
Unravelling the Supramolecular Driving Forces in the Formation of CO2-Responsive Pseudopeptidic Low-Molecular-Weight Hydrogelators

 , ,
, ,  , and
, and
Abstract
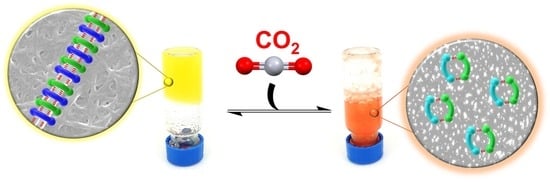
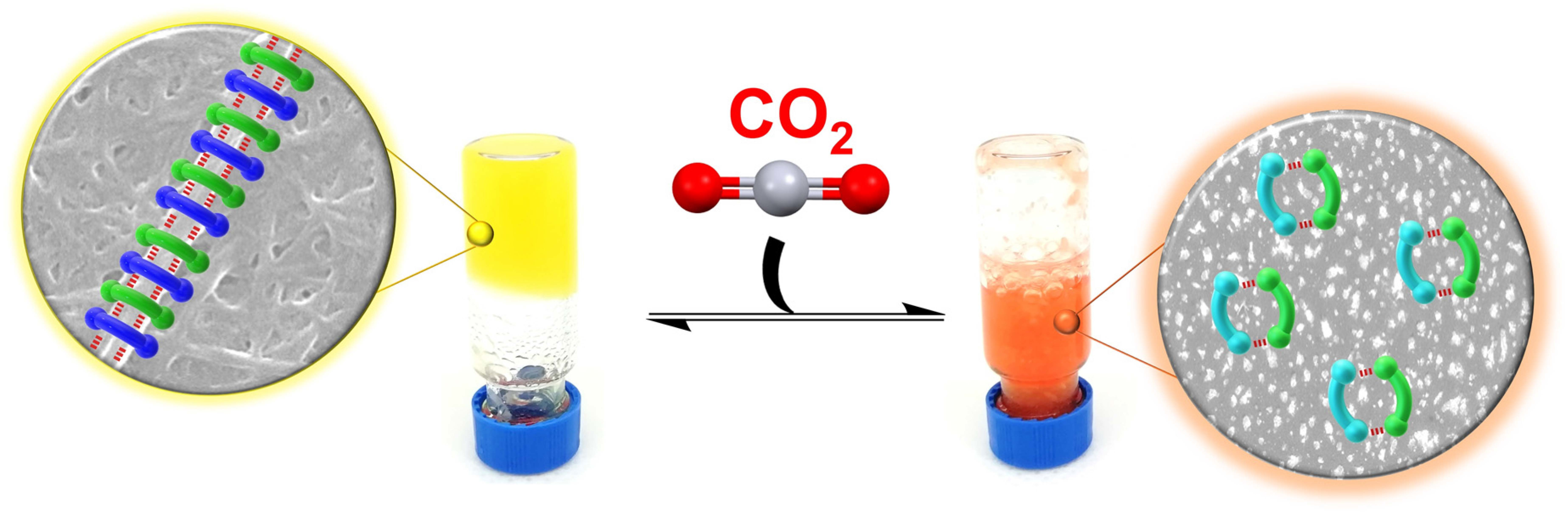
:
1. Introduction
2. Results and Discussion
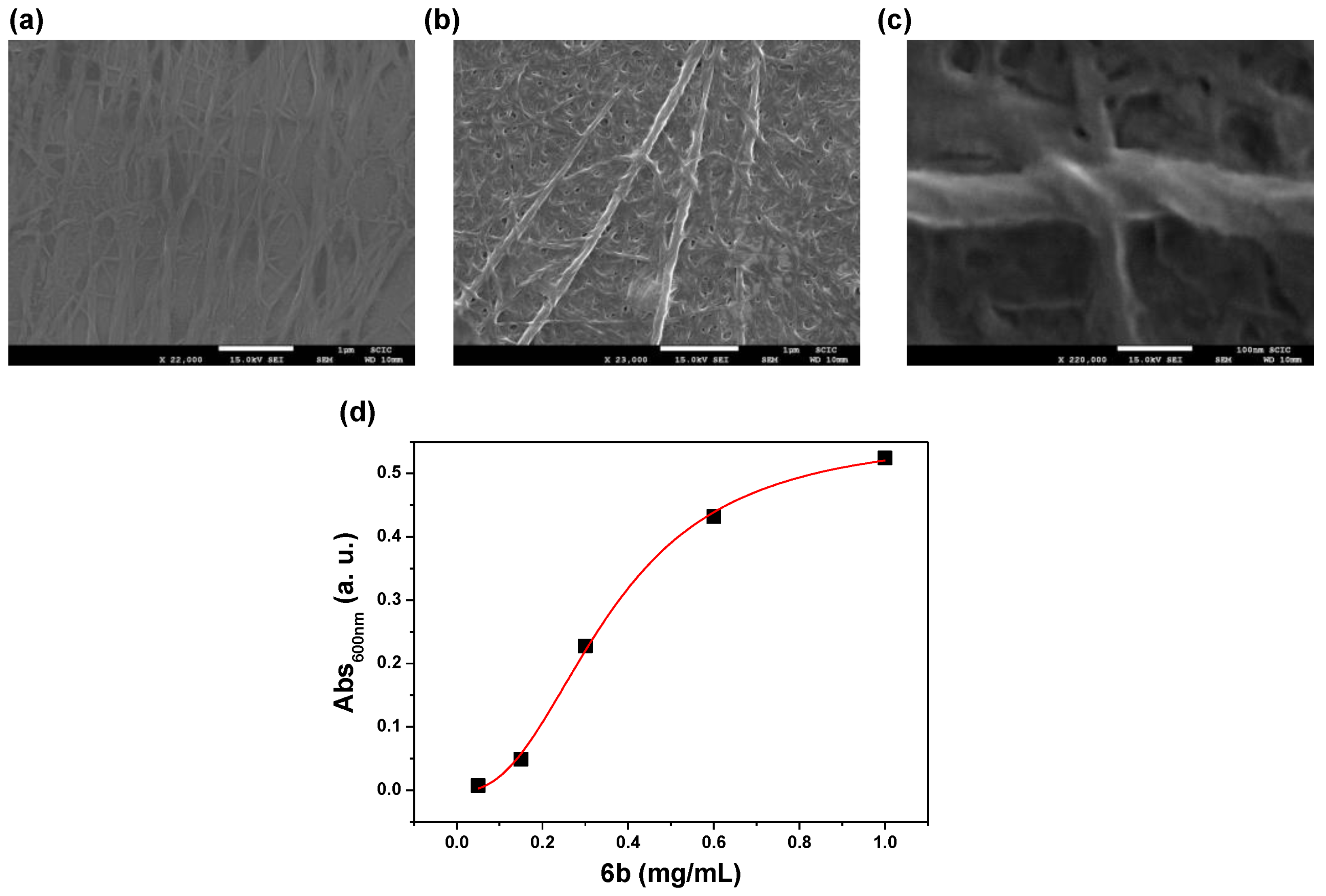
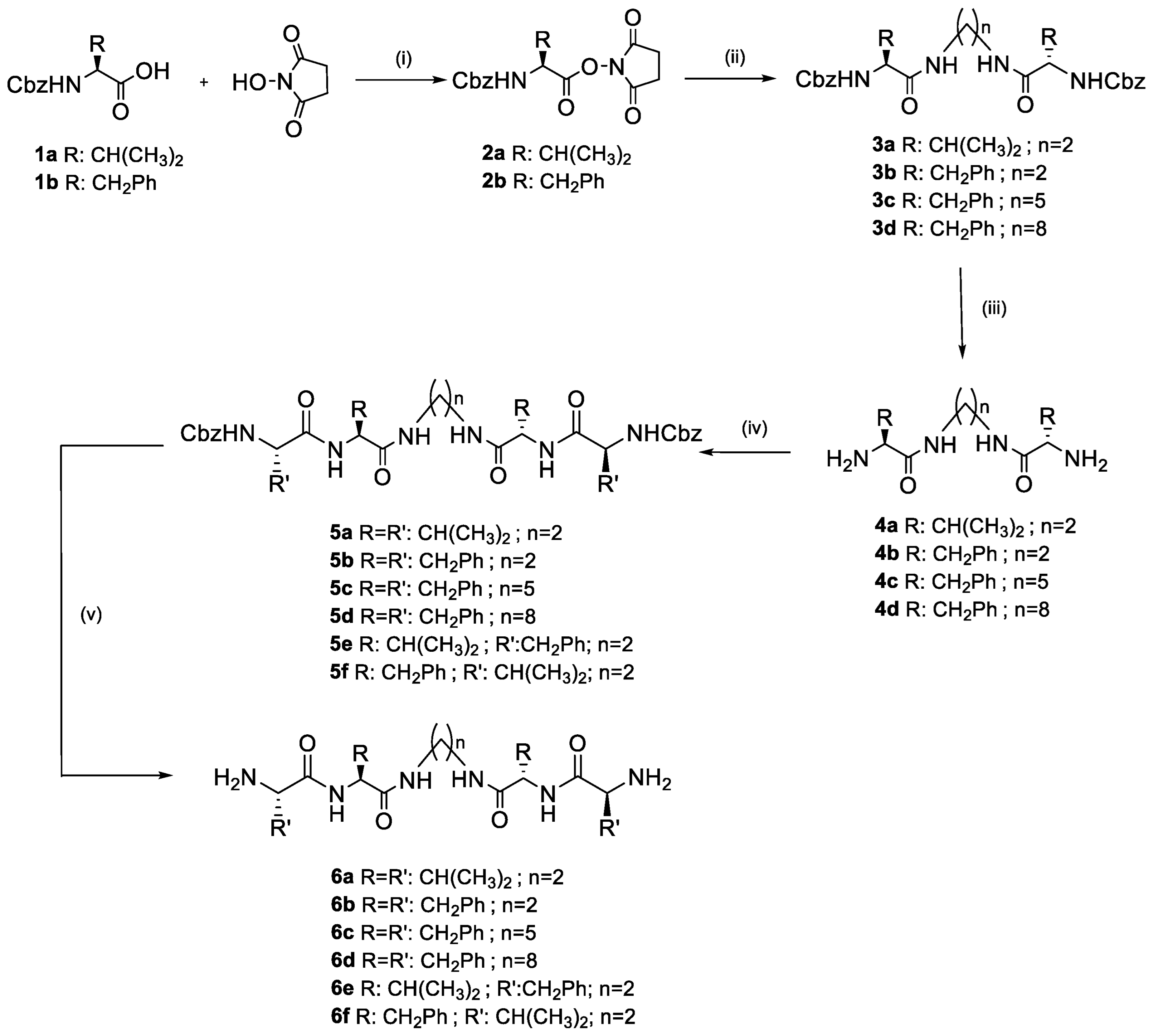
2.1. Synthesis of the Pseudopeptidic Compounds and Gelation Properties of LMWGs
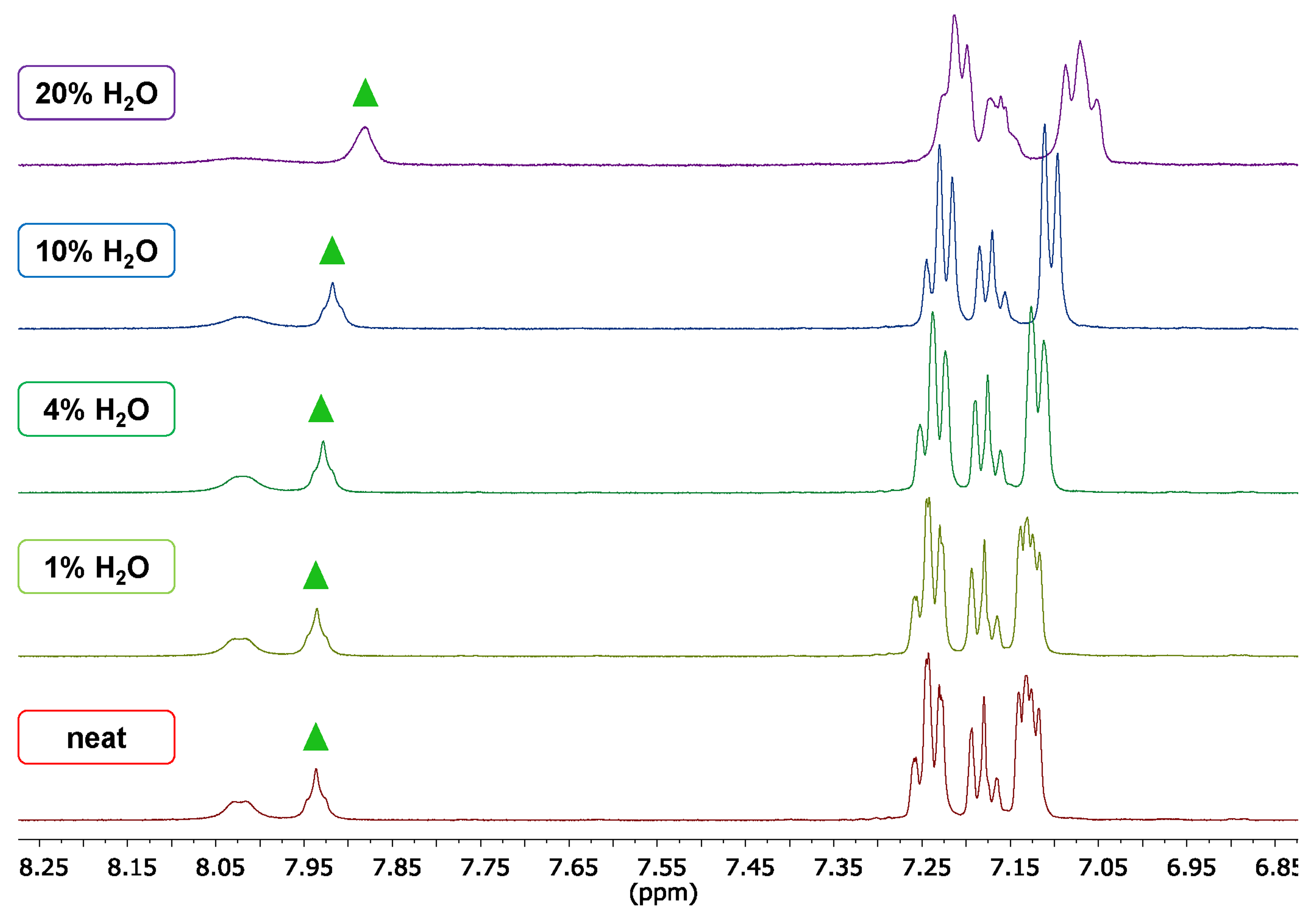
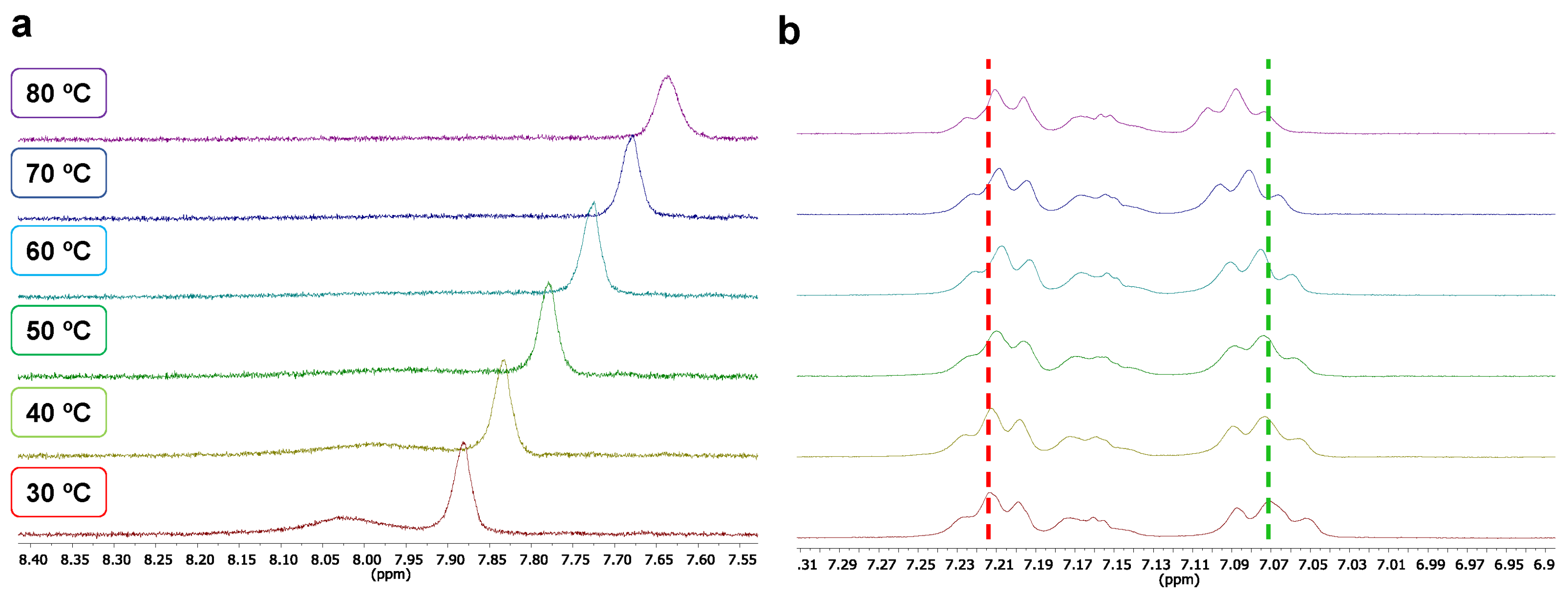
2.2. Supramolecular Driving Forces
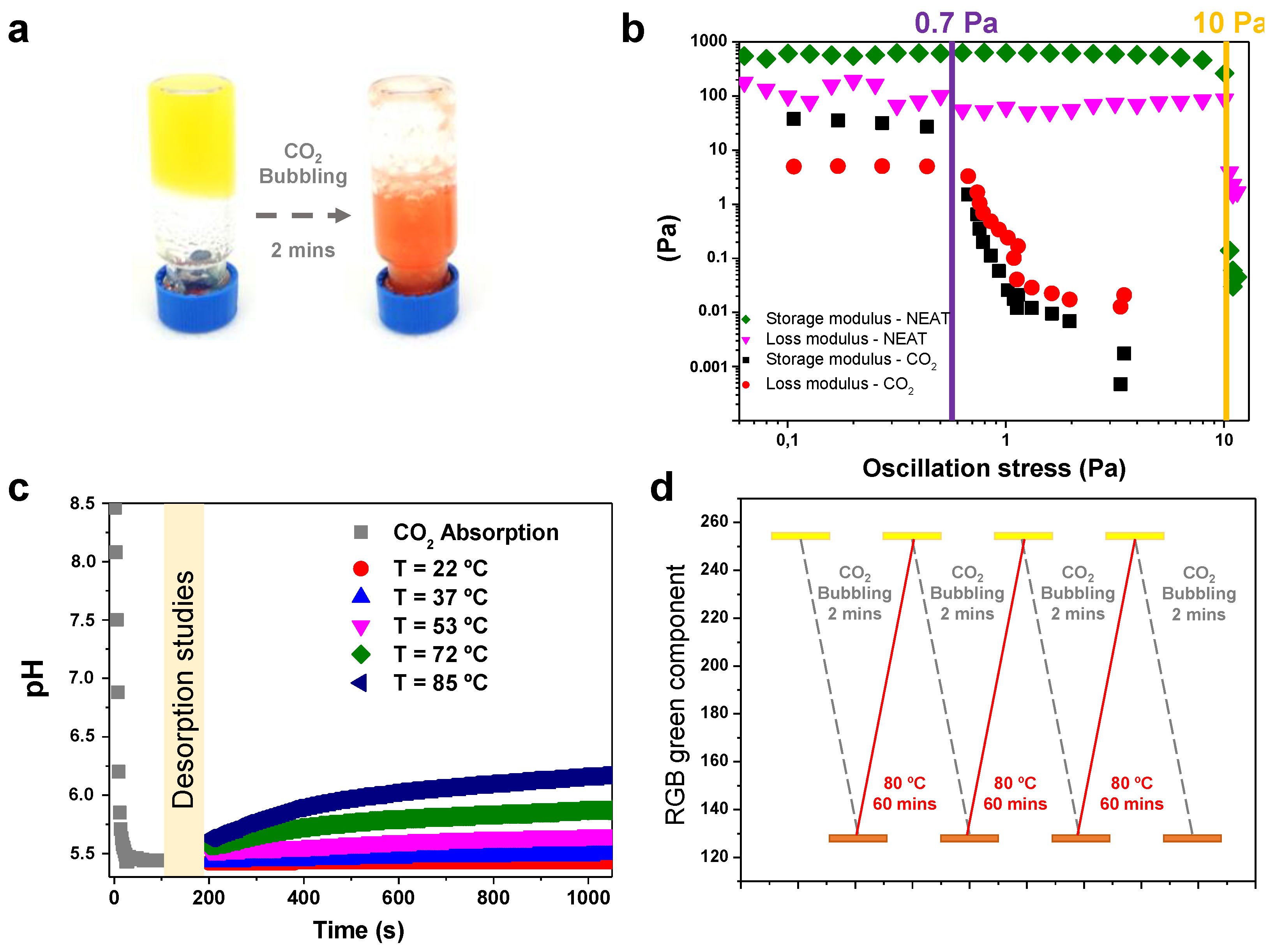
2.3. CO2 Stimulus Responsiveness
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Gelation Experiments
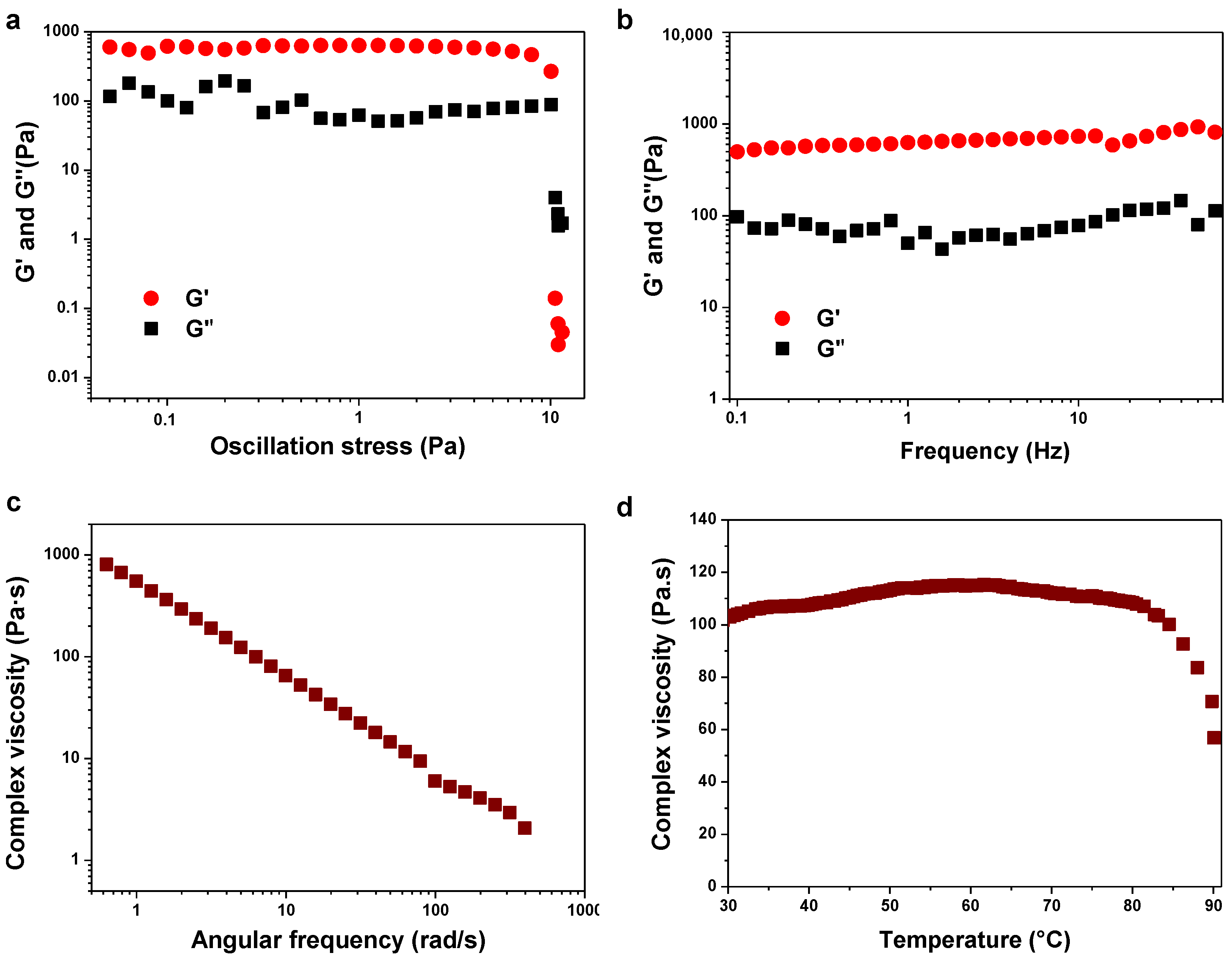
4.3. Rheological Experiments
4.4. Molecular Modelling
4.5. Synthetic Protocols
4.5.1. Synthesis of Cbz-N Protected Tetra-Pseudopeptidic Intermediates (5)
Synthesis of 5a
Synthesis of 5b
Synthesis of 5c
Synthesis of 5d
Synthesis of 5e
Synthesis of 5f
4.5.2. Synthesis of Tetra-Pseudopeptidic Compounds (6)
Synthesis of 6a
Synthesis of 6b
Synthesis of 6c
Synthesis of 6d
Synthesis of 6e
Synthesis of 6f
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Dastidar, P. Supramolecular gelling agents: Can they be designed? Chem. Soc. Rev. 2008, 37, 2699–2715. [Google Scholar] [CrossRef] [PubMed]
- Buerkle, L.E.; Rowan, S. Supramolecular gels formed from multi-component low molecular weight species. Chem. Soc. Rev. 2012, 41, 6089–6102. [Google Scholar] [CrossRef] [PubMed]
- Draper, E.R.; Adams, D.J. How should multicomponent supramolecular gels be characterised? Chem. Soc. Rev. 2018, 47, 3395–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chivers, P.R.A.; Smith, D.K. Shaping and structuring supramolecular gels. Nat. Rev. Mater. 2019, 4, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Hakala, T.A.; Schnaider, L.; Bernardes, G.J.L.; Gazit, E.; Knowles, T.P.J. Biomimetic peptide self-assembly for functional materials. Nat. Rev. Chem. 2020, 4, 615–634. [Google Scholar] [CrossRef]
