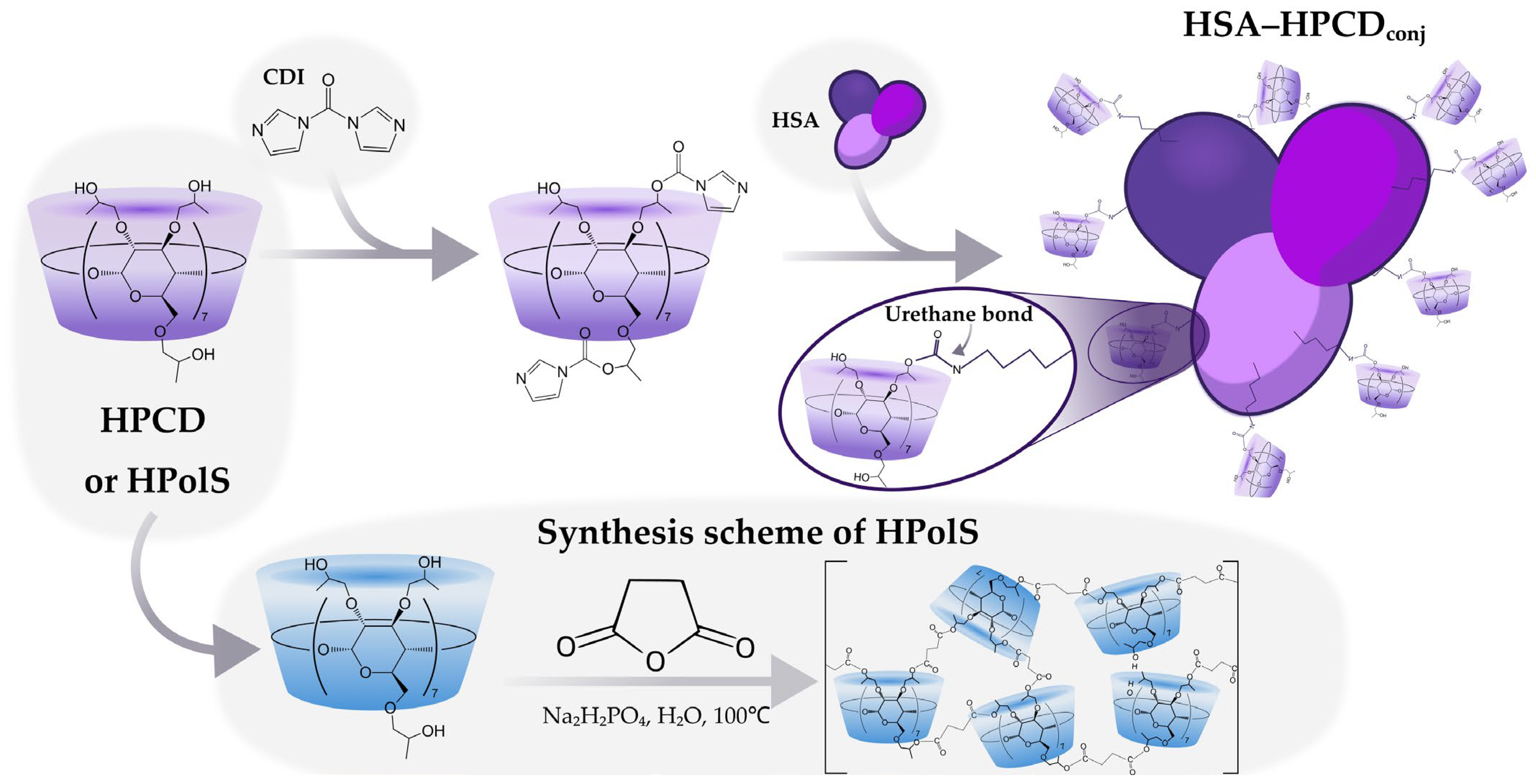
2.2.1. Synthesis of HPCD-Based Polymer Linked by Succinic Anhydride (HPolS)
2.2.2. Synthesis of HSA Conjugates with HPCD or HPolS
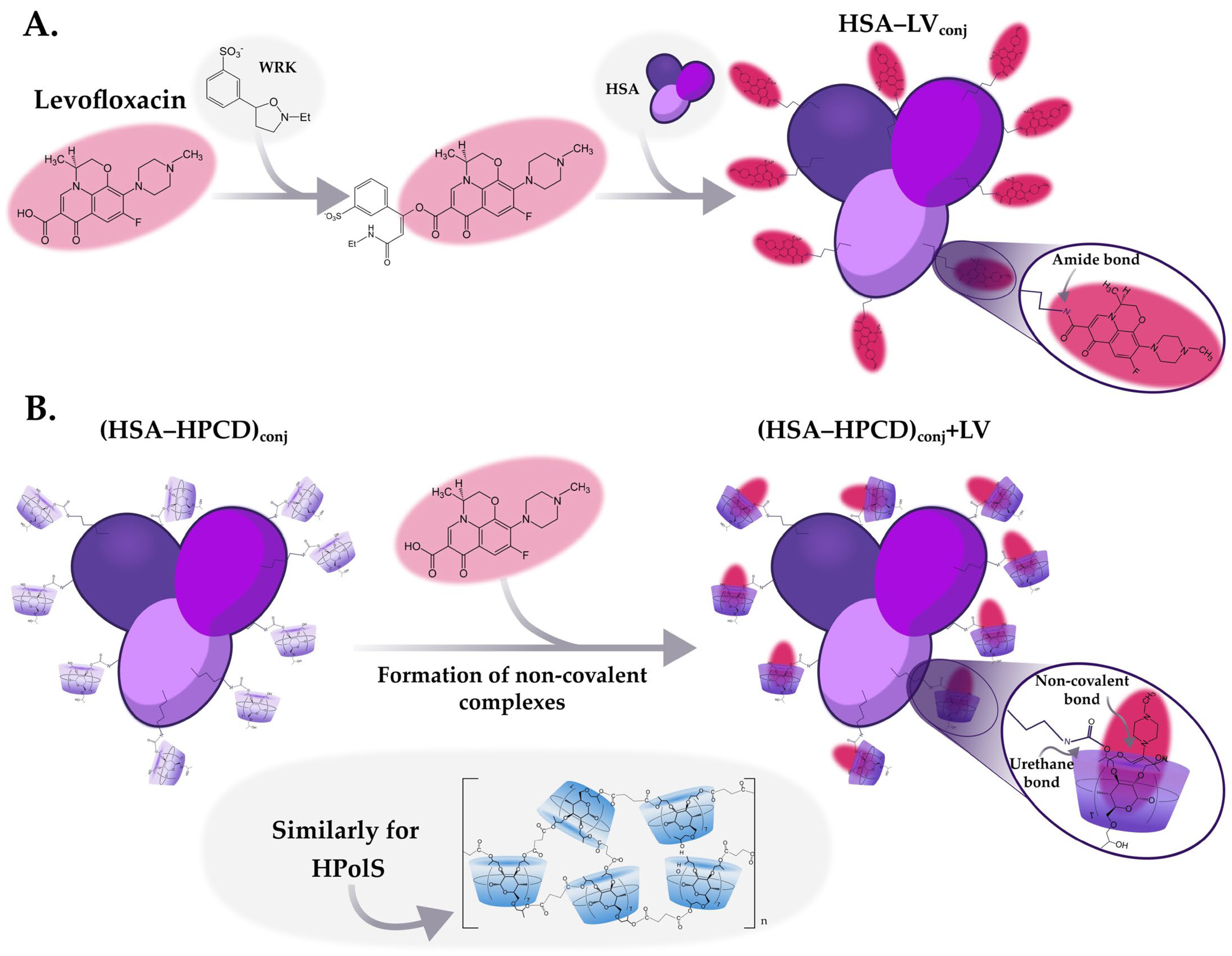
2.2.3. Synthesis of HSA Conjugates with LV
2.2.4. Obtaining Non-Covalent Complexes with LV
2.2.5. UV-Vis Spectrophotometry
2.2.6. FTIR Spectroscopy
2.2.7. Circular Dichroism Spectroscopy
2.2.8. Fluorescence Spectroscopy
2.2.9. Release Kinetic Studies
2.2.10. Evaluation of Antibacterial Activity In Vitro Studies
2.2.11. Hemolysis Assay
3. Results and Discussion
3.1. Synthesis of HSA Grafted by Monomeric and Polymeric HPCD
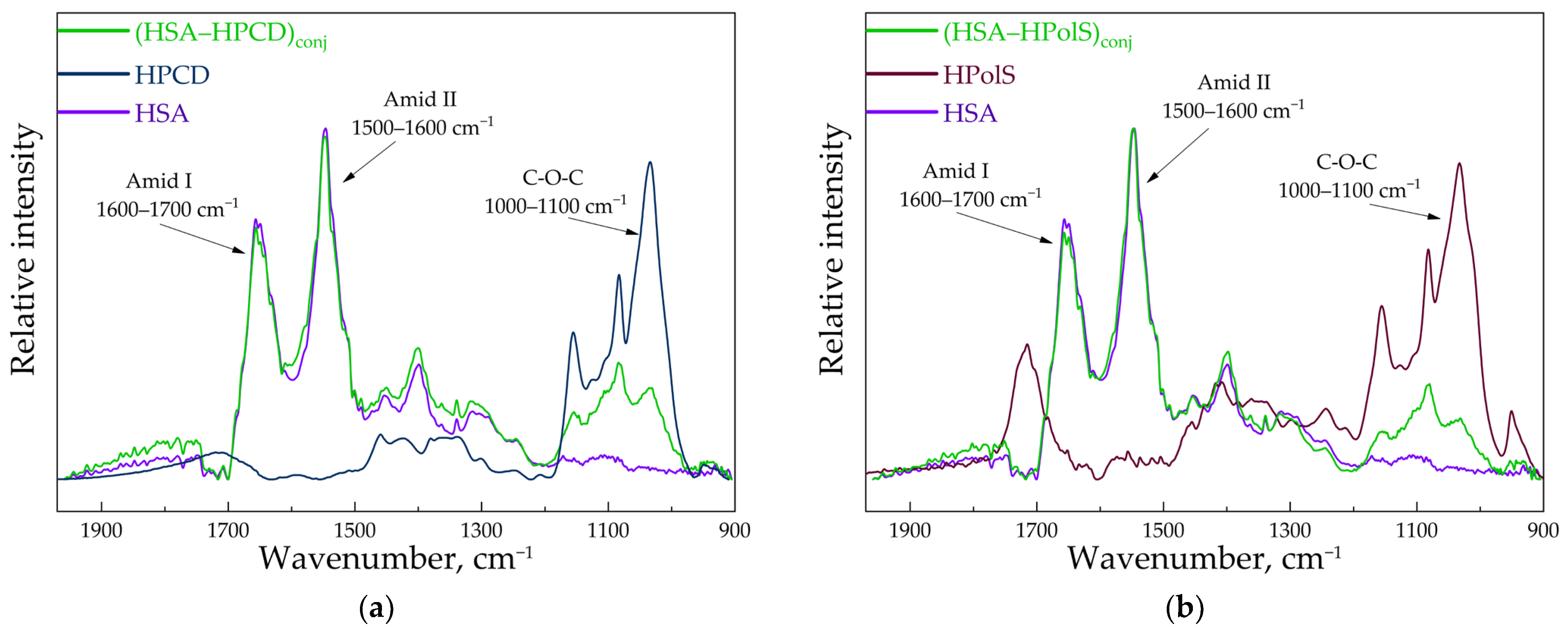
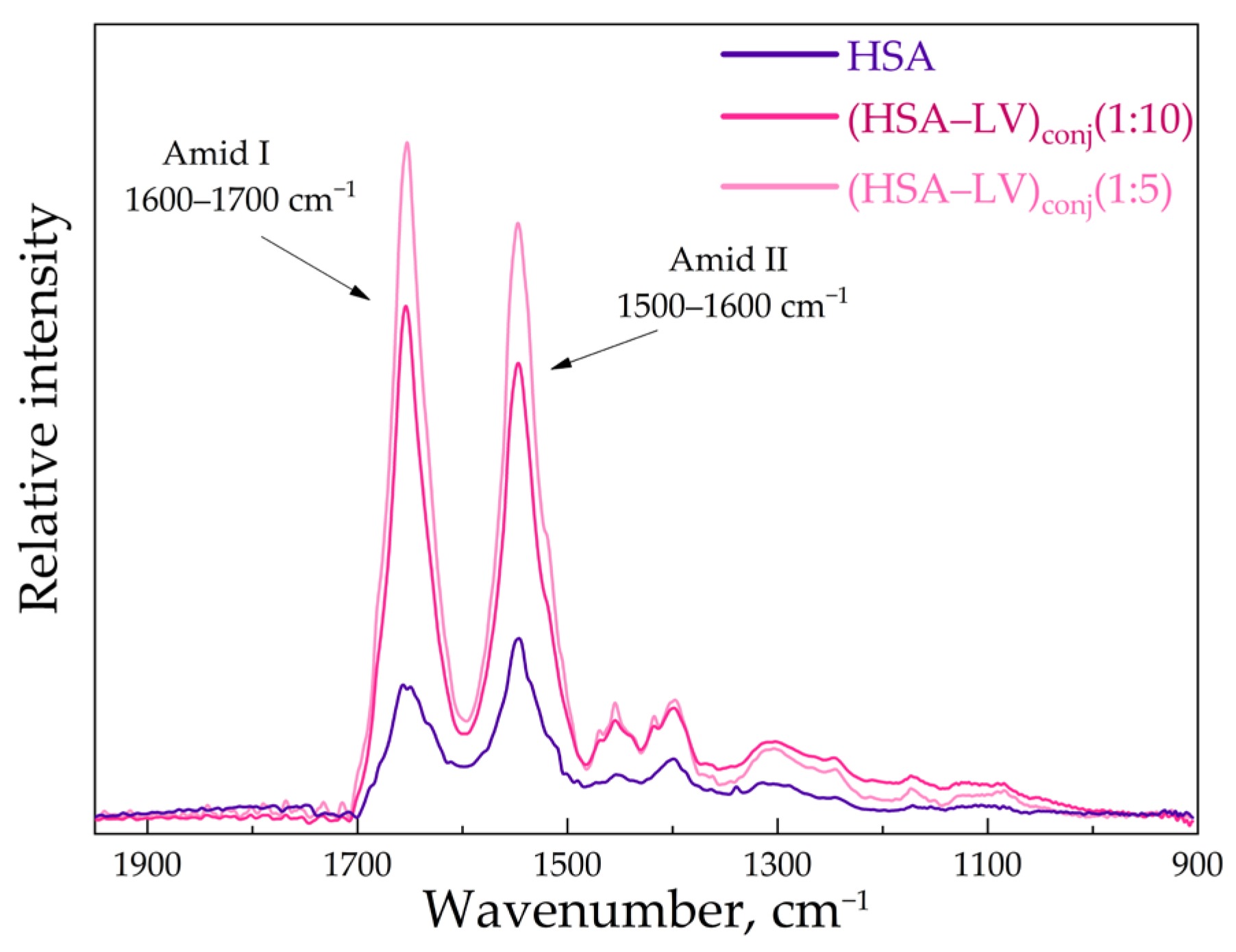
3.2. FTIR and Structure Features of Synthesized Molecules
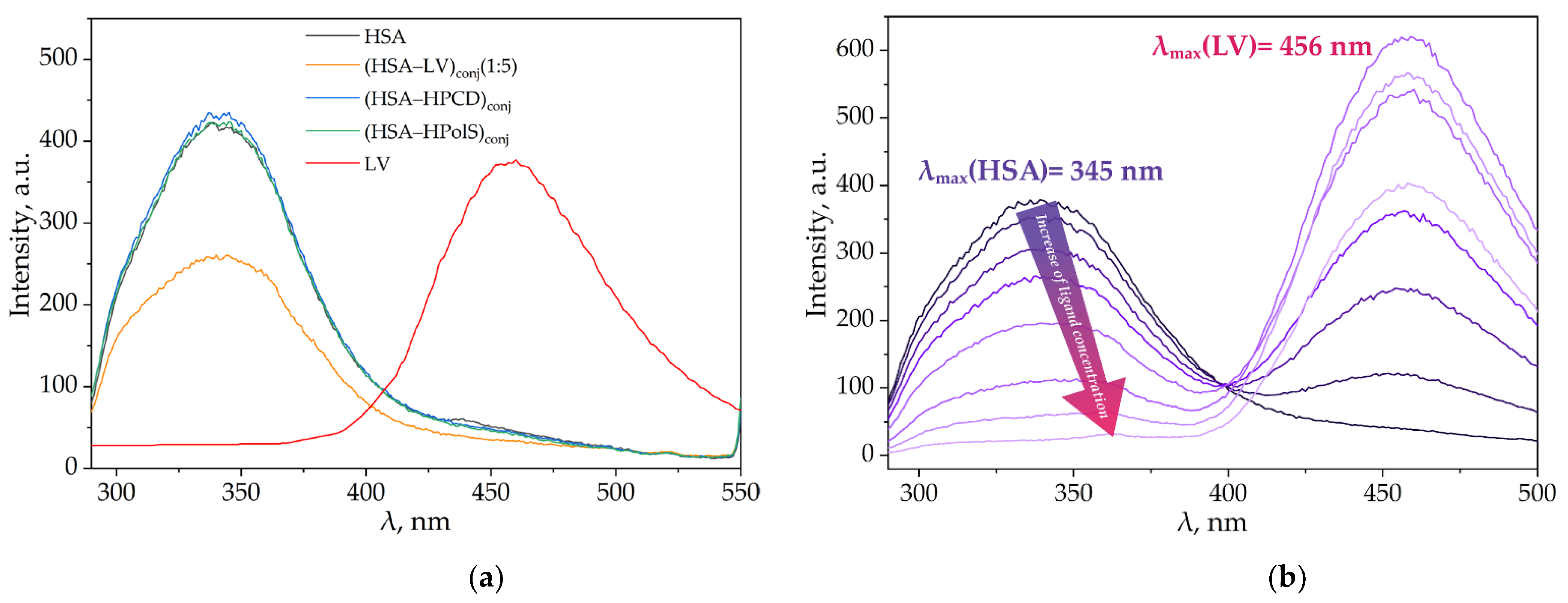
3.3. Fluorescence Spectroscopy
3.3.1. Impact of HPCD, HPolS, and Conjugate Formation on the Fluorescence Spectra of HSA and LV
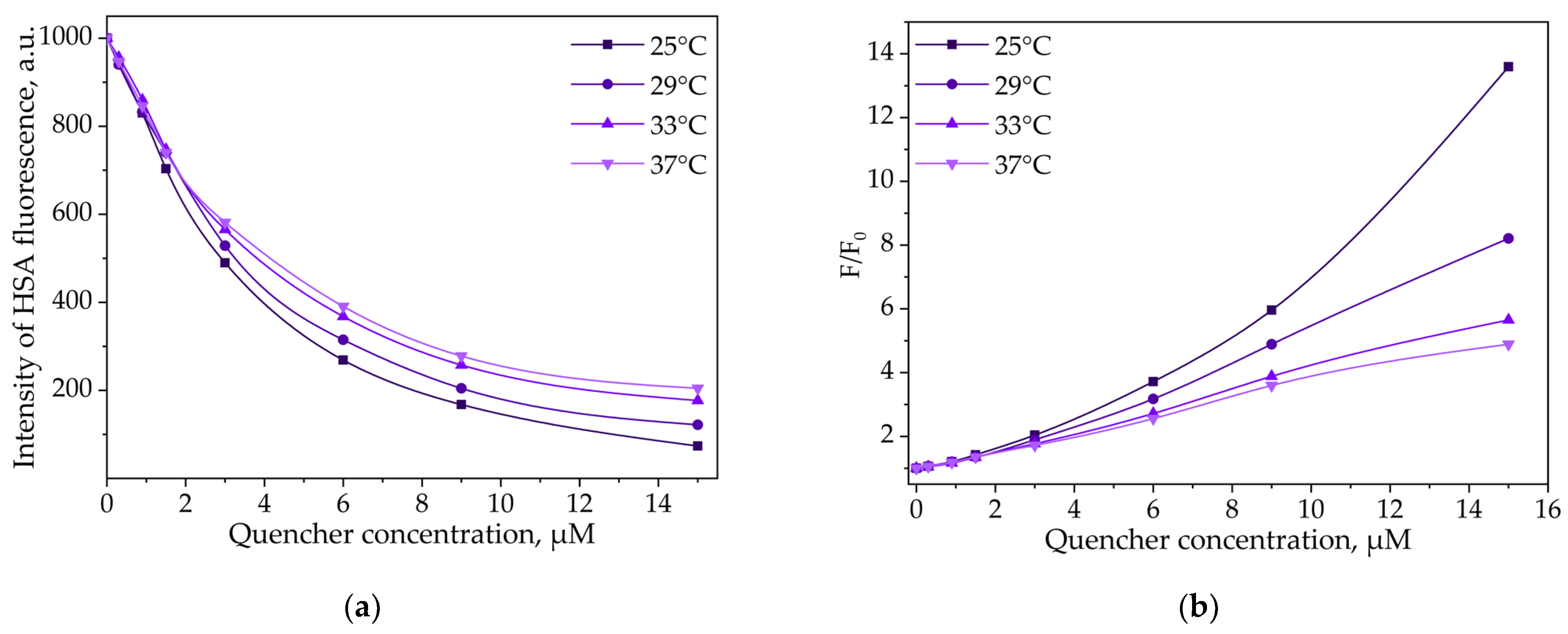
3.3.2. Thermodynamic Parameters and Mechanisms of the Complex Formation of HSA and LV
HSA and LV Interactions
HSA and LV+CD Inclusion Complex Interactions
(HSA−HPCD)conj and (HSA−HPolS)conj Non-Covalent Interactions with LV
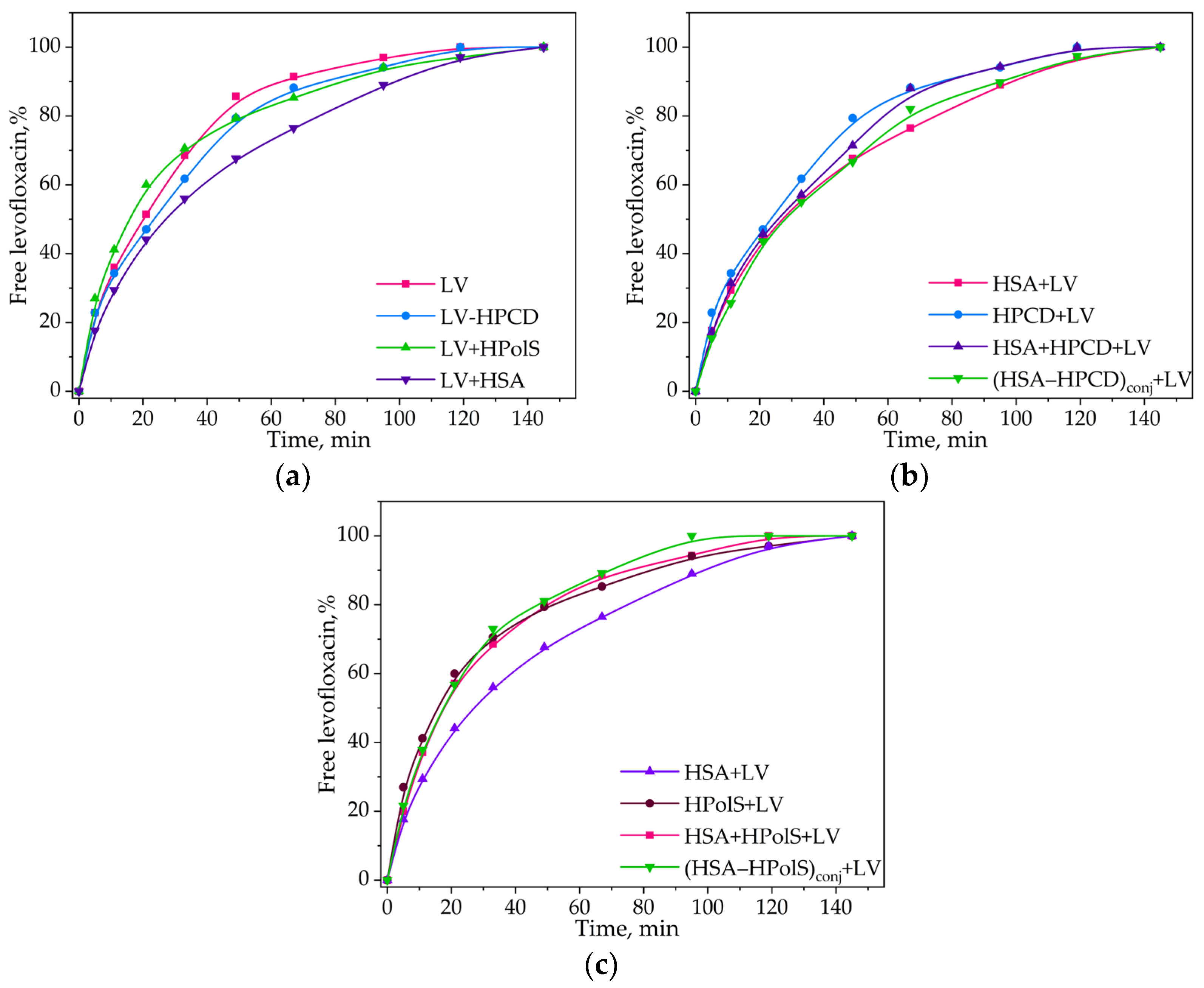
3.4. Kinetics of Levofloxacin Release
3.5. Conjugates Safety
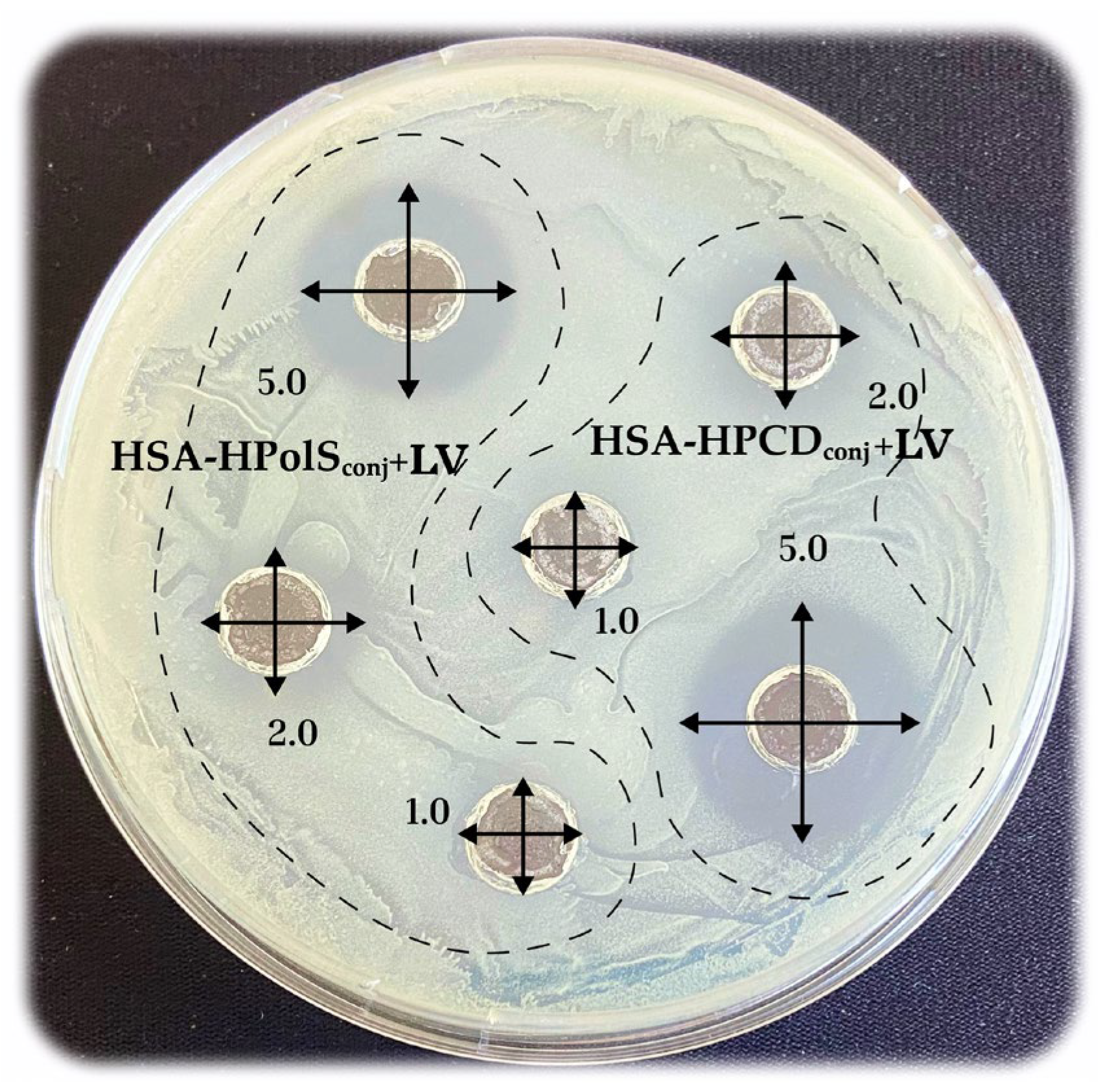
3.6. Antibacterial Activity of Conjugates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.-R.; Suntharalingam, K.; Johnstone, T.C.; Yoo, H.; Lin, W.; Brooks, J.G.; Lippard, S.J. Pt(IV) Prodrugs Designed to Bind Non-Covalently to Human Serum Albumin for Drug Delivery. J. Am. Chem. Soc. 2014, 136, 8790–8798. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, T.; Gan, L.-S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, Y.; Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. [Google Scholar] [CrossRef] [PubMed]
- Trotta, F.; Caldera, F.; Cavalli, R.; Soster, M.; Riedo, C.; Biasizzo, M.; Uccello Barretta, G.; Balzano, F.; Brunella, V. Molecularly imprinted cyclodextrin nanosponges for the controlled delivery of L-DOPA: Perspectives for the treatment of Parkinson’s disease. Expert Opin. Drug Deliv. 2016, 13, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Davis, R.; Bryson, H.M. Levofloxacin. Drugs 1994, 47, 677–700. [Google Scholar] [CrossRef]
- Van Bambeke, F.; Michot, J.M.; Van Eldere, J.; Tulkens, P.M. Quinolones in 2005: An update. Clin. Microbiol. Infect. 2005, 11, 256–280. [Google Scholar] [CrossRef]
