iNOS Gene Ablation Prevents Liver Fibrosis in Leptin-Deficient ob/ob Mice

 , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Blood Measurements
2.3. Intrahepatic Lipid Content
2.4. Cell Cultures
2.5. RNA Extraction and Real-Time PCR
2.6. Quantification and Characterization of Fibrotic Depots
2.7. Statistical Analysis
3. Results
3.1. Lack of iNOS Gene Ameliorates the Obese Phenotype of ob/ob Mice
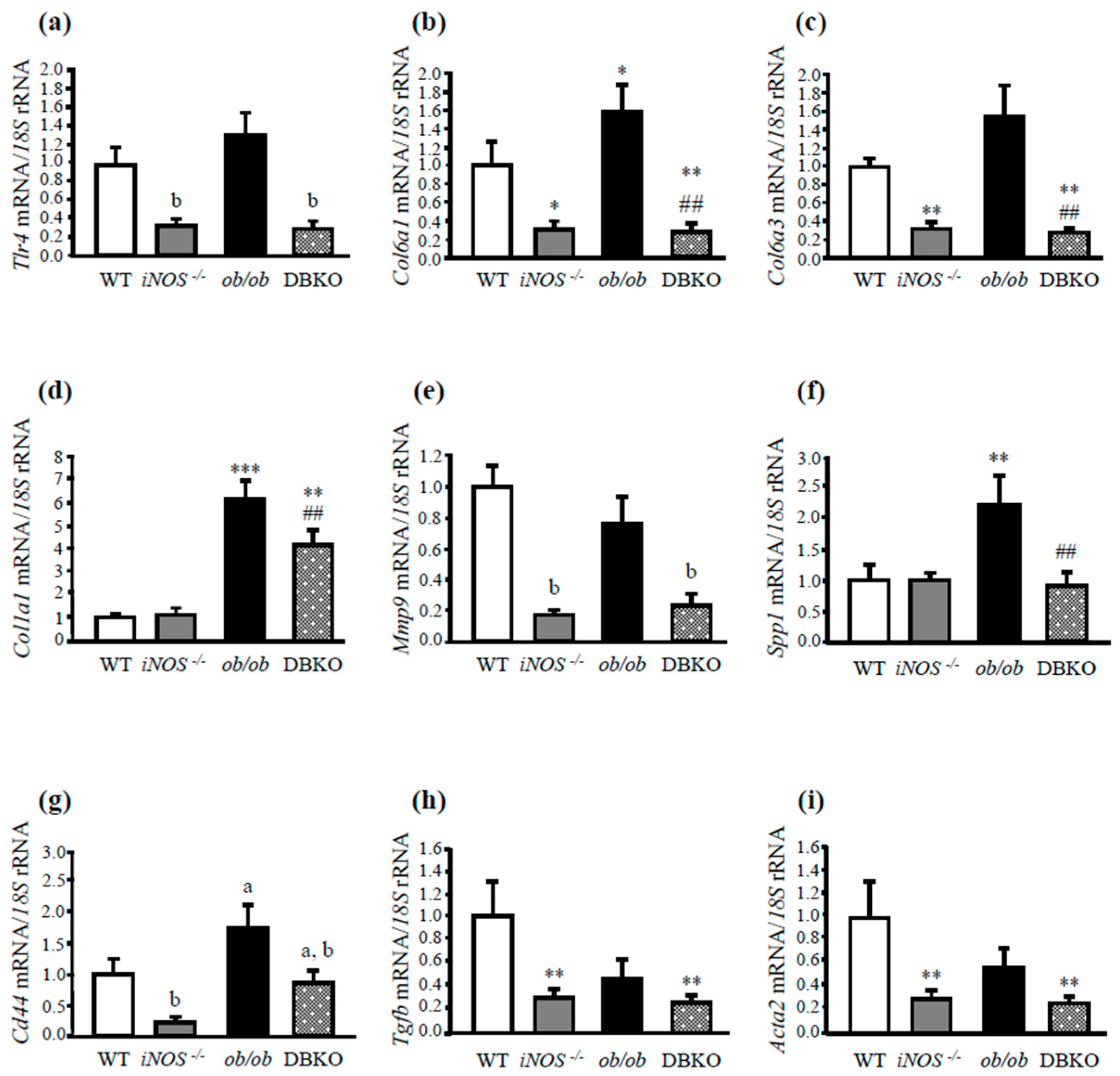
3.2. ob/ob Mice Lacking iNOS Display Changes in the Expression of Molecules Involved in Liver Inflammation
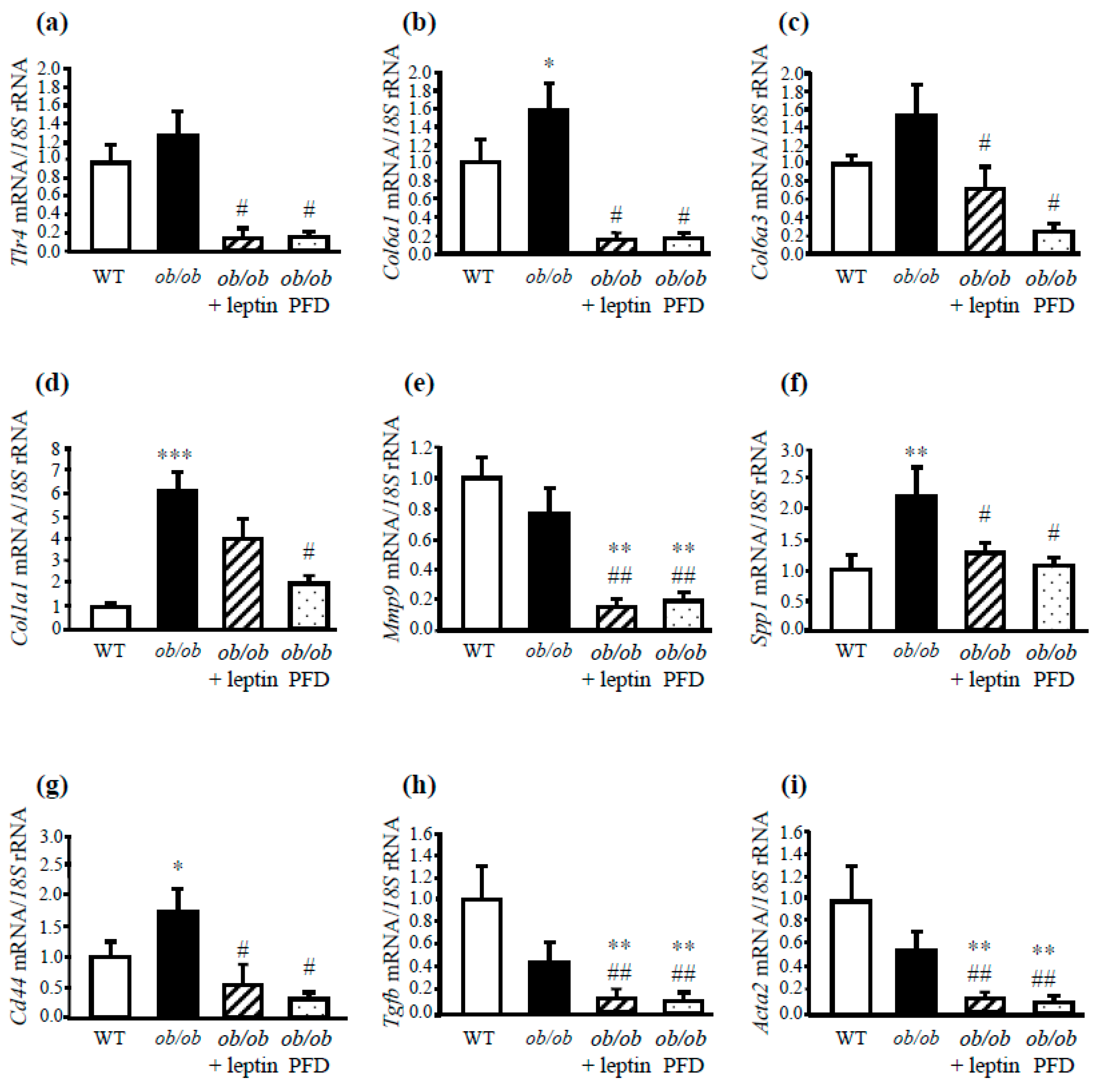
3.3. Leptin Administration Protects from Inflammation and Fibrosis in the Liver of ob/ob Mice
3.4. Leptin Treatment Increases Inflammatory and Fibrotic Genes in AML12 Hepatocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blond, E.; Disse, E.; Cuerq, C.; Drai, J.; Valette, P.J.; Laville, M.; Thivolet, C.; Simon, C.; Caussy, C. EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease in severely obese people: Do they lead to over-referral? Diabetologia 2017, 60, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell Mol. Life Sci. 2011, 68, 3175–3199. [Google Scholar] [CrossRef] [PubMed]
- El-Karef, A.; Yoshida, T.; Gabazza, E.C.; Nishioka, T.; Inada, H.; Sakakura, T.; Imanaka-Yoshida, K. Deficiency of tenascin-C attenuates liver fibrosis in immune-mediated chronic hepatitis in mice. J. Pathol. 2007, 211, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Van Eyken, P.; Sciot, R.; Desmet, V.J. Expression of the novel extracellular matrix component tenascin in normal and diseased human liver. An immunohistochemical study. J. Hepatol. 1990, 11, 43–52. [Google Scholar] [CrossRef]
- Marra, F. Leptin and liver fibrosis: A matter of fat. Gastroenterology 2002, 122, 1529–1532. [Google Scholar] [CrossRef] [PubMed]
- Bertolani, C.; Marra, F. Role of adipocytokines in hepatic fibrosis. Curr. Pharm. Des. 2010, 16, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Ezquerro, S.; Méndez-Gimenez, L.; Becerril, S.; Frühbeck, G. Revisiting the adipocyte: A model for integration of cytokine signaling in the regulation of energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E691–E714. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ambrosi, J.; Salvador, J.; Paramo, J.A.; Orbe, J.; de Irala, J.; Díez-Caballero, A.; Gil, M.J.; Cienfuegos, J.A.; Frübeck, G. Involvement of leptin in the association between percentage of body fat and cardiovascular risk factors. Clin. Biochem. 2002, 35, 315–320. [Google Scholar] [CrossRef]