- Wang, D.X.; Wang, M.X. Exploring Anion−π Interactions and Their Applications in Supramolecular Chemistry. Acc. Chem. Res. 2020, 53, 1364–1380. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, I.M.; Niu, S.Q.; Hall, M.B.; Zaric, S.O. Role of aromatic amino acids in amyloid self-assembly. Int. J. Biol. Macromol. 2020, 156, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Piras, C.C.; Smith, D.K. Sequential Assembly of Mutually Interactive Supramolecular Hydrogels and Fabrication of Multi-Domain Materials. Chem. Eur. J. 2019, 25, 11318–11326. [Google Scholar] [CrossRef]
- Cheng, W.; Ding, Z.; Zheng, X.; Lu, Q.; Kong, X.; Zhou, X.; Lumand, G.; Kaplan, D.L. Injectable hydrogel systems with multiple biophysical and biochemical cues for bone regeneration. Biomater. Sci. 2020, 8, 2537–2548. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Wu, J. pH-Sensitive nanogels for drug delivery in cancer therapy. Biomater. Sci. 2021, 9, 574–589. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Delivery Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Mayr, J.; Saldias, C.; Diaz, D.D. Release of small bioactive molecules from physical gels. Chem. Soc. Rev. 2018, 47, 1484–1515. [Google Scholar] [CrossRef] [PubMed]
- Guo, V.H.; Bae, J.; Fang, Z.W.; Li, P.P.; Zhao, F.; Vu, G.H. Hydrogels and Hydrogel-Derived Materials for Energy and Water Sustainability. Chem. Rev. 2020, 120, 7642–7707. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.A.; Leung, F.K.C.; Stuart, M.C.A.; Kajitani, T.; Fukushima, T.; van der Giessen, E.; Feringa, B.L. Artificial muscle-like function from hierarchical supramolecular assembly of photoresponsive molecular motors. Nat. Chem. 2018, 10, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Nele, V.; Wojciechowski, J.P.; Armstrong, J.P.K.; Stevens, M.M. Tailoring Gelation Mechanisms for Advanced Hydrogel Applications. Adv. Funct. Mater. 2020, 30, 2002759. [Google Scholar] [CrossRef]