- Wolfson, J.S.; Hooper, D.C. Fluoroquinolone antimicrobial agents. Clin. Microbiol. Rev. 1989, 2, 378–424. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Bannerjee, S.; Bhati, L.; Pandey, S.; Pandey, P.; Sriwastawa, B. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Le-Deygen, I.M.; Skuredina, A.A.; Uporov, I.V.; Kudryashova, E.V. Thermodynamics and molecular insight in guest–host complexes of fluoroquinolones with β-cyclodextrin derivatives, as revealed by ATR-FTIR spectroscopy and molecular modeling experiments. Anal. Bioanal. Chem. 2017, 409, 6451–6462. [Google Scholar] [CrossRef] [PubMed]
- Yakupova, L.R.; Skuredina, A.A.; Markov, P.O.; Le-Deygen, I.M.; Kudryashova, E.V. Cyclodextrin Polymers as a Promising Drug Carriers for Stabilization of Meropenem Solutions. Appl. Sci. 2023, 13, 3608. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Yakupova, L.R.; Kopnova, T.Y.; Le-Deygen, I.M.; Belogurova, N.G.; Kudryashova, E.V. Cyclodextrins and Their Polymers Affect Human Serum Albumin’s Interaction with Drugs Used in the Treatment of Pulmonary Infections. Pharmaceutics 2023, 15, 1598. [Google Scholar] [CrossRef] [PubMed]
- Varshney, A.; Sen, P.; Ahmad, E.; Rehan, M.; Subbarao, N.; Khan, R.H. Ligand Binding Strategies of Human Serum Albumin: How Can the Cargo be Utilized? Chirality 2010, 22, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-W.; Wang, K.; Zhang, X.-X. Study of the interactions between fluoroquinolones and human serum albumin by affinity capillary electrophoresis and fluorescence method. Anal. Chim. Acta 2007, 603, 101–110. [Google Scholar] [CrossRef]
- Missoun, F.; de los Ríos, A.P.; Ortiz-Martínez, V.; Salar-García, M.J.; Hernández-Fernández, J.; Hernández-Fernández, F.J. Discovering Low Toxicity Ionic Liquids for Saccharomyces cerevisiae by Using the Agar Well Diffusion Test. Processes 2020, 8, 1163. [Google Scholar] [CrossRef]
- Le-Deygen, I.M.; Skuredina, A.A.; Mamaeva, P.V.; Kolmogorov, I.M.; Kudryashova, E.V. Conjugates of Chitosan with β-Cyclodextrins as Promising Carriers for the Delivery of Levofloxacin: Spectral and Microbiological Studies. Life 2023, 13, 272. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Kopnova, T.Y.; Tychinina, A.S.; Golyshev, S.A.; Le-Deygen, I.M.; Belogurova, N.G.; Kudryashova, E.V. The New Strategy for Studying Drug-Delivery Systems with Prolonged Release: Seven-Day In Vitro Antibacterial Action. Molecules 2022, 27, 8026. [Google Scholar] [CrossRef]
- Hearn, M.T.W. 1,1′-Carbonyldiimidazole-mediated immobilization of enzymes and affinity ligands. Methods Enzymol. 1987, 135, 102–117. [Google Scholar]
- Métro, T.-X.; Martinez, J.; Lamaty, F. 1,1′-Carbonyldiimidazole and Mechanochemistry: A Shining Green Combination. ACS Sustain. Chem. Eng. 2017, 5, 9599–9602. [Google Scholar] [CrossRef]