- Frühbeck, G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes 1999, 48, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Gómez-Ambrosi, J. Modulation of the leptin-induced white adipose tissue lipolysis by nitric oxide. Cell Signal. 2001, 13, 827–833. [Google Scholar] [CrossRef]
- Rodríguez, A.; Fortuño, A.; Gómez-Ambrosi, J.; Zalba, G.; Díez, J.; Frühbeck, G. The inhibitory effect of leptin on angiotensin II-induced vasoconstriction in vascular smooth muscle cells is mediated via a nitric oxide-dependent mechanism. Endocrinology 2007, 148, 324–331. [Google Scholar]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Sáinz, N.; Ramírez, B.; Collantes, M.; Peñuelas, I.; Gómez-Ambrosi, J.; Frübeck, G. Deletion of inducible nitric-oxide synthase in leptin-deficient mice improves brown adipose tissue function. PLoS ONE 2010, 5, e10962. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Gómez-Ambrosi, J.; Salvador, J. Leptin-induced lipolysis opposes the tonic inhibition of endogenous adenosine in white adipocytes. FASEB J. 2001, 15, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Shiuchi, T.; Nakagami, H.; Iwai, M.; Takeda, Y.; Cui, T.; Chen, R.; Minokoshi, Y.; Horiuchi, M. Involvement of bradykinin and nitric oxide in leptin-mediated glucose uptake in skeletal muscle. Endocrinology 2001, 142, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.D.; Mielenz, M.; Taub, D.D.; Parvizi, N. Leptin induces growth hormone secretion from peripheral blood mononuclear cells via a protein kinase C- and nitric oxide-dependent mechanism. Endocrinology 2003, 144, 5595–5603. [Google Scholar] [CrossRef] [PubMed]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Méndez-Gimenez, L.; Ramírez, B.; Sainz, N.; Llorente, M.; Unamuno, X.; Gómez-Ambrosi, J.; Frübeck, G. Targeted disruption of the iNOS gene improves adipose tissue inflammation and fibrosis in leptin-deficient ob/ob mice: Role of tenascin C. Int. J. Obes. (Lond.) 2018, 42, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Sainz, N.; Rodríguez, A.; Catalán, V.; Becerril, S.; Ramírez, B.; Gómez-Ambrosi, J.; Frübeck, G. Leptin administration downregulates the increased expression levels of genes related to oxidative stress and inflammation in the skeletal muscle of ob/ob mice. Mediat. Inflamm. 2010, 2010, 784343. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Alonso, R.; Marzo, F.; Santidrián, S. A modified method for the indirect quantitative analysis of phytate in foodstuffs. Anal. Biochem. 1995, 225, 206–212. [Google Scholar]