- Fleming, S.; Ulijn, R.V. Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev. 2014, 43, 8150–8177. [Google Scholar] [CrossRef]
- Dou, X.-Q.; Feng, C.-L. Amino Acids and Peptide-Based Supramolecular Hydrogels for Three-Dimensional Cell Culture. Adv. Mater. 2017, 29, 1604062. [Google Scholar] [CrossRef]
- Ramalhete, S.M.; Nartowski, K.P.; Sarathchandra, N.; Foster, J.S.; Round, A.N.; Angulo, J.; Lloyd, G.O.; Khimyak, Y.Z. Supramolecular Amino Acid Based Hydrogels: Probing the Contribution of Additive Molecules using NMR Spectroscopy. Chem. Eur. J. 2017, 23, 8014–8024. [Google Scholar] [CrossRef] [Green Version]
- Das, T.; Häring, M.; Haldar, D.; Díaz, D.D. Phenylalanine and derivatives as versatile low-molecular-weight gelators: Design, structure and tailored function. Biomater. Sci. 2018, 6, 38–59. [Google Scholar] [CrossRef]
- Giraud, T.; Hoschtettler, P.; Pickaert, G.; Averlant-Petit, M.-C.; Stefan, L. Emerging low-molecular weight nucleopeptide-based hydrogels: State of the art, applications, challenges and perspectives. Nanoscale 2022, 14, 4908–4921. [Google Scholar] [CrossRef]
- Ulijn, R.V.; Smith, A.M. Designing peptide based nanomaterials. Chem. Soc. Rev. 2008, 37, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Zanna, N.; Merlettini, A.; Tomasini, C. Self-healing hydrogels triggered by amino acids. Org. Chem. Front. 2016, 3, 1699–1704. [Google Scholar] [CrossRef]
- Steed, J.W. Anion-tuned supramolecular gels: A natural evolution from urea supramolecular chemistry. Chem. Soc. Rev. 2010, 39, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Valls, A.; Castillo, A.; Porcar, R.; Hietala, S.; Altava, B.; García-Verdugo, E.; Luis, S.V. Urea-Based Low-Molecular-Weight Pseudopeptidic Organogelators for the Encapsulation and Slow Release of (R)-Limonene. J. Agric. Food Chem. 2020, 68, 7051–7061. [Google Scholar] [CrossRef]
- Rubio, J.; Alfonso, I.; Burguete, M.I.; Luis, S.V. Interplay between hydrophilic and hydrophobic interactions in the self-assembly of a gemini amphiphilic pseudopeptide: From nano-spheres to hydrogels. Chem. Commun. 2012, 48, 2210–2212. [Google Scholar] [CrossRef] [Green Version]