- Stöllner, D.; Scheller, F.W.; Warsinke, A. Activation of Cellulose Membranes with 1,1′-Carbonyldiimidazole or 1-Cyano-4-dimethylaminopyridinium tetrafluoroborate as a Basis for the Development of Immunosensors. Anal. Biochem. 2002, 304, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Faridnouri, H.; Ghourchian, H.; Hashemnia, S. Direct electron transfer enhancement of covalently bound tyrosinase to glassy carbon via Woodward’s reagent K. Bioelectrochemistry 2011, 82, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.N.; Colman, R.W. A Novel Method for Chemical Modification of Functional Groups Other Than a Carboxyl Group in Proteins byN-Ethyl-5-phenylisooxazolium-3′-sulfonate (Woodward’s Reagent-K): Inhibition of ADP-Induced Platelet Responses Involves Covalent Modification of Aggreg. Anal. Biochem. 1996, 240, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, N.V.; Zhdanov, D.D.; Sokolov, N.N.; Aleksandrova, S.S.; Pokrovskaya, M.V.; Kudryashova, E.V. Rhodospirillum rubrum L-Asparaginase Conjugates with Polyamines of Improved Biocatalytic Properties as a New Promising Drug for the Treatment of Leukemia. Appl. Sci. 2023, 13, 3373. [Google Scholar] [CrossRef]
- Paoli, P.; Fiaschi, T.; Cirri, P.; Camici, G.; Manao, G.; Cappugi, G.; Raugei, G.; Moneti, G.; Ramponi, G. Mechanism of acylphosphatase inactivation by Woodward’s reagent K. Biochem. J. 1997, 328, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Sinha, U.; Brewer, J.M. A spectrophotometric method for quantitation of carboxyl group modification of proteins using Woodward’s Reagent K. Anal. Biochem. 1985, 151, 327–333. [Google Scholar] [CrossRef]
- Dunn, B.M.; Anfinsen, C.B.; Shrager, R.I. Kinetics of Woodward’s Reagent K Hydrolysis and Reaction with Staphylococcal Nuclease. J. Biol. Chem. 1974, 249, 3717–3723. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Le-Deygen, I.M.; Belogurova, N.G.; Kudryashova, E.V. Effect of cross-linking on the inclusion complex formation of derivatized β-cyclodextrins with small-molecule drug moxifloxacin. Carbohydr. Res. 2020, 498, 108183. [Google Scholar] [CrossRef]
- Yakupova, L.R.; Kopnova, T.Y.; Skuredina, A.A.; Le-Deygen, I.M.; Shustrov, P.N.; Novoselov, A.M.; Kudryashova, E.V. The Formation of β-Cyclodextrin Complexes with Levofloxacin and Ceftriaxone as an Approach to the Regulation of Drugs’ Pharmacokinetic. Colloid J. 2023, 85, 114–127. [Google Scholar] [CrossRef]
- Richner, G.; Puxty, G. Assessing the Chemical Speciation during CO 2 Absorption by Aqueous Amines Using in Situ FTIR. Ind. Eng. Chem. Res. 2012, 51, 14317–14324. [Google Scholar] [CrossRef]
- Furer, V.L.; Vandyukova, I.I.; Vandyukov, A.E.; Fuchs, S.; Majoral, J.P.; Caminade, A.M.; Kovalenko, V.I. Vibrational spectra study of fluorescent dendrimers built from the cyclotriphosphazene core with terminal dansyl and carbamate groups. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 462–470. [Google Scholar] [CrossRef]