- Rodríguez, A.; Catalán, V.; Gómez-Ambrosi, J.; García-Navarro, S.; Rotellar, F.; Valentí, V.; Silva, C.; Gil, M.J.; Salvador, J.; Burrell, M.A.; et al. Insulin- and leptin-mediated control of aquaglyceroporins in human adipocytes and hepatocytes is mediated via the PI3K/Akt/mTOR signaling cascade. J. Clin. Endocrinol. Metab. 2011, 96, E586–E597. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin. Endocrinol. (Oxf.) 2008, 68, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Rotellar, F.; Silva, C.; Rodríguez, A.; Salvador, J.; Gil, M.J.; Cienfuegos, J.A.; Frühbeck, G. Validation of endogenous control genes in human adipose tissue: Relevance to obesity and obesity-associated type 2 diabetes mellitus. Horm. Metab. Res. 2007, 39, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; . Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1a induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Patouraux, S.; Rousseau, D.; Bonnafous, S.; Lebeaupin, C.; Luci, C.; Canivet, C.M.; Schenck, A.S.; Bertola, A.; Saint-Paul, M.C.; Iannelli, A.; Gugenheim, J.; et al. CD44 is a key player in non-alcoholic steatohepatitis. J. Hepatol. 2017, 67, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Sáinz, N.; Rodríguez, A.; Catalán, V.; Becerril, S.; Ramírez, B.; Gómez-Ambrosi, J.; Frühbeck, G. Leptin administration favors muscle mass accretion by decreasing FoxO3a and increasing PGC-1a in ob/ob mice. PLoS ONE 2009, 4, e6808. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Gómez-Ambrosi, J. Control of body weight: A physiologic and transgenic perspective. Diabetologia 2003, 46, 143–172. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Investig. 2002, 109, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Muruzábal, F.J.; Frühbeck, G.; Gómez-Ambrosi, J.; Archanco, M.; Burrell, M.A. Immunocytochemical detection of leptin in non-mammalian vertebrate stomach. Gen. Comp. Endocrinol. 2002, 128, 149–152. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Deretzi, G. The potential adverse role of leptin resistance in nonalcoholic fatty liver disease: A hypothesis based on critical review of the literature. J. Clin. Gastroenterol. 2011, 45, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-β (TGF-β) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Sakaida, I.; Uchida, K.; Terai, S.; Okita, K. Leptin enhances TNF-a production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci. 2005, 77, 1502–1515. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, R.; Jones-Carson, J.; Reed, D.A.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C.; Fantuzzi, G. Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicity: Role of tumor necrosis factor a and IL-18. Proc. Natl. Acad. Sci. USA 2000, 97, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Sofat, N.; Kashiwagi, M.; Orend, G. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Vilarino-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Cousin, B.; Bascands-Viguerie, N.; Kassis, N.; Nibbelink, M.; Ambid, L.; Casteilla, L.; Pénicaud, L. Cellular changes during cold acclimatation in adipose tissues. J. Cell Physiol. 1996, 167, 285–289. [Google Scholar] [CrossRef]
- Bankoglu, E.E.; Seyfried, F.; Rotzinger, L.; Nordbeck, A.; Corteville, C.; Jurowich, C.; Germer, C.T.; Otto, C.; Stopper, H.; et al. Impact of weight loss induced by gastric bypass or caloric restriction on oxidative stress and genomic damage in obese Zucker rats. Free Radic. Biol. Med. 2016, 94, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Tateya, S.; Rizzo, N.O.; Handa, P.; Cheng, A.M.; Morgan-Stevenson, V.; Daum, G.; Clowes, A.W.; Morton, G.J.; Schwartz, M.W.; Kim, F. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes 2011, 60, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918.e6–927.e6. [Google Scholar] [CrossRef] [PubMed]
- Anavi, S.; Eisenberg-Bord, M.; Hahn-Obercyger, M.; Genin, O.; Pines, M.; Tirosh, O. The role of iNOS in cholesterol-induced liver fibrosis. Lab. Investig. 2015, 95, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Zhou, J. Hypoxia-inducible factor-1a under the control of nitric oxide. Methods Enzymol. 2007, 435, 463–478. [Google Scholar] [PubMed]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Lancha, A.; Rodríguez, A.; Catalán, V.; Becerril, S.; Sainz, N.; Ramírez, B.; Burrell, M.A.; Salvador, J.; Frühbeck, G.; Gómez-Ambrosi, J. Osteopontin deletion prevents the development of obesity and hepatic steatosis via impaired adipose tissue matrix remodeling and reduced inflammation and fibrosis in adipose tissue and liver in mice. PLoS ONE 2014, 9, e98398. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Jeong, S.; Xia, Q.; Kong, X. Role of Osteopontin in Liver Diseases. Int. J. Biol. Sci. 2016, 12, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Nagoshi, S. Osteopontin: Versatile modulator of liver diseases. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2014, 44, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Marroquin, C.E.; Wai, P.Y.; Kuo, P.C. Nitric oxide-dependent osteopontin expression induces metastatic behavior in HepG2 cells. Dig. Dis. Sci. 2005, 50, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Duarte, S.; Tsuchihashi, S.; Busuttil, R.W.; Coito, A.J. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. Am. J. Pathol. 2009, 174, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Escuredo, J.M.; Gómez-Ambrosi, J.; Catalán, V.; Domingo, P.; Giralt, M.; Frühbeck, G.; Villarroya, F. Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients. Int. J. Obes. (Lond.) 2015, 39, 121–129. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type | iNOS-/- | ob/ob | DBKO | |
|---|---|---|---|---|
| Body weight (g) | 24.0 ± 0.4 | 23.1 ± 0.4 | 44.9 ± 1.5 | 43.1 ± 0.8 |
| Liver weight (g) | 1.04 ± 0.02 | 1.00 ± 0.03 | 3.15 ± 0.12 | 2.97 ± 0.11 |
| FFA (mmol/L) | 0.74 ± 0.08 | 0.58 ± 0.03 | 1.07 ± 0.06*** | 0.85 ± 0.06 |
| TG (mg/dL) | 66.3 ± 4.4 | 77.1 ± 3.9 | 97.3 ±5.3** | 90.4 ± 0.1 † |
| Glycerol (mg/dL) | 0.025 ± 0.001 | 0.022 ± 0.003 | 0.036 ±.0.001*** | 0.030 ± 0.003***, †† |
| Intrahepatic TG (mg/g) | 19.0 ± 2.2 | 16.8 ± 1.5 | 28.5 ± 3.0 | 24.5 ± 2.6 |
| Glucose (mg/dL) | 83 ± 5 | 77± 2 | 410 ±42*** | 96 ± 5***, †† |
| Insulin (ng/mL) | 0.42 ± 0.04 | 0.32 ± 0.04 | 9.66 ± 0.61*** | 8.89 ± 1.17***,††† |
| HOMA | 1.6 ± 0.2 | 1.1 ± 0.2 | 172.3.0 ± 21.5*** | 34.5 ± 5.2***, †† |
| Adiponectin (μg/mL) | 22.6 ± 4.1 | 25.6 ± 3.1 | 16.6 ± 0.9*** | 20.1 ± 5.0**,†† |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becerril, S.; Rodríguez, A.; Catalán, V.; Ramírez, B.; Unamuno, X.; Gómez-Ambrosi, J.; Frühbeck, G. iNOS Gene Ablation Prevents Liver Fibrosis in Leptin-Deficient ob/ob Mice. Genes 2019, 10, 184. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10030184
Becerril S, Rodríguez A, Catalán V, Ramírez B, Unamuno X, Gómez-Ambrosi J, Frühbeck G. iNOS Gene Ablation Prevents Liver Fibrosis in Leptin-Deficient ob/ob Mice. Genes. 2019; 10(3):184. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10030184
Chicago/Turabian StyleBecerril, Sara, Amaia Rodríguez, Victoria Catalán, Beatriz Ramírez, Xabier Unamuno, Javier Gómez-Ambrosi, and Gema Frühbeck. 2019. "iNOS Gene Ablation Prevents Liver Fibrosis in Leptin-Deficient ob/ob Mice" Genes 10, no. 3: 184. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10030184