- Valls, A.; Burguete, M.I.; Kuret, L.; Altava, B.; Luis, S.V. Open chain pseudopeptides as hydrogelators with reversible and dynamic responsiveness to pH, temperature and sonication as vehicles for controlled drug delivery. J. Mol. Liq. 2022, 348, 118051. [Google Scholar] [CrossRef]
- Raza, F.; Zhu, Y.; Chen, L.; You, X.; Zhang, J.; Khan, A.; Waseem Khan, M.; Hasnat, M.; Zafar, H.; Wu, J.; et al. Paclitaxel-loaded pH responsive hydrogel based on self-assembled peptides for tumor targeting. Biomater. Sci. 2019, 7, 2023–2036. [Google Scholar] [CrossRef]
- Cardoso, A.Z.; Alvarez, A.E.A.; Cattoz, B.N.; Griffiths, P.C.; King, S.M.; Frith, W.J.; Adams, D.J. The influence of the kinetics of self-assembly on the properties of dipeptide hydrogels. Faraday Discuss. 2013, 166, 101–116. [Google Scholar] [CrossRef]
- Shaikh, H.; Rho, J.Y.; Macdougall, L.J.; Gurnani, P.; Lunn, A.M.; Yang, J.; Huband, S.; Mansfield, E.D.H.; Peltier, R.; Perrier, S. Hydrogel and Organogel Formation by Hierarchical Self-Assembly of Cyclic Peptides Nanotubes. Chem. Eur. J. 2018, 24, 19066–19074. [Google Scholar] [CrossRef]
- Chu, C.-W.; Stricker, L.; Kirse, T.M.; Hayduk, M.; Ravoo, B.J. Light-Responsive Arylazopyrazole Gelators: From Organic to Aqueous Media and from Supramolecular to Dynamic Covalent Chemistry. Chem. Eur. J. 2019, 25, 6131–6140. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Heilshorn, S.C. Adaptable Hydrogel Networks with Reversible Linkages for Tissue Engineering. Adv. Mater. 2015, 27, 3717. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Liao, W.-C.; Sohn, Y.S.; Fadeev, M.; Cecconello, A.; Nechushtai, R.; Willner, I. Stimuli-Responsive Nucleic Acid-Based Polyacrylamide Hydrogel-Coated Metal–Organic Framework Nanoparticles for Controlled Drug Release. Adv. Funct. Mater. 2018, 28, 1705137. [Google Scholar] [CrossRef]