- Lingegowda, D.C.; Kumar, J.K.; Prasad, A.D.; Zarei, M.; Gopal, S. FTIR spectroscopic studies on cleome gynandra-Comparative analysis of functional group before and after extraction. Rom. J. Biophys. 2012, 22, 137–143. [Google Scholar]
- Tretiakova, D.; Le-Deigen, I.; Onishchenko, N.; Kuntsche, J.; Kudryashova, E.; Vodovozova, E. Phosphatidylinositol Stabilizes Fluid-Phase Liposomes Loaded with a Melphalan Lipophilic Prodrug. Pharmaceutics 2021, 13, 473. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Meyer, J.D.; Brown, J.L.; Manning, M.C.; Carpenter, J.F. Comparative fourier transform infrared and circular dichroism spectroscopic analysis of α1-proteinase inhibitor and ovalbumin in aqueous solution. Arch. Biochem. Biophys. 2000, 383, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Haris, P.I.; Severcan, F. FTIR spectroscopic characterization of protein structure in aqueous and non-aqueous media. J. Mol. Catal. B Enzym. 1999, 7, 207–221. [Google Scholar] [CrossRef]
- Aleem, O.; Kuchekar, B.; Pore, Y.; Late, S. Effect of β-cyclodextrin and hydroxypropyl β-cyclodextrin complexation on physicochemical properties and antimicrobial activity of cefdinir. J. Pharm. Biomed. Anal. 2008, 47, 535–540. [Google Scholar] [CrossRef]
- Zhao, D.; Zhao, L.; Zhu, C.-S.; Huang, W.-Q.; Hu, J.-L. Water-insoluble β-cyclodextrin polymer crosslinked by citric acid: Synthesis and adsorption properties toward phenol and methylene blue. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 195–201. [Google Scholar] [CrossRef]
- Iranfar, H.; Rajabi, O.; Salari, R.; Chamani, J. Probing the Interaction of Human Serum Albumin with Ciprofloxacin in the Presence of Silver Nanoparticles of Three Sizes: Multispectroscopic and ζ Potential Investigation. J. Phys. Chem. B 2012, 116, 1951–1964. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Wang, Y.-Q.; Jiang, M.-L. A fluorimetric study of the interaction of C.I. Solvent Red 24 with haemoglobin. Dye. Pigment. 2009, 82, 156–163. [Google Scholar] [CrossRef]
- Wei, Y.; Li, J.; Dong, C.; Shuang, S.; Liu, D.; Huie, C.W. Investigation of the association behaviors between biliverdin and bovine serum albumin by fluorescence spectroscopy. Talanta 2006, 70, 377–382. [Google Scholar] [CrossRef]
- Seedher, N.; Agarwal, P. Complexation of fluoroquinolone antibiotics with human serum albumin: A fluorescence quenching study. J. Lumin. 2010, 130, 1841–1848. [Google Scholar] [CrossRef]
- Kaur, A.; Khan, I.A.; Banipal, P.K.; Banipal, T.S. Deciphering the complexation process of a fluoroquinolone antibiotic, levofloxacin, with bovine serum albumin in the presence of additives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 191, 259–270. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Kopnova, T.Y.; Belogurova, N.G.; Kudryashova, E.V. Encapsulation of Ciprofloxacin into a Cyclodextrin Polymer Matrix: The Complex Formation with Human Serum Albumin and In Vitro Studies. Chemistry 2023, 5, 1942–1960. [Google Scholar] [CrossRef]
- Yakupova, L.R.; Kopnova, T.Y.; Skuredina, A.A.; Kudryashova, E.V. Effect of Methyl-β-Cyclodextrin on the Interaction of Fluoroquinolones with Human Serum Albumin. Russ. J. Bioorg. Chem. 2022, 48, 163–172. [Google Scholar] [CrossRef]
- Yakupova, L.R.; Skuredina, A.A.; Kopnova, T.Y.; Kudryashova, E.V. In Vitro Biological Properties of Cyclodextrin-Based Polymers: Interaction with Human Serum Albumin, Red Blood Cells and Bacteria. Polysaccharides 2023, 4, 343–357. [Google Scholar] [CrossRef]
- Hu, Y.J.; Ou-Yang, Y.; Zhang, Y.; Liu, Y. Affinity and specificity of ciprofloxacin-bovine serum albumin interactions: Spectroscopic approach. Protein J. 2010, 29, 234–241. [Google Scholar] [CrossRef]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of Protein Association Reactions: Forces Contributing to Stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Seedher, N.; Agarwal, P. Competitive binding of fluoroquinolone antibiotics and some other drugs to human serum albumin: A luminescence spectroscopic study. Luminescence 2013, 28, 562–568. [Google Scholar] [CrossRef]
- Poureshghi, F.; Ghandforoushan, P.; Safarnejad, A.; Soltani, S. Interaction of an antiepileptic drug, lamotrigine with human serum albumin (HSA): Application of spectroscopic techniques and molecular modeling methods. J. Photochem. Photobiol. B Biol. 2017, 166, 187–192. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N. A spectral deciphering of the binding interaction of an intramolecular charge transfer fluorescence probe with a cationic protein: Thermodynamic analysis of the binding phenomenon combined with blind docking study. Photochem. Photobiol. Sci. 2011, 10, 980–991. [Google Scholar] [CrossRef]
- Zhang, G.; Ma, Y. Mechanistic and conformational studies on the interaction of food dye amaranth with human serum albumin by multispectroscopic methods. Food Chem. 2013, 136, 442–449. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N.; Bhattacharya, S.C. Binding of ciprofloxacin to bovine serum albumin: Photophysical and thermodynamic aspects. J. Photochem. Photobiol. B Biol. 2017, 172, 11–19. [Google Scholar] [CrossRef]