- Doring, A.; Birnbaum, W.; Kuckling, D. Responsive hydrogels—Structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science. Chem. Soc. Rev. 2013, 42, 7391–7420. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Boissiere, O.; Kumar, S.; Tong, X.; Tremblay, L.; Zhao, Y. Two-Way CO2-Switchable Triblock Copolymer Hydrogels. Macromolecules 2012, 45, 7440–7445. [Google Scholar] [CrossRef]
- Zhang, L.; Qian, J.; Fan, Y.; Feng, W.; Tao, Z.; Yang, H. A facile CO2 switchable nanocomposite with reversible transition from sol to self-healable hydrogel. RSC Adv. 2015, 5, 62229–62234. [Google Scholar] [CrossRef]
- Jie, K.; Zhou, Y.; Yao, Y.; Shi, B.; Huang, F. CO2-Responsive Pillar[5]arene-Based Molecular Recognition in Water: Establishment and Application in Gas-Controlled Self-Assembly and Release. J. Am. Chem. Soc. 2015, 137, 10472–10475. [Google Scholar] [CrossRef]
- Nagasawa, Y.; Seida, Y.; Gotoh, T.; Furuya, E. Influence of Hydrophobicity of Backbone Polymer in Thermo-Responsive Hydrogel with Immobilized Amine on Cycle Capacity for Absorption and Recovery of CO2. Polymers 2019, 11, 1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.G.; Zhang, M.; Zhu, X.X. CO2-Switchable Self-Healing Host–Guest Hydrogels. Macromolecules 2017, 50, 9696–9701. [Google Scholar] [CrossRef]
- Satav, S.S.; Bhat, S.; Thayumanavan, S. Feedback Regulated Drug Delivery Vehicles: Carbon Dioxide Responsive Cationic Hydrogels for Antidote Release. Biomacromolecules 2010, 11, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Weiss, R.G. Chemically Reversible Organogels: Aliphatic Amines as “Latent” Gelators with Carbon Dioxide. J. Am. Chem. Soc. 2001, 123, 10393–10394. [Google Scholar] [CrossRef]
- Rao, A.B.; Rubin, E.S. A Technical, Economic, and Environmental Assessment of Amine-Based CO2 Capture Technology for Power Plant Greenhouse Gas Control. Environ. Sci. Technol. 2002, 36, 4467–4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allix, F.; Curcio, P.; Nghi Pham, Q.; Pickaert, G.; Jamart-Gregoire, B. Evidence of Intercolumnar π−π Stacking Interactions in Amino-Acid-Based Low-Molecular-Weight Organogels. Langmuir 2010, 26, 16818–16827. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Adler-Abramovich, L.; Gazit, E.; Verma, S. Spacer driven morphological twist in Phe-Phe dipeptide conjugates. Tetrahedron 2013, 69, 2004–2009. [Google Scholar] [CrossRef]
- Becerril, J.; Bolte, M.; Burguete, M.I.; Galindo, F.; García-España, E.; Luis, S.V.; Miravet, J.F. Efficient Macrocyclization of U-Turn Preorganized Peptidomimetics: The Role of Intramolecular H-Bond and Solvophobic Effects. J. Am. Chem. Soc. 2003, 125, 6677–6686. [Google Scholar] [CrossRef] [PubMed]
- Gorla, L.; Martí-Centelles, V.; Altava, B.; Burguete, M.I.; Luis, S.V. The role of the side chain in the conformational and self-assembly patterns of C2-symmetric Val and Phe pseudopeptidic derivatives. CrystEngComm 2019, 21, 2398–2408. [Google Scholar] [CrossRef]
- Becerril, J.; Escuder, B.; Miravet, J.F.; Gavara, R.; Luis, S.V. Understanding the Expression of Molecular Chirality in the Self-Assembly of a Peptidomimetic Organogelator. Eur. J. Org. Chem. 2005, 2005, 481–485. [Google Scholar] [CrossRef]
- Murata, K.; Aoki, M.; Suzuki, T.; Harada, T.; Kawabata, H.; Komori, T.; Ohseto, F.; Ueda, K.; Shinkai, S. Thermal and Light Control of the Sol-Gel Phase Transition in Cholesterol-Based Organic Gels. Novel Helical Aggregation Modes as Detected by Circular Dichroism and Electron Microscopic Observation. J. Am. Chem. Soc. 1994, 116, 6664–6676. [Google Scholar] [CrossRef]
- Yan, C.; Pochan, D. Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 2010, 39, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- El-Husseiny, H.M.; Mady, E.A.; Hamabe, L.; Abugomaa, A.; Shimada, K.; Yoshida, T.; Tanaka, T.; Yokoi, A.; Elbadawy, M.; Tanaka, R. Smart/stimuli-responsive hydrogels: Cutting-edge platforms for tissue engineering and other biomedical applications. Mater. Today Bio 2022, 13, 100186. [Google Scholar] [CrossRef]