- Tabassum, S.; Al-Asbahy, W.M.; Afzal, M.; Arjmand, F. Synthesis, characterization and interaction studies of copper based drug with Human Serum Albumin (HSA): Spectroscopic and molecular docking investigations. J. Photochem. Photobiol. B Biol. 2012, 114, 132–139. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Kopnova, T.Y.; Le-deygen, I.M.; Kudryashova, E. V Physical and Chemical Properties of the Guest–Host Inclusion Complexes of Cyprofloxacin with β-Cyclodextrin Derivatives. Mosc. Univ. Chem. Bull. 2020, 75, 218–224. [Google Scholar] [CrossRef]
- Sahiner, N.; Sagbas, S.; Sahiner, M.; Blake, D.A.; Reed, W.F. Polydopamine particles as nontoxic, blood compatible, antioxidant and drug delivery materials. Colloids Surf. B Biointerfaces 2018, 172, 618–626. [Google Scholar] [CrossRef]
- Sahiner, N.; Sagbas, S.; Aktas, N. Preparation of macro-, micro-, and nano-sized poly(Tannic acid) particles with controllable degradability and multiple biomedical uses. Polym. Degrad. Stab. 2016, 129, 96–105. [Google Scholar] [CrossRef]
- Szente, L.; Singhal, A.; Domokos, A.; Song, B. Cyclodextrins: Assessing the Impact of Cavity Size, Occupancy, and Substitutions on Cytotoxicity and Cholesterol Homeostasis. Molecules 2018, 23, 1228. [Google Scholar] [CrossRef]
- Sparreboom, A.; Scripture, C.D.; Trieu, V.; Williams, P.J.; De, T.; Yang, A.; Beals, B.; Figg, W.D.; Hawkins, M.; Desai, N. Comparative Preclinical and Clinical Pharmacokinetics of a Cremophor-Free, Nanoparticle Albumin-Bound Paclitaxel (ABI-007) and Paclitaxel Formulated in Cremophor (Taxol). Clin. Cancer Res. 2005, 11, 4136–4143. [Google Scholar] [CrossRef] [PubMed]
- Chemmanur, A.T.; Wu, G.Y. Drug evaluation: Albuferon-alpha--an antiviral interferon-alpha/albumin fusion protein. Curr. Opin. Investig. Drugs 2006, 7, 750–758. [Google Scholar] [PubMed]
- Arnaouteli, S.; Bamford, N.C.; Stanley-Wall, N.R.; Kovács, Á.T. Bacillus subtilis biofilm formation and social interactions. Nat. Rev. Microbiol. 2021, 19, 600–614. [Google Scholar] [CrossRef] [PubMed]
- An, Y.H.; Friedman, R.J. Concise review of mechanisms of bacterial adhesion to biomaterial surfaces. J. Biomed. Mater. Res. 1998, 43, 338–348. [Google Scholar] [CrossRef]
- Bridgett, M.J.; Davies, M.C.; Denyer, S.P. Control of staphylococcal adhesion to polystyrene surfaces by polymer surface modification with surfactants. Biomaterials 1992, 13, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival Mechanisms of Clinically Relevant Microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef]
- Paulsson, M.; Kober, M.; Freij-Larsson, C.; Stollenwerk, M.; Wesslén, B.; Ljungh, Å. Adhesion of staphylococci to chemically modified and native polymers, and the influence of preadsorbed fibronectin, vitronectin and fibrinogen. Biomaterials 1993, 14, 845–853. [Google Scholar] [CrossRef]
- Brokke, P.; Dankert, J.; Carballo, J.; Feijen, J. Adherence of Coagulase-Negative Staphylococci onto Polyethylene Catheters in vitro and in vivo: A Study on the Influence of various Plasma Proteins. J. Biomater. Appl. 1991, 5, 204–226. [Google Scholar] [CrossRef]
- Reynolds, E.C.; Wong, A. Effect of adsorbed protein on hydroxyapatite zeta potential and Streptococcus mutans adherence. Infect. Immun. 1983, 39, 1285–1290. [Google Scholar] [CrossRef]
- del Prado, G.; Ruiz, V.; Naves, P.; Rodríguez-Cerrato, V.; Soriano, F.; del Carmen Ponte, M. Biofilm formation by Streptococcus pneumoniae strains and effects of human serum albumin, ibuprofen, N-acetyl-l-cysteine, amoxicillin, erythromycin, and levofloxacin. Diagn. Microbiol. Infect. Dis. 2010, 67, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, R.; Ishima, Y.; Chuang, V.T.G.; Watanabe, H.; Shimizu, T.; Ando, H.; Okuhira, K.; Otagiri, M.; Ishida, T.; Maruyama, T. The Therapeutic Effect of Human Serum Albumin Dimer-Doxorubicin Complex against Human Pancreatic Tumors. Pharmaceutics 2021, 13, 1209. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Trieu, V.; Damascelli, B.; Soon-Shiong, P. SPARC Expression Correlates with Tumor Response to Albumin-Bound Paclitaxel in Head and Neck Cancer Patients. Transl. Oncol. 2009, 2, 59–64. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HPCD | HPolS |
|---|---|---|
| ζ-potential, mV | 0 | −0.6 ± 0.02 |
| Hydrodynamic radius, nm | 0.15 | 200 ± 20 |
| Ka with LV, M−1 | (1.0 ± 0.3) × 103 | (1.0 ± 0.1) × 106 |
| α-Helix | β-Structures | Random | |
|---|---|---|---|
| HSA | 64 ± 2% | 19 ± 1% | 17 ± 1% |
| (HSA–HPCD)conj | 56 ± 2% | 20 ± 1% | 24 ± 1% |
| HSA–(HPolS)conj | 54 ± 2% | 20 ± 1% | 26 ± 1% |
| Parameter | (HSA−LV)conj 1:5 | (HSA−LV)conj 1:10 | (HSA−HPCD)conj | (HSA−HPolS)conj |
|---|---|---|---|---|
| Molecular weight, kDa | 66.8 | 68.6 | 88.1 | 80.1 |
| LV, % (w/w) | 2.7% | 5.4% | − | − |
| Molar ratio 1 | 1:5 | 1:10 | 1:15 | 1:10 |
| T, K | n | Ka, 105 M−1 | ΔG, kJ/mol | ΔH, kJ/mol | ΔS, J/mol/K | |
|---|---|---|---|---|---|---|
| HSA+LV [32] | 298 | 1.34 ± 0.07 | 23.4 ± 0.5 | −36.3 ± 0.1 | −57.7 ± 0.8 | −71 ± 2 |
| 302 | 1.33 ± 0.02 | 17.9 ± 0.3 | −36.1 ± 0.1 | |||
| 306 | 1.32 ± 0.03 | 13.1 ± 0.4 | −35.8 ± 0.1 | |||
| 310 | 1.33 ± 0.05 | 9.9 ± 0.2 | −35.6 ± 0.1 | |||
| HSA+(LV+HPCD) [32] | 298 | 1.51 ± 0.08 | 110 ± 1.2 | −40.1 ± 0.2 | −217.6 ± 1.4 | −596 ± 4 |
| 302 | 1.41 ± 0.04 | 34.0 ± 0.5 | −37.7 ± 0.4 | |||
| 306 | 1.33 ± 0.03 | 12.6 ± 0.2 | −35.7 ± 0.4 | |||
| 310 | 1.24 ± 0.03 | 3.5 ± 0.5 | −32.9 ± 0.3 | |||
| (HSA+HPCD)conj+LV | 298 | 1.37 ± 0.01 | 17 ± 3 | −35.6 ± 0.1 | −187 ± 7 | −510 ± 20 |
| 302 | 1.29 ± 0.02 | 6 ± 1 | −33.5 ± 0.1 | |||
| 306 | 1.23 ± 0.02 | 2.7 ± 0.7 | −31.8 ± 0.1 | |||