- Lin, C.; Skufca, J.; Partch, R.E. New insights into prediction of weak π–π complex association through proton-nuclear magnetic resonance analysis. BMC Chem. 2020, 14, 66. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfonso, I.; Burguete, M.I.; Galindo, F.; Luis, S.V.; Vigara, L. Molecular Rotors as Simple Models to Study Amide NH-Aromatic Interactions and Their Role in the Folding of Peptide-like Structures. J. Org. Chem. 2007, 72, 7947–7956. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Crawshaw, J.P.; Maitland, G.C.; Martin Trusler, J.P.; Vega-Mazza, D. The pH of CO2-Saturated Water at Temperatures Between 308 K and 423 K at Pressures up to 15 MPa. J. Supercrit. Fluids 2013, 82, 129–137. [Google Scholar] [CrossRef]
- Wang, M.; Lawal, A.; Stephenson, P.; Sidders, J.; Ramshaw, C. Post-combustion CO2 capture with chemical absorption: A state-of-the-art review. Chem. Eng. Res. Des. 2011, 89, 1609–1624.A. [Google Scholar] [CrossRef] [Green Version]
- Heldebrant, D.J.; Koech, P.K.; Ang, M.T.C.; Liang, C.; Rainbolt, J.E.; Yonker, C.R.; Jessop, P.G. Reversible zwitterionic liquids, the reaction of alkanol guanidines, alkanol amidines, and diamines with CO2. Green Chem. 2010, 12, 713–721. [Google Scholar] [CrossRef]
- Said, R.B.; Kolle, J.M.; Essalah, K.; Tangour, B.; Sayari, A. A Unified Approach to CO2–Amine Reaction Mechanisms. ACS Omega 2020, 5, 26125–26133. [Google Scholar] [CrossRef] [PubMed]
- Note: Studies in D2O or D2O/DMSO-d6 mixtures could not be performed due to gel formation.
- Hampe, E.M.; Rudkevich, D.M. Exploring reversible reactions between CO2 and amines. Tetrahedron 2003, 59, 9619–9625. [Google Scholar] [CrossRef]
- Deppmeier, B.J.; Driessen, A.J.; Hehre, T.S.; Hehre, W.J.; Johnson, J.A.; Klunzinger, P.E.; Leonard, J.M.; Pham, I.N.; Pietro, W.J.; Jianguo, Y. Calculated Using Spartan08 Software at the MMFFaq Level of Theory; Spartan ‘08, build 132 (Mar 27 2009); Wavefunction Inc.: Irvine, CA, USA, 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Comp. | Solvent(s) | Proportion (v/v) | Conc. (mg/mL) | Result |
|---|---|---|---|---|---|
| 1 | 6a | H2O:DMSO | 90:10 | 1 | S |
| 2 | 6b | H2O:DMSO | 90:10 | 1 | G 4 |
| 3 | 6c | H2O:DMSO | 90:10 | 1 | wG 4 |
| 4 | 6c | H2O:DMSO | 90:10 | 3 | G 4 |
| 5 | 6d | H2O:DMSO | 90:10 | 1 | wG 2 |
| 6 | 6d | H2O:DMSO | 90:10 | 5 | wG 3 |
| 7 | 6e | H2O:DMSO | 90:10 | 1 | S |
| 8 | 6e | H2O:DMSO | 90:10 | 5 | S |
| 9 | 6f | H2O:DMSO | 90:10 | 1 | S |
| 10 | 6f | H2O:DMSO | 90:10 | 5 | wG 3 |
| 11 | 6e + 6f | H2O:DMSO | 90:10 | 0.5 + 0.5 | wG |
| 12 | 6b | H2O:DMSO | 50:50 | 1 | wG 3 |
| 13 | 6a | H2O:EtOH | 90:10 | 1 | I |
| 14 | 6b | H2O:EtOH | 90:10 | 1 | I |
| 15 | 6b | H2O:EtOH | 70:30 | 1 | wG 3 |
| 16 | 6b | H2O:EtOH | 70:30 | 0.5 | wG 3 |
| 17 | 6b | H2O:EtOH | 50:50 | 1 | G 3 |
| 18 | 6a | H2O:EtOH | 50:50 | 1 | S |
| 19 | 6a | H2O | - | 1 | I 2 |
| 20 | 6b | H2O | - | 1 | G 2 |
| Vial | Buffer (mM) | pH | Result 1 |
|---|---|---|---|
| 1 | - 2 | 1.2 | S |
| 2 | Citrate (20) | 4.4 | wG |
| 3 | Citrate (20) | 6.0 | wG |
| 4 | Phosphate (30) | 7.1 | wG |
| 5 | HEPES (20) | 8.1 | G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteve, F.; Villanueva-Antolí, A.; Altava, B.; García-Verdugo, E.; Luis, S.V. Unravelling the Supramolecular Driving Forces in the Formation of CO2-Responsive Pseudopeptidic Low-Molecular-Weight Hydrogelators. Gels 2022, 8, 390. https://0-doi-org.brum.beds.ac.uk/10.3390/gels8060390
Esteve F, Villanueva-Antolí A, Altava B, García-Verdugo E, Luis SV. Unravelling the Supramolecular Driving Forces in the Formation of CO2-Responsive Pseudopeptidic Low-Molecular-Weight Hydrogelators. Gels. 2022; 8(6):390. https://0-doi-org.brum.beds.ac.uk/10.3390/gels8060390
Chicago/Turabian StyleEsteve, Ferran, Alexis Villanueva-Antolí, Belén Altava, Eduardo García-Verdugo, and Santiago V. Luis. 2022. "Unravelling the Supramolecular Driving Forces in the Formation of CO2-Responsive Pseudopeptidic Low-Molecular-Weight Hydrogelators" Gels 8, no. 6: 390. https://0-doi-org.brum.beds.ac.uk/10.3390/gels8060390