| 310 | 1.12 ± 0.01 | 0.9 ± 0.1 | −29.4 ± 0.1 | |||
| (HSA+HPolS)conj+LV | 298 | 1.21 ± 0.05 | 3.3 ± 1.9 | −31.48 ± 0.3 | −149 ± 8 | −400 ± 20 |
| 302 | 1.14 ± 0.05 | 1.4 ± 0.8 | −29.73 ± 0.3 | |||
| 306 | 1.09 ± 0.02 | 0.68 ± 0.12 | −28.33 ± 0.1 | |||
| 310 | 1.01 ± 0.04 | 0.31 ± 0.13 | −26.68 ± 0.2 |
| T, K | n | Ka, 105 M−1 | ΔG, kJ/mol | ΔH, kJ/mol | ΔS, J/mol/K | |
|---|---|---|---|---|---|---|
| present study | 310 | 1.33 ± 0.05 | 9.9 ± 0.2 | −35.6 ± 0.1 | −57.7 ± 0.8 | −71 ± 2 |
| present study 1 | 298 | 1 | 0.6 ± 0.1 | −27 ± 1 | −38 ± 3 | −37 ± 10 |
| Zhang et al. 2 [18] | 298 | 1.04 | 0.49 | −27.0 | −42.2 | −62.6 |
| Zhang et al. 3 [18] | −27.4 | −54.7 | −91.6 |
| The Presence of HSA | LV | LV+HPCD | LV+HPolS | |
|---|---|---|---|---|
| tg α 2 | − | 2.4 ± 0.2 | 2.2 ± 0.2 | 2.9 ± 0.2 |
| free | 2.1 ± 0.2 | 2.2 ± 0.2 | 2.9 ± 0.2 | |
| conjugate 3 | 2.1 ± 0.2 | 2.7 ± 0.2 |
| Strain | Time, h | 109 CFU 1 | ||||
|---|---|---|---|---|---|---|
| LV | (HSA−HPCD)conj+LV | (HSA−HPolS)conj+LV | HSA−LV 1:5 | HSA−LV 1:10 | ||
| E. coli | 24 | 1.6 ± 0.2 | 2.0 ± 0.2 | 0.2 ± 0.2 | 1.0 ± 0.1 | 1.1 ± 0.1 |
| 48 | 2.1 ± 0.2 | 2.3 ± 0.2 | 0.7 ± 0.1 | 1.7 ± 0.2 | 1.6 ± 0.1 | |
| B. subtilis | 24 | 0.9 ± 0.1 | 0.4 ± 0.1 | 0.8 ± 0.1 | 0.45 ± 0.05 | 1.1 ± 0.1 |
| 48 | 1.0 ± 0.1 | 0.5 ± 0.1 | 0.9 ± 0.1 | 0.51 ± 0.05 | 1.2 ± 0.1 | |
| Strain | LV | (HSA−HPCD)conj+LV | (HSA−HPolS)conj+LV | (HSA−LV)conj 1:5 | (HSA−LV)conj 1:10 |
|---|---|---|---|---|---|
| E. coli ATCC 25922 | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | 9 ± 1 | 9 ± 1 |
| B. subtilis ATCC 6633 | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.8 ± 0.1 | 3 ± 1 | 5 ± 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopnova, T.Y.; Yakupova, L.R.; Belogurova, N.G.; Kudryashova, E.V. Human Serum Albumin Grafted by Monomeric and Polymeric β-Cyclodextrin as Drug Delivery System for Levofloxacin with Improved Pharmacological Properties. Future Pharmacol. 2024, 4, 139-162. https://0-doi-org.brum.beds.ac.uk/10.3390/futurepharmacol4010010
Kopnova TY, Yakupova LR, Belogurova NG, Kudryashova EV. Human Serum Albumin Grafted by Monomeric and Polymeric β-Cyclodextrin as Drug Delivery System for Levofloxacin with Improved Pharmacological Properties. Future Pharmacology. 2024; 4(1):139-162. https://0-doi-org.brum.beds.ac.uk/10.3390/futurepharmacol4010010
Chicago/Turabian StyleKopnova, Tatiana Yu, Linara R. Yakupova, Natalya Georgievna Belogurova, and Elena Vadimovna Kudryashova. 2024. "Human Serum Albumin Grafted by Monomeric and Polymeric β-Cyclodextrin as Drug Delivery System for Levofloxacin with Improved Pharmacological Properties" Future Pharmacology 4, no. 1: 139-162. https://0-doi-org.brum.beds.ac.uk/10.3390/futurepharmacol4